Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(23):4536-4546. doi:10.7150/jca.26681 This issue Cite
Research Paper
The Combination of Omega-3 Stearidonic Acid and Docetaxel Enhances Cell Death over Docetaxel Alone in Human Prostate Cancer Cells
Mahmoud Mansour1 ![]() , Sabrina van Ginkel1, John C. Dennis1, Brandon Mason3, Isra Elhussin2, Kodye Abbott1, Satyanarayana R Pondugula1, Temesgen Samuel2, Edward Morrison1
, Sabrina van Ginkel1, John C. Dennis1, Brandon Mason3, Isra Elhussin2, Kodye Abbott1, Satyanarayana R Pondugula1, Temesgen Samuel2, Edward Morrison1
1. Department of Anatomy, Physiology, and Pharmacology, College of Veterinary Medicine, Auburn University, Auburn, AL;
2. Department of Pathobiology, College of Veterinary Medicine, Tuskegee University, Tuskegee, AL;
3. VCA Alameda East Veterinary Hospital, 9770 East Alameda Ave, Denver, CO 80247.
Received 2018-4-14; Accepted 2018-8-26; Published 2018-11-11
Abstract
Background: Docetaxel (DOC), or Taxotere, is an anthracycline antibiotic used to treat multiple types of cancer. It is a first-line chemotherapy treatment for patients with metastasized, hormone-resistant prostate cancer (PCa) or for patients with high-risk, localized PCa that could benefit from early chemotherapy treatment. Previously, we showed that stearidonic acid (SDA), an omega-3 fatty acid, enhances the cytotoxicity of doxorubicin (DOX) in human PCa cells. This observation suggests that PCa therapies using SDA and chemotherapeutic drugs in combination offer attractive possibilities for developing treatments that ameliorate toxic side effects of some commonly used chemotherapy drugs.
Objectives: We used androgen-resistant PC3 and DU 145 cells derived from human prostate cancer to quantify the effects of combined SDA and DOC on proliferation/viability and on the production of pro-apoptotic caspases 9 and 3. We also compared the effects of SDA with those of BAY, a pharmacological inhibitor of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-ĸB), in androgen-sensitive LNCaP cells. Finally, we qualitatively and quantitatively assessed the drug combination on androgen receptor (AR) and peroxisome proliferator-activated receptor gamma (PPARγ) expression in LNCaP and PC3 cells, respectively.
Methods: The half maximal inhibitory concentration (IC50) and combination indices of SDA and DOC in PC3 and DU 145 cells were determined using the MTT cell viability assay. To quantify the effects of SDA and BAY on NF-ĸB activity, we used luciferase reporter assays in LNCaP cells that were stably transduced with lentiviral vectors carrying NF-ĸB response element sequence upstream of the luciferase gene sequence. AR and PPARγ expression were assessed by western blotting and immunocytochemistry. We considered caspase 9 and 3 cleavage to be apoptosis markers and determined the drug combination effect on the extent of that cleavage by western blot analysis.
Results: The cytotoxic effects of DOC were synergistically enhanced by SDA when the two were added to DU145 and PC3 cell cultures. Combination index (CI) analyses based on the Chou-Talalay method and mass action law showed synergistic interaction with CI <1. SDA suppressed TNFα-induced NF-κB activity similarly to BAY. The SDA/DOC combination down regulated testosterone (T)-induced AR and troglitazone-induced PPARγ protein expression when compared to using the drugs singly. Similarly, the SDA/DOC combination induced caspase 9 and 3 production and cleavage suggesting apoptosis induction. Like our DOX studies, this work provides proof-of-concept for using SDA and DOC in combination to reduce the dose, and therefore the toxicity, of DOC and possibly increasing the survival benefit in DOC clinical translation studies.
Keywords: stearidonic acid, docetaxel, prostate cancer, drug combination, peroxisome proliferator-activated receptor gamma, nuclear factor kappa-light-chain-enhancer of activated B cells, testosterone, caspase 9, caspase 3.
Introduction
Prostate cancer treatment protocols include active surveillance, also called watchful waiting; hormone therapy with such compounds as luteinizing hormone-releasing hormone agonists; external or interstitial radiation therapy; cryotherapy, which involves in vivo freezing of cancer cells; chemotherapy with such drugs as DOC; and radical prostatectomy [1]. No single therapy is effective for many patients and treatment frequently involves combined therapies because the cancers develop resistance to the treatment(s). Despite the intense research effort, optimal treatment for Castration-Resistant Prostate Cancer (CRPC) with minimal side effects is lacking. High morbidity and mortality rates remain a significant challenge especially in older patients.
DOC, an injectable antimitotic drug used as a first line therapy in advanced CRPC, inhibits mitosis by binding microtubules. DOC is usually given in combination with prednisone or mitoxantrone for men with symptomatic CRPC [2-4]. Although patients experience significant survival and palliative benefits, DOC's dose-limiting side effects increase patients' anguish. These effects include hypersensitivity reactions, fluid retention, mucositis, neuropathy, myalgia, alopecia, nausea, nail changes, and vomiting [5]. The reduction of drug side effects and chemoresistance by combination therapy remains, therefore, an important research effort [6].
Previous studies showed the antitumor and health-promoting effects of fish derived (n-3) long-chain polyunsaturated fatty acids [7-9]. For example, treatment of PCa cells with eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) induces cell cycle arrest and apoptosis [10] and decreases prostate tumor growth in vivo [11]. The omega-3 fatty acid SDA (18:4 [n-3]), an EPA and DHA precursor, decreases the proliferation index and increases apoptosis in xenograft PCa models [8, 12]. Tumor suppression by EPA and DHA appears to act via modulation of the nuclear transcription factors androgen receptor (AR) [13], nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and peroxisome proliferator-activated receptor-γ (PPAR γ) [14].
SDA (18:4[n-3]) is present in plants of the Boraginaceae family (e.g. seed oils from echium and borage). Previous work showed potential health benefits of SDA and other omega-3 polyunsaturated fatty acids including inhibition of cancer cell growth, enhancement of tumor sensitivity to radiation or chemotherapy or reduction of side effects [8, 9, 15-17]. We showed that SDA in combination with DOX (or Adriamycin, an anthracycline antibiotic), a chemotherapeutic drug used in treatment of numerous cancers, increases cell death in PCa cell lines compared to treatment with DOX alone [17]. Here, we report that DOC toxicity to human PCa cells is enhanced when it is combined with SDA. This observation is encouraging especially because SDA, like EPA and DHA [18-20], also increases EPA accumulation in the heart and has lipid lowering effects [21, 22]. SDA's cardiovascular protective benefits and its mitigation of DOC's side effects may be exploited in CRPC treatment.
Materials and Methods
Cell culture and reagents
Human prostate adenocarcinoma cell lines, PC3, DU145, LNCaP, immortalized normal prostate epithelial RWPE-1 cells, and 3-(4, 5-dimethylthiazol-2-yl)-2-5-diphnyltetrazolium bromide (MTT) cell viability kits were obtained from the American Type Culture Collection (ATCC, Manassas, VA). DMEM and RPMI-1640 with HEPES and L-Glutamine (with and without phenol red) and phosphate buffered saline (PBS) without calcium and magnesium were purchased from Lonza (Walkersville, MD). Keratinocyte-SFM defined media with growth promoting supplement for propagation of RWPE-1 was purchased from Gibco Life Technologies (Carlsbad, CA). Advantage and charcoal-treated fetal calf serums (FCS) was purchased from Atlanta Biologicals (Lawrenceville, GA). The PPARγ agonist Troglitazone and SDA (#90320) were obtained from Cayman Chemical (Ann Arbor, MI). Trypsin-EDTA, penicillin-streptomycin antibiotics, and cell culture supplies were purchased from VWR International, LCC (Atlanta, GA). The following were purchased from Sigma (St Louis, MO): staurosporine (S6942), testosterone (T1500), DMSO (D8418), DOC (01885), Bay 11-7280 (B5556), and TNFα (SRP2102).Western blotting primary antibodies obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX) were: anti-AR (Sc-7305), anti-PPARγ (Sc-7273), and anti-caspase 3 (Sc-7272). Anti-caspase 9 (ab69514) and goat anti-rabbit IgG H&L (HRP) (ab6721) were purchased from Abcam (Cambridge, MA). Goat HRP- conjugated anti-mouse IgG was purchased from VWR (AC2115). DyLight 488-conjugated goat anti-rabbit IgG (Thermo Scientific, Rockford, IL) and Alexa Flour 568-conjugated goat anti-mouse IgG (Invitrogen-Life technologies, Grand Island, NY) were used to visualize primary binding in immunocytochemistry assays.
Cell maintenance
LNCaP, PC3, and DU145 cells were grown in DMEM or RPMI-1640 with HEPES and L-Glutamine media supplemented with 10% (v/v) FCS and 1% (v/v) streptomycin-penicillin (Life Technologies, Grand Island, NY). RWPE-1 cells were maintained in keratinocyte-SFM media containing bovine pituitary extract and human recombinant epidermal growth factor. Cell cultures were maintained in T-75 vented-cap tissue culture flasks at 37°C in a humidified incubator with a 5% CO2. For assays, cells were suspended with 0.25% Trypsin and counted using a TC-10 automated cell counter (Bio-Rad, Hercules, CA).
MTT cell viability assay
Cells in logarithmic-phase growth were trypsinized and counted using a TC-10 automated cell counter. The effect of drug treatments on viability and/or proliferation was assessed using an MTT cell proliferation assay kit (ATCC® 30-1010K). Cells were seeded at 2 x104 cells per well in 96-well plates and allowed to adhere overnight. The next day, the media were replaced with fresh media containing the appropriate drugs and following 72 hours incubation, 10 μl yellow tetrazolium MTT (5mg/ml) was added to each well. Plates were then incubated for an additional 3-4 hours at 370C. Intracellular purple formazan crystals formed by metabolically active cells, are dissolved by the addition of 100 μl/well of ready-to-use detergent buffer (10% SDS in 0.01N HCl) over 5-6 hours at room temperature in dark. Color absorbance (OD) was determined at 570 nm using SpectraMax-Plus-384 microplate reader (Molecular Devices, Sunnyvale, CA). Each experiment was repeated three times.
Drug combination
DOC or SDA was dissolved in ethanol or DMSO and further diluted in charcoal-treated, phenol red free RPMI-1640 media to desired concentrations. Drug mixtures for calculation of CI values were based on the median effect analysis method [23]. Ten two-fold serial dilutions of working concentrations were prepared in charcoal-treated RPMI-1640 with five concentrations above and five concentrations below the calculated IC50 for each drug in each cell line. These ratios corresponded to 0.03, 0.06, 0.12, 0.25, 0.5, 1, 2, 4, 8, 16, 32 times the IC50 for each drug in each cell line. SDA/DOC mixtures were made using those ratios for each drug. Each concentration was tested in 8 replicas and the results were confirmed in at least three independent experiments.
NF-ĸB promoter activity assays
To test the effect of SDA and BAY on NF-κB activity, androgen-sensitive LNCaP cells were transduced with lentiviral vectors carrying NF-κB response element sequence upstream of the luciferase gene sequence. Cells were treated with vehicle, SDA, or TNFα alone or with TNFα in combination with SDA or Bay 11-7280. Firefly luciferase activity was measured using the Neolite Luciferase Assay System (PerkinElmer, Downers Grove, IL).
Invasion assay
PC3 cell migration was performed using 12-well Matrigel invasion chambers (Corning® BioCoatTM Matrigel® Invasion Chamber, Discovery Labware, Inc., Bedford, MA). 4.5 X 104 cells in 500 µl of DMEM containing 1% antibiotic with or without 50 µM SDA (Cayman Chemical, Ann Arbor, MI) were added to each of the upper chambers. The lower chambers contained 750 µl of DMEM media with 10% fetal bovine serum (FBS) and 1% antibiotic. The chambers were incubated at 37°C and 5% CO2 for 48 hours. The non-migrating cells were removed from the upper chamber with a cotton swab. The migrated cells in the Matrigel on the lower part of the chamber were fixed with 4% formaldehyde, permeabilized with 100% methanol, and stained with 0.5% crystal violet. Stained cells in four fields per Matrigel membrane were counted at 20X magnification.
Immunocytochemistry (ICC)
Cells were grown on Lab-Tek II 8-well chamber slides (VWR, Suwanee, GA) for 24 hours at 3 x105 cells per well. Appropriate treatments were added and the next day cells were rinsed with phosphate buffered saline (PBS) and fixed in buffered 4% paraformaldehyde (30 minutes). After fixation, cells were rinsed in PBS and treated with absolute methanol for 10 minutes. Following equilibration in PBS, the cells were incubated for 10 minutes in blocking solution that was 5% normal goat serum, 2% bovine serum albumin (BSA) in PBS, pH 7.3 then incubated with primary antibody appropriately diluted in blocking solution overnight at room temperature. In each experiment, one well for each treatment group and cell line received no primary antibody to detect non-specific secondary antibody binding. Next day, the cells were washed and treated with secondary antibody appropriately diluted in blocker at room temperature for one hour. The slides were washed and mounted with Vectashield (Vector Laboratories, Burlingame, CA) that contained DAPI (Vector laboratories) to label cell nuclei. Slides were sealed with clear nail polish and examined with a Nikon Eclipse C1 2000-E confocal microscope. Z-stacks were acquired and rendered with Nikon EZ-C1 3.91 software. To facilitate signal intensity comparisons between treatment groups, images were acquired and processed as follows: the gain for each laser was adjusted to the positive control treatment well, which was scanned first. That value was then used for each z-stack acquisition for each treatment. The control well z-stack was rendered, and the saturation and dark levels were adjusted until a good quality image was obtained. Those values were used for subsequently rendered images of each treatment group.
Western blot analysis
For detection of initiator caspase 9 and effector caspase 3 production by western blotting, 5 x 105 PC3 cells were grown in 6 well plate and allowed to attach overnight. Next day, the cells were treated with the appropriate drug concentrations for 6 to 8 hours. Cells assayed for PPARγ and AR protein expression were treated for 48 hours. Attached and suspended cells were collected and lysed on ice with cold RIPA lysis buffer (Sigma, R0278) for 30 minutes and western blotting was performed as previously described [24]. Aliquots containing 25-µg of protein from treated or untreated cells were run on 10% precast SDS-PAGE gels (Bio-Rad) then transferred to nitrocellulose membranes using the Bio-Rad protocol. Membranes were blocked with 5% non-fat dry milk in TBS-T (1% Tween-20) buffer, probed with primary antibodies diluted appropriately in block buffer, then washed in TBS-T. Membranes were then incubated with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies and signal was detected using the enhanced chemiluminescence system (E2400, Denville). Relative protein amounts were determined with Un-Scan-It software (Silk Scientific, Inc., Orem, UT, USA).
Data analyses
Data from luciferase and MTT viability assays were expressed as the mean ± SE. Data from the transfection assays were analyzed by one-way ANOVA followed by Dunnett's post hoc test (Graph Pad Prism 5.0, San Diego, CA). P<0.05 was considered statistically significant. MTT data were analyzed by GraphPad Prism® to produce dose-response curves for SDA and DOC using nonlinear analysis. Combination index (CI) values were determined by the third generation "CompuSyn" software written by Nick Mart of MIT using the median-effect method [25]. The CI index was calculated using the following formula:
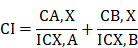
Where CA, X and CB, X are the concentrations of DOC and SDA used in combination to achieve X% drug effect. The ICX, A and ICX, B are the concentrations for single drugs (DOC or SDA) that achieve the same effect. Synergy is defined as CI <1; additivity is defined as CI = 1; and antagonism is defined as CI> 1.
To calculate drug effect, the mean OD values for each drug concentration was subtracted from the mean OD values of cells treated with vehicle and the resulting fractions (between 0-100%) were plotted against drug concentrations in logarithmic scale.
Results
Determination of IC50 for SDA and DOC in LNCaP, PC3, and DU-145 cells
To identify the SDA:DOC interaction as synergism, additive effect, or antagonism, we calculated the dose-effect curves for SDA and DOC applied singly to androgen-sensitive LNCaP and androgen-resistant PC3 and DU145 cells. This step generates the IC50--the drug concentration causing 50% cell growth inhibition for each drug--that is requisite for the CI calculation. CI<1 indicates synergism, CI=1 indicates additive effect, and CI>1 indicates antagonism.
Ten two-fold serial dilutions of each drug were used in the in vitro MTT experiments and data showed that SDA or DOC treatments inhibited proliferation of LNCaP, PC3 and DU145 cells with variable IC50 values. SDA IC50s were 556, 110, 150 µM in LNCaP, PC3 and DU145, respectively. DOC values were 296, 117, 507 nM respectively for the three cell lines (Fig. 1). Both drugs inhibited cell viability/proliferation of PC3 and DU145 cells to a greater degree compared to LNCaP cells.
Calculation of IC50 for SDA and DOC using MTT-dose response curves expressed as the log of inhibition vs viability/proliferation of LNCaP, PC3, and DU 145 cells. Dilutions of SDA or DOC are two-fold. SDA IC50s were 556.2, 110.6, 150 µM and DOC IC50s were 296.4, 117, 507.6 nM for LNCaP, PC3 and DU145 cells, respectively.
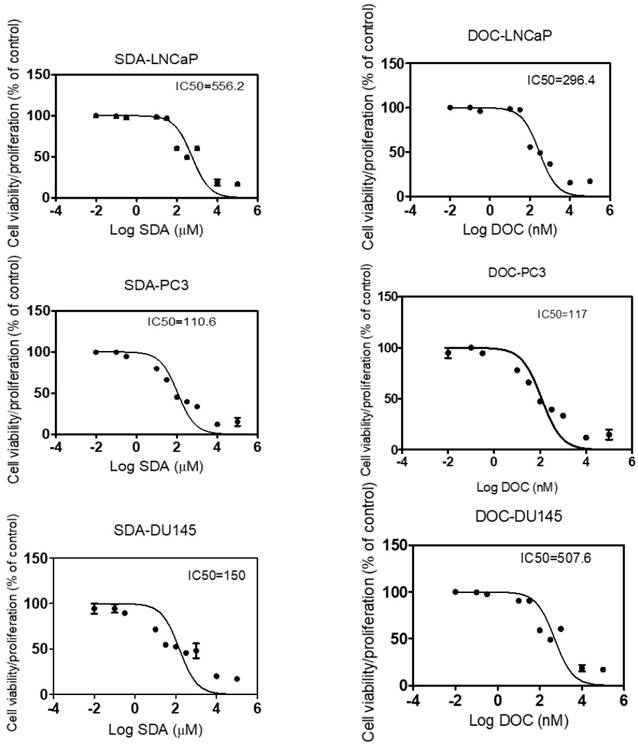
Nontoxic concentrations of SDA and DOC did not affect proliferation of RWPE-1 prostate epithelial cells
To determine the anti-proliferative effect of SDA and DOC on cells derived from normal prostate epithelium, we exposed RWPE-1 cells to concentrations of SDA or DOC alone and in combination. Concentrations of SDA and/or DOC were derived from the IC50 values calculated for each cell line and the two were mixed in constant ratios of DOC: SDA. For each cell line these values were: LNCaP (1:1878), PC3 (1:940), and DU145 PC3 (1:295). Treatment of RWPE-1 with DOC plus SDA did not significantly affect cell viability. SDA in concentrations ≤ 200 μM had no significant toxic effects on RWPE-1 when used alone. DOC or SDA alone caused significant cell death at 8 nM and 400 µM, respectively (Fig. 2).
MTT RWPE-1 proliferation results of three experiments. Nontoxic SDA concentrations have no significant effect on RWPE-1 viability when used alone or combined with DOC in constant ratios of DOC: SDA in LNCaP (1:1878), PC3 (1:295), and DU145 (1:940) cells. Significant DOC toxicity occurred at ≥ 8 nM and SDA toxicity was observed at ≥ 400 µM.
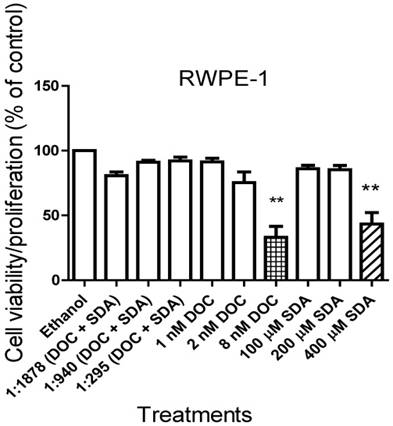
SDA combined with low doses of DOC synergistically inhibited androgen-independent PC3 cell growth
We conducted cell viability assays on PC3 cells using the IC50 concentrations for each drug singly or in combination. The IC50 concentration of SDA or DOC caused approximately 50% cell death whereas SDA combined with DOC at the IC50 concentration for each caused more than 75% cell death (Fig. 3).
MTT cell proliferation assays showed that SDA potentiated DOC inhibition of PC3 cell growth. SDA synergistically enhanced DOC's cytotoxicity to PC3 cells when the two drugs were combined at the IC50 concentration equivalent for each drug.
Combination of SDA and DOC synergistically caused death of androgen-independent PC3 and DU 145 prostate cancer cells
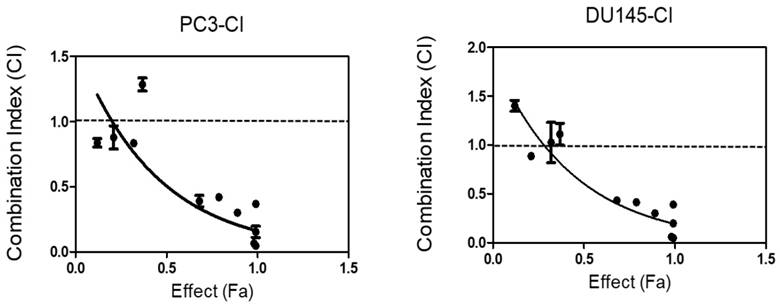
To determine if SDA augments DOC-induced cell death in a synergistic or additive manner, we performed dose-effect MTT experiments using androgen-independent PC3 and DU 145 cells. Isobologram and CI analyses showed that the CI for drug combinations was <1 in both cell lines (Table 1 and Fig 4). These observations demonstrated that the combination of SDA and DOC in low doses caused significantly greater cell growth inhibition compared with either drug alone.
CI values for SDA plus DOC in androgen-resistant PC3 and DU145 cells at ED50, ED75, ED90 and ED95.
| Combination index (CI) at 50-95% effect | |||||
|---|---|---|---|---|---|
| Cell line | Ratio (IC50a/IC50b) | ED50 | ED75 | ED90 | ED95 |
| PC3 | 1:950 | 0.43829 | 0.29954 | 0.28227 | 0.29464 |
| DU145 | 1:295 | 0.65394 | 0.47177 | 0.22676 | 0.10819 |
CI < 1 indicates synergism; CI > 1 indicates antagonism; and CI = 1 indicates additive effect. CI is obtained by dividing SDA IC50a/ DOC IC50b.
CI analysis of SDA and DOC in androgen-independent PC3 and DU145 cells based on the Chou and Talalay combination index theorem. Stimulation at Fa ˂ 0.5 showed data points with CI >1, which indicate antagonism. This effect is less relevant to therapy than high Fa ˃ 0.5 since cancer cell death in small numbers is less useful.
SDA enhances the anti-apoptotic activity of DOC in PC3 cells
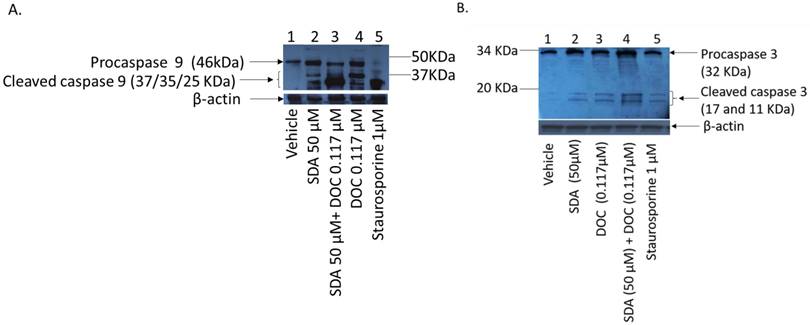
DOC induces apoptosis in PCa cells via caspase dependent and independent mechanisms [26-28]. We asked if SDA affected apoptosis induction in PC3 cells and we used staurosporine (sigma, S6942) as positive controls. Staurosporine, an alkaloid extracted from the bacterium Streptomyces staurosporeus, induces apoptosis through both caspase-dependent and caspase-independent mechanisms [29]. Treatment of PC3 cells with SDA induces cleavage of procaspase 9 and activation of initiator caspase 9 (Fig 5-a), which is a pivotal step in the cascade leading to the initiator caspase 3 activation (Fig 5-b) and induction of apoptosis. The combination of SDA with DOC enhances that cleavage.
(A) Western blot analysis of caspase cleavage in PC3 cells. (A) Ethanol vehicle (lane1); SDA at 50 μM (lane 2); combination of 50 μM SDA and 0.117μM DOC (lane 3); 0.117μM DOC (lane 4); Staurosporine (lane 5, positive control); Both SDA and DOC induced pro-caspase 9 cleavage (lanes 2 and 4) but the combination of SDA and DOC augmented procaspase 9 cleavage (lane 3). (B) Cleavage of procaspase 3 and activation of initiator caspase 3. Ethanol vehicle (lane1), SDA or DOC alone (lanes 2 and 3) and SDA and DOC in combination (lane 4). Staurosporine (lane 5, positive control).
SDA inhibited PC3 human cell invasion/migration in vitro
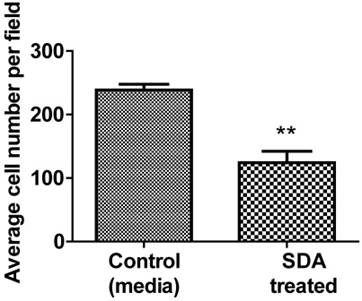
We used Matrigel invasion chambers to determine the effect of SDA on PC3 cell invasion. Following treatment with 50 µM SDA (1/2 of ICD50), PC3 cell migration was significantly inhibited over the 48-hour incubation period (Fig. 6).
Representative data of Matrigel invasion chamber assay. Pretreatment of PC3 androgen-independent human PCa cells with 50 μM SDA caused 52% invasion reduction compared to the control (P<0.0048).
SDA inhibited TNFα-induced NF-ĸB upregulation
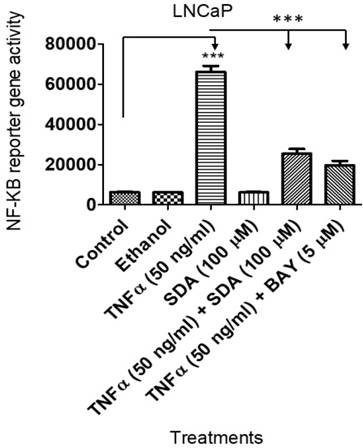
We showed previously that SDA suppresses NF-ĸB activity in androgen dependent LNCaP cells [17] . Here, we wanted to compare SDA effects with those of Bay 11-7082 (Bay), a pharmacological inhibitor of NF-ĸB. To that end, we transduced LNCaP cells with a lentiviral vector carrying the NF-ĸB response element and the firefly luciferase reporter gene. Experimental cells received TNFα with SDA or with Bay. Both SDA and Bay significantly reduced the reporter gene activity (Fig. 7). Twelve-hours after the treatment, NF-ĸB activated reporter gene activity was determined by measuring firefly luciferase activity, (p<0.001).
Luciferase assay showing that SDA inhibits TNFα activation/induction of NF-κB in LNCaP cells. Treatment groups received media, the carrier ethanol, TNFα, SDA, TNFα plus SDA, or TNFα plus BAY 11-7082.
SDA plus DOC synergistically down regulate troglitazone-induced PPARγ expression in PC3 cells
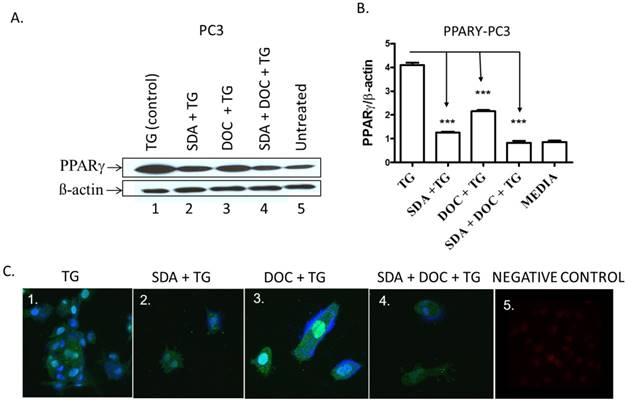
One pathway by which EPA and DHA omega-3 fatty acids from fish induce apoptosis in cancer cells is via PPARγ activation [30, 31]. We hypothesized that SDA alone or in combination with DOC could activate PPARγ in PC3 cells that express high PPARγ levels compared to normal prostate epithelial cells [24]. TG-induced PPARγ was downregulated by either SDA or DOC and to a greater degree by SDA and DOC in combination as shown quantitatively by western blotting (Fig.8 A and B) and qualitatively by ICC (Fig. 8C).
(A-C) SDA and DOC synergistically inhibited TG-induced PPARγ protein expression in PC3 cells. (A) PC3 cells were treated with TG PPARγ agonist (lane 1); SDA plus TG (lane 2); DOC plus TG (lane 3); SDA plus DOC plus TG (lane 4); media (lane 5). (B) Quantitative analysis of western blots (n=3) in A. (C) Qualitative analysis of PPARγ by ICC of treatments in (A). Nuclei were labeled with DAPI. Positive control showing nuclear PPARγ signal (panel 1); SDA+TG (panel 2); DOC+TG (panel 3); SDA+DOC+TG (panel 4); no anti-PPARγ antibody (panel 5).These data showed an inhibition of TG-induced PPARγ by SDA or DOC and as well as the two in combination.
SDA plus DOC synergistically down regulates testosterone-induced AR expression in LNCaP cells
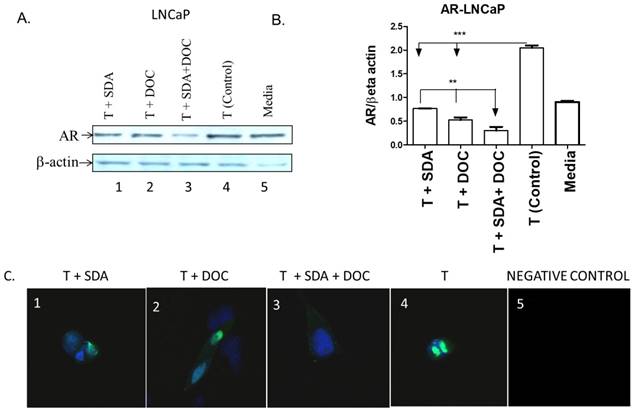
Mechanistic studies showed that fish-sourced omega-3 fatty acids inhibit the growth of androgen-dependent LNCaP cells by promoting the degradation of AR [32]. We hypothesized that SDA would counter AR protein expression induced by treating LNCaP cells with T. We selected LNCaP cells to address this hypothesis because we [24] and others previously showed that LNCaP cells, in contrast to PC3 cells, express high AR levels. We used western blotting to quantitatively assess the effect of SDA and DOC (Fig. 9A and B) and to qualitatively assess that effect by ICC (Fig. 9C). Both SDA and DOC significantly downregulated T-induced AR protein expression and the two in combination down regulated AR expression significantly more than either compound alone.
SDA plus DOC down regulated T-induced AR protein expression in LNCaP cells. (A) LNCaP cells were treated with T plus SDA (lane 1); T plus DOC (lane 2); T plus SDA plus DOC (lane 3); T (lane 4) or media (lane 5). (B) Quantitative analysis of western blots (n=3). (C) Qualitative analysis of AR by immunocytochemistry of treatments in (A). LNCaP cells were treated with drugs and labeled for AR. T+SDA (panel 1); T+ DOC (panel 2); T+SDA+DOC (panel 3); positive control showing strong nuclear AR localization (panel 4); no anti-AR antibody (panel 5). Panels 1-3 show reduced nuclear AR signal suggesting inhibition of T-induced AR by SDA, DOC, and the two in combination.
Discussion
We treated cells from four human prostate derived cells lines with SDA and DOC, singly or in combination, to document the effects those drugs have on the several cell types. SDA or SDA plus DOC in non-toxic concentrations have minimal effects on RWPE-1 cells, which are derived from normal human prostate epithelial cells. SDA and DOC together have synergistic effects on LNCaP, PC3, and DU 145 cells as determined by the Chou-Taladay method [33]. SDA and DOC induce apoptosis initiator caspase 9 and effector caspase 3. SDA and DOC in combination suppress troglitazone-induced PPARγ in PC3 cells and the testosterone-induced androgen receptor in LNCaP cells. SDA alone suppresses PC3 cell invasion and migration and block TNFα-induced NF-ĸB activity in stably transduced LNCaP cells. The toxic effect of SDA on PC3 and DU 145 cells and lack of significant SDA toxicity to normal RWPE-1 cells suggest that SDA produces cancer specific anti-proliferative effects.
The mechanism by which SDA induces cell death in the PCa cell lines studied here is unclear. A likely mechanism is interference with NF-ĸB, a constitutively active transcription factor in many cancers including PCa [34, 35]. We therefore asked what effect SDA had on NF-ĸB activity. The rationale for the comparison stems from the observation that NF-ĸB inhibition by the apoptosis-inducing antitumor drug nitrosylcobalamin (NO-Cbl) potentiates the anticancer effect of Apo2L/TRAIL, which is a TNFrelated apoptosisinducing ligand that triggers fast apoptosis in many cancer cell types but not in normal cells [36]. Further, some chemotherapeutic agents, including DOC, induce NF-ĸB activity in PCa cells and contribute to DOC-chemotherapeutic resistance (28). A recent study showed that down regulation of NF-ĸB by plant-derived compounds similar to SDA such as genistein, an isoflavone in soybeans, potentiate the chemotherapeutic effects of DOC in several cancer-derived cells, including androgen-resistant PC3 cells [37]. Our data show that SDA compares favorably with BAY, a pharmacological inhibitor of NF-ĸB. This finding provides a rationale for using SDA in combination therapy for prostate--and possibly other--cancer(s) that express high NF-ĸB levels.
NF-ĸB signaling is a well-established conduit for resistance to apoptosis and for propagation of angiogenesis and invasiveness in androgen-resistant PCa cells [38, 39]. Inhibition of NF-ĸB by SDA may have contributed to the decreased cell migration/ invasion seen in the androgen resistant PC3 cells. The potentiation of cell death by SDA and DOC in combination may also be mediated by SDA-induced NF-ĸB inhibition; direct induction of apoptosis; and inhibition of P-gp by SDA [15]. P-gp is an efflux transporter of toxic compounds from cells and by inhibiting P-gp, SDA would cause increased DOC accumulation within cancer cells and would thereby potentiate DOC's antitumor activity.
The Isobologram and CI analyses showed that the CI for drug combinations was <1 in both the PC3 and DU 145 cell lines except for one combination point in PC3 cells and 3 points in DU 145 cells. These data showed CI slightly higher than 1 but at Fa ˂ 0.5. As indicated in the legend for Fig. 4, this effect is less relevant to therapy than high Fa, i.e. ˃ 0.5 since cancer cell death in small numbers is less useful in therapy. Nonetheless, these data should be interpreted with caution because the results are from in vitro studies.
The synergistic effects of SDA and DOC in cell killing and the decrease in PC3 cell migration by SDA cannot be entirely explained by SDA's inhibition of NF-ĸB. It is conceivable that SDA treatment blunted other tumor promoting factors associated with cell migration or the development of resistance to DOC. For example, we reported previously that SDA treated canine lymphoid tumor cells show enhanced sensitivity to vincristine and DOX, which are widely used chemotherapeutic drugs for treatment of lymphoma and other cancers [15]. Further, the decreased cell migration/invasion of SDA treated PC3 cells likely involves inhibiting genes responsible for initiating cell migration and invasiveness like E-cadherin. This possibility requires further investigation.
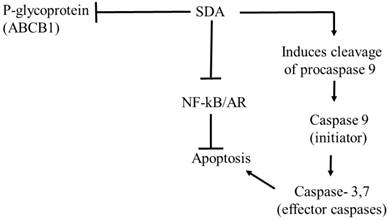
The piscine omega-3 fatty acid DHA induces apoptosis by activating PPARγ [40, 41]. We therefore hypothesized that SDA likewise induces cell death via PPARγ and subsequent apoptosis [42]. Contradicting the hypothesis, our data show an inhibition of TG-induced PPARγ by SDA treatment. Supporting the accuracy of that observation is the report that SDA does not activate PPARγ in transfected hepatocyte cells [17] . Based on these observations, we presume that SDA reverses the suboptimal effect of, and resistance to, DOC by several mechanisms that include potentiation of apoptosis, inhibition of tumor promoting nuclear factors NF-κB and AR, and increased expression of the P-gp efflux transporter (see Fig. 10).
Proposed pathway of antitumor effects of SDA in PCa cells. The antitumor effect of SDA seems to be mediated by several mechanisms that include: (1) inhibition of NF-κB and AR nuclear factors that promote PCa cell growth and inhibit apoptosis; (2) production of apoptosis initiator caspase 9 and subsequent activation of the effector caspase 3, which suggests apoptosis through the intrinsic mitochondrial pathway; (3) inhibition of P-glycoprotein (P-gp) or ABCB1 drug efflux transporter encoded by the MDR1 gene causing increased intracellular accumulation of DOC. The effect of SDA on P-gp is supported by our group's previous study [15] that showed inhibition of P-gp in HEK293 cells that stably expressed P-gp.
Our data also show that SDA treatment down regulates AR protein expression in LNCaP cells treated with T, an AR agonist. This finding agrees with previous reports showing the DHA-induced inhibition of hormone-dependent PCa cell growth through degradation of the AR protein [32]. The greater downregulation of AR receptor protein by SDA-DOC combination compared to each drug alone suggests utility of this combination for both androgen deprivation therapy and for chemotherapy. For example, recent data from the National Institutes of Health showed that the median survival in men given androgen deprivation therapy (ADT) plus DOC was 13.6 months longer than with ADT alone with 85 PCa deaths in the combination group compared to 114 in the ADT-alone group [43].
Overall, the capacity of SDA to down regulate three nuclear factors (NF-κB, PPARγ, and AR) in this study is noteworthy and deserves further study. In a variety of human cancers including PCa, the major perpetrators mediating development of chemoresistance are upregulation of NF-ĸB [44, 45], continued signaling from the AR [46], and multidrug resistance genes [47]. Previously, our group showed that SDA-mediated chemosensitization of VCR and DOX was associated with SDA inhibition of P-gp, an ATP-binding cassette transporter that functions as cell protector by extruding toxins and xenobiotics from cells. Inhibiting P-gp efflux activity by SDA will lead to enhanced accumulation of the chemotherapeutic drug(s) and consequently increased anticancer efficacy.
In summary, findings from this study showed that the combination of SDA, a plant-based omega-3 dietary fatty acid, and DOC has synergistic and differential killing effect on human PCa-derived cells. The cytotoxic effects of SDA or SDA plus DOC combinations on cells derived from normal human prostate epithelial cells were minimal. These data suggest that SDA has cancer dependent anti-proliferative effects and could be combined with DOC to reduce the dose of DOC necessary to produce significant human PCa cell death. Dietary supplementation with fish- or plant-based omega-3 fatty acids alone is unlikely to treat or prevent cancer [48]. However, the combination of SDA and chemotherapy could lower the toxicity of chemotherapy by allowing lower doses of toxic chemotherapeutic drugs. The use of a dietary supplement to reduce the toxicity of chemotherapy is deeply appealing because of its potentially wide applicability to common cancers like breast, ovarian, colon, lung cancer, and PCa where chemotherapy is the mainstay treatment. Our data warrant further study in an animal model to investigate the anti-proliferative effects of SDA, both alone and in combination with DOC, on prostate tumors.
Acknowledgements
This work is supported by funding through Auburn University Research Initiative in Cancer (AURIC) and Auburn University Intramural Grants Program (IGP) to M. Mansour.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Middleton RG, Thompson IM, Austenfeld MS, Cooner WH, Correa RJ, Gibbons RP. et al. Prostate cancer clinical guidelines panel summary report on the management of clinically localized prostate cancer. The American Urological Association. J Urol. 1995;154:2144-8
2. Petrylak DP, Tangen CM, Hussain MH, Lara PN Jr, Jones JA, Taplin ME. et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513-20
3. Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN. et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502-12
4. Berthold DR, Pond GR, Soban F, de Wit R, Eisenberger M, Tannock IF. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. J Clin Oncol. 2008;26:242-5
5. Pronk LC, Stoter G, Verweij J. Docetaxel (Taxotere): single agent activity, development of combination treatment and reducing side-effects. Cancer Treat Rev. 1995;21:463-78
6. Lee BY, Hochgräfe F, Lin H-M, Castillo L, Wu J, Raftery MJ. et al. Phosphoproteomic profiling identifies focal adhesion kinase as a mediator of docetaxel resistance in castrate-resistant prostate cancer. Mol Cancer Ther. 2014;13:190-201
7. Berquin IM, Edwards IJ, Chen YQ. Multi-targeted therapy of cancer by omega-3 fatty acids. Cancer Lett. 2008;269:363-77
8. Whelan J. Dietary stearidonic acid Is a long chain (n-3) polyunsaturated fatty acid with potential health benefits. J Nutr. 2009;139:5-10
9. Hardman WE. (n-3) Fatty acids and cancer therapy. J Nutr. 2004;134:3427S-30S
10. Siddiqui RA, Shaikh SR, Sech LA, Yount HR, Stillwell W, Zaloga GP. Omega 3-fatty acids: health benefits and cellular mechanisms of action. Mini Rev Med Chem. 2004;4:859-71
11. Berquin IM, Min Y, Wu R, Wu J, Perry D, Cline JM. et al. Modulation of prostate cancer genetic risk by omega-3 and omega-6 fatty acids. J Clin Invest. 2007;117:1866-75
12. Kelavkar UP, Hutzley J, Dhir R, Kim P, Allen KG, McHugh K. Prostate tumor growth and recurrence can be modulated by the omega-6:omega-3 ratio in diet: athymic mouse xenograft model simulating radical prostatectomy. Neoplasia. 2006;8:112-24
13. Friedrichs W, Ruparel SB, Marciniak RA, deGraffenried L. Omega-3 fatty acid inhibition of prostate cancer progression to hormone independence is associated with suppression of mTOR signaling and androgen receptor expression. Nutr Cancer. 2011;63:771-7
14. Calder PC. Mechanisms of action of (n-3) fatty acids. J Nutr. 2012;142:592S-9S
15. Pondugula SR, Ferniany G, Ashraf F, Abbott KL, Smith BF, Coleman ES. et al. Stearidonic acid, a plant-based dietary fatty acid, enhances the chemosensitivity of canine lymphoid tumor cells. Biochem Biophys Res Commun. 2015;460:1002-7
16. Hardman WE, Moyer MP, Cameron IL. Consumption of an omega-3 fatty acids product, INCELL AAFA™, reduced side-effects of CPT-11 (irinotecan) in mice. Br J Cancer. 2002;86:983
17. Trebelhorn C, Dennis JC, Pondugula S, Samuel T, Coleman E, Flannery P. et al. Plant-based omega-3 stearidonic acid (SDA) enhances antitumor activity of doxorubicin (DOX) in human prostate cancer cell lines. J Cancer Res Ther. 2014;2:132-43
18. Harris WS, Miller M, Tighe AP, Davidson MH, Schaefer EJ. Omega-3 fatty acids and coronary heart disease risk: clinical and mechanistic perspectives. Atherosclerosis. 2008;197:12-24
19. von Schacky C, Harris WS. Cardiovascular benefits of omega-3 fatty acids. Cardiovasc Res. 2007;73:310-5
20. Hansen SN, Harris WS. New evidence for the cardiovascular benefits of long chain omega-3 fatty acids. Curr Atheroscler Rep. 2007;9:434-40
21. Harris WS. Expert opinion: omega-3 fatty acids and bleeding-cause for concern? Am J Cardiol. 2007;99:44C-6C
22. Harris WS, DiRienzo MA, Sands SA, George C, Jones PG, Eapen AK. Stearidonic acid increases the red blood cell and heart eicosapentaenoic acid content in dogs. Lipids. 2007;42:325-33
23. Chou T-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010;70:440-6
24. Mansour M, Schwartz D, Judd R, Akingbemi B, Braden T, Morrison E. et al. Thiazolidinediones/PPARgamma agonists and fatty acid synthase inhibitors as an experimental combination therapy for prostate cancer. Int J Oncol. 2011;38:537-46
25. Chou T-C, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Biol Regul. 1984;22:27-55
26. Muenchen HJ, Poncza PJ, Pienta KJ. Different docetaxel-induced apoptotic pathways are present in prostate cancer cell lines LNCaP and PC-3. Urology. 2001;57:366-70
27. Mediavilla-Varela M, Pacheco FJ, Almaguel F, Perez J, Sahakian E, Daniels TR. et al. Docetaxel-induced prostate cancer cell death involves concomitant activation of caspase and lysosomal pathways and is attenuated by LEDGF/p75. Mol Cancer. 2009;8:68
28. Fabbri F, Amadori D, Carloni S, Brigliadori G, Tesei A, Ulivi P. et al. Mitotic catastrophe and apoptosis induced by docetaxel in hormone-refractory prostate cancer cells. J Cell Physiol. 2008;217:494-501
29. Belmokhtar CA, Hillion J, Segal-Bendirdjian E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene. 2001;20:3354-62
30. Edwards IJ, O'Flaherty JT. Omega-3 fatty acids and PPARgamma in cancer. PPAR Res. 2008;2008:358052
31. Sun H, Berquin IM, Owens RT, O'Flaherty JT, Edwards IJ. Peroxisome Proliferator-activated Receptor γ-mediated up-regulation of syndecan-1 by n-3 fatty acids promotes apoptosis of human breast cancer cells. Cancer Res. 2008;68:2912-9
32. Hu Z, Qi H, Zhang R, Zhang K, Shi Z, Chang Y. et al. Docosahexaenoic acid inhibits the growth of hormone-dependent prostate cancer cells by promoting the degradation of the androgen receptor. Mol Med Rep. 2015;12:3769-74
33. Chou T-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621-81
34. Chaturvedi MM, Sung B, Yadav VR, Kannappan R, Aggarwal BB. NF-kappaB addiction and its role in cancer: 'one size does not fit all'. Oncogene. 2011;30:1615-30
35. Jin R SJ, Edwards JR, DeGraff DJ, Lee C, Park SI, Matusik RJ. Activation of NF-kappa B signaling promotes growth of prostate cancer cells in bone. PLoS ONE. 2013;8:e60983
36. Chawla-Sarkar M, Bauer JA, Lupica JA, Morrison BH, Tang Z, Oates RK. et al. Suppression of NF-κB survival signaling by nitrosylcobalamin sensitizes neoplasms to the anti-tumor effects of Apo2L/TRAIL. J Biol Chem. 2003;278:39461-9
37. Li Y, Ahmed F, Ali S, Philip PA, Kucuk O, Sarkar FH. Inactivation of nuclear factor κB by soy isoflavone genistein contributes to increased apoptosis induced by chemotherapeutic agents in human cancer cells. Cancer Res. 2005;65:6934-42
38. Karin M. Nuclear factor-[kappa]B in cancer development and progression. Nature. 2006;441:431-6
39. Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6:203-8
40. Edwards IJ, Sun H, Hu Y, Berquin IM, O'Flaherty JT, Cline JM. et al. In vivo and in vitro regulation of syndecan 1 in prostate cells by n-3 polyunsaturated fatty acids. J Biol Chem. 2008;283:18441-9
41. D'Eliseo D, Velotti F. Omega-3 fatty acids and cancer cell cytotoxicity: Implications for multi-targeted cancer therapy. J Clin Med. 2016;5:15
42. Tachibana K, Yamasaki D, Ishimoto K, Doi T. The role of PPARs in cancer. PPAR Res. 2008;2008:102737
43. Sweeney CJ, Chen Y-H, Carducci M, Liu G, Jarrard DF, Eisenberger M. et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N Engl J Med. 2015;373:737-46
44. Yeh PY, Chuang S-E, Yeh K-H, Song YC, Cheng A-L. Involvement of nuclear transcription factor-κB in low-dose doxorubicin-induced drug resistance of cervical carcinoma cells. Biochem Pharmacol. 2003;66:25-33
45. Yeh PY, Chuang S-E, Yeh K-H, Song YC, Ea C-K, Cheng A-L. Increase of the resistance of human cervical carcinoma cells to cisplatin by inhibition of the MEK to ERK signaling pathway partly via enhancement of anticancer drug-induced NFκB activation. Biochem Pharmacol. 2002;63:1423-30
46. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nature Reviews Clinical Oncology. 2017
47. Liem AA, Chamberlain MP, Wolf CR, Thompson AM. The role of signal transduction in cancer treatment and drug resistance. Eur J Surg Oncol. 2002;28:679-84
48. Berquin IM, Edwards IJ, Chen YQ. Multi-targeted therapy of cancer by omega-3 fatty acids. Cancer letters. 2008;269:363-77
Author contact
![]() Corresponding author: Phone: 334-844-6741, Fax: 334-844-4542, e-mail: mansomaedu, Present Address: Department of Anatomy, Physiology and Pharmacology, College of Veterinary Medicine, 1130 Wire Road, Auburn University, Auburn AL 36849
Corresponding author: Phone: 334-844-6741, Fax: 334-844-4542, e-mail: mansomaedu, Present Address: Department of Anatomy, Physiology and Pharmacology, College of Veterinary Medicine, 1130 Wire Road, Auburn University, Auburn AL 36849