Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(18):3257-3262. doi:10.7150/jca.25930 This issue Cite
Review
Functional roles of Speckle-Type Poz (SPOP) Protein in Genomic stability
Xi Wei1,#, Joshua Fried2,5#, Ying Li3, Linfei Hu4, Ming Gao4, Sheng Zhang1, Bo Xu 2,5,6 ![]()
1. Department of Diagnostic and Therapeutic Ultrasonography, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center of Cancer, Key Laboratory of Cancer Prevention and Therapy, Tianjin, China
2. Department of Oncology, Southern Research Institute and Cancer Cell Biology Program, University of Alabama at Birmingham Graduate School, Birmingham, AL, 35205
3. The Third Department of Breast Cancer, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center of Cancer, Key Laboratory of Cancer Prevention and Therapy, Tianjin, China
4. Department of Thyroid and Cervical Tumor, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center of Cancer, Key Laboratory of Cancer Prevention and Therapy, Tianjin, China.
5. Cancer Cell Biology Program, University of Alabama at Birmingham Comprehensive Cancer Center Birmingham, AL 35205, USA.
6. Key Laboratory of Breast Cancer Prevention and Therapy, Ministry of Education, National Clinical Research Center of Cancer, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center for Cancer, Tianjin, China
#Xi Wei and Joshua Fried contributed equally to this work.
Received 2018-3-7; Accepted 2018-7-27; Published 2018-9-7
Abstract
Understanding the functional significance of the essential elements in maintaining genomic stability provides insights into the process of tumor initiation and progression, and predicts therapeutic responses. One such element that has recently attracted significant attention is the Speckle-Type Poz Protein (SPOP), an E3 ubiquitin ligase adaptor protein. SPOP is frequently mutated or has altered expression in various cancers, including prostate, renal and endometrial. SPOP is involved in the regulation of proteasome-mediated degradation of several oncoproteins. Moreover, recent data also indicate SPOP's direct involvement in the DNA damage response. SPOP mutants induce alternations in the DNA damage repair pathway by promoting the error-prone Non-homologous end joining (NHEJ) pathway. SPOP has been linked with significant functions in cellular signaling pathways and cancer suppression. This mini-review will discuss recent findings regarding SPOP's role in genomic stability in the pathological setting.
Keywords: SPOP, genomic stability, cancer, DNA damage response
Introduction
Cancer initiation and progression are propelled by the malfunctioning of critical cellular processes. These erroneous functions cause oncogenic phenotypes that can be classified into one of several categories, commonly referred to as the “Hallmarks of Cancer” (1). These hallmarks are driven by oncogenic mutations in genes that give rise to altered expression of the associated proteins, which are involved in the regulation of cellular functions. Mutations within the genome of a cellular lineage contribute to genome instability. Of these mutations in cancer, some are more universal; whereas others are unique to or are far more common in certain cancer subtypes.
Accumulating evidence indicates that the gene encoding SPOP is one such frequent mutation in prostate cancer (2). SPOP was discovered in 1997 by Nagai et al and named for the nuclear speckles it formed and its homology to the Poz domain (3). Soon after its discovery SPOP's function as an E3 ubiquitin ligase adaptor protein was elucidated (4, 5). These studies showed that SPOP interacted with CUL3 to mediate ubiquitination of target substrates. Multiple early SPOP publications hinted that it had an anti-tumor function (5-8). Other studies, mostly in drosophila noted that the fly homolog of SPOP played a role in hedgehog signaling (9-11).
The first investigation focusing on SPOP's role in tumor formation and progression was not published until 2009 by Liu et al (12). Beginning with the landmark paper by Barbieri et al in 2012 highlighting both the frequency and uniqueness of SPOP mutation in prostate cancer, interest in the protein rose dramatically. Since then multiple studies have been published focusing on SPOP's role in cancer. Meanwhile, evidence exists in the literature that SPOP can act as either a tumor suppressor or promoter depending on the context. Mutation of the SPOP gene and/or altered expression of the protein are associated with cancer formation and progression of varieties of carcinomas including prostate, breast, gastric, renal, and gliomas. In this review, we provide a look at the SPOP protein, its role in prostate and other cancers, and the potential clinical impact of SPOP mutation.
SPOP gene and protein
A. SPOP structure
In 2009, Zhuang et al purified wild-type SPOP, and found it is composed of 374 amino acids and two domains: an N-terminal part containing residues 28-166 (SPOPMATH) and a C-terminal part containing residues172-329 (SPOP BTB) (13). The MATH region mediates substrate binding. Intriguingly, in prostate cancer all of the mutations are localized to this domain (2). The BTB domain facilitates the formation of a 2:2 complex with the CUL3 N-terminal domain. Through these interactions, SPOP participates in ubiquitination and protein degradation (14, 15).
Interestingly, the crystallographic and small-angle X-ray scattering analyses (SAXS) data indicate that the most striking feature of the SPOP structure is the dimeric and asymmetric arrangement of substrate-binding MATH domain and a 55o orientation BTB/3-box domain (13). The MATH domain is located in the middle of the V-shaped groove composed by the two protomers in the BTB domain. The dimeric SPOP BTB domain assembles with CUL3, a subtype of the Cullin gene, generating a dimeric ubiquitin ligase composed of two substrate-binding sites and two catalytic cores. Because of this dimeric structure, SPOP domains can recruit substrates and elongate ubiquitin chains to simple various and flexible orientations for higher avidity and more conformational options for mediating ubiquitination (16). However, some substrates, such as DEK and TRIM24, demonstrated a decrease in ubiquitination due to heteromeric complexes of wild-type and mutant SPOP protein (17).
B. SPOP function
SPOP binds to CUL3 via the BTB domain to form a complex for ubiquitinating target proteins (5). Table 1 lists the published SPOP substrates. Ubiquitin is a small regulatory protein that can attach itself to proteins and label them for proteasomal degradation (18). Ubiquitin ligases play an important role in maintaining genome stability and cell cycle control (19). During the ubiquitination process, a ubiquitin protein interacts with the substrate domain and Ub-E3 ligase, a substrate enzyme, to modulate the Ub system (20). E3 ligases can be grouped into the RING domain or the closely related U-box domain. The RING domain combined with the Cullin family can provide a scaffold for ubiquitin ligases (E3s) to form Cullin-RING ligases (CRLs) (21, 22).
List of known SPOP substrates
| SPOP Substrates | |
|---|---|
| Protein Name | Protein Function |
| MacroH2.A | Chromatin Organization / Accessibility |
| PDX1 | Insulin / Glucose Transport |
| Daxx | Transcription Repression / Apoptosis regulation |
| ERa | Hormone Signaling / Growth / Development |
| HHIP | Hedgehog Signaling / Development |
| Gli2/3 | Hedgehog Signaling / Development |
| SRC3 | Hormone Signaling |
| AR | Hormone Signaling / Growth / Development |
| SUFU | Hedgehog Signaling / Development |
| DUSP7 | Tyrosine Phosphotase / Multiple Pathways |
| PTEN | Phosphotase / Metabolism |
| DDIT3 | ER Stress |
| DEK | mRNA Processing |
| ERG | Transcription factor Multiple Pathways |
| SENP7 | Senescence |
| PR | Hormone Signaling / Growth / Development |
| TRIM24 | Transcriptional Control of Nuclear Receptors / Multiple Pathways |
| SETD2 | Epigenetic Regulation |
| CDC20 | Cell Cycle Regulation |
| Sirt2 | Deacetylase |
| EgIN2 | Oxygen Response |
| C-Myc | Transcription Factor / Multiple Pathways |
| INF2 | Mitochondrial Dynamics |
| HDAC6 | Epigenetic Regulation |
| BRD4 | Chromatin Reader |
| PDL1 | Apoptosis / Immune Response |
| MMP2 | ECM regulation |
SPOP in prostate cancer
Two studies published in 2011 and 2012 showed that SPOP is frequently mutated in prostate cancer (2, 23). The 2012 study by Barbieri et al was especially interesting because it showed that prostate tumors with SPOP mutation did not have the very prevalent TMPRSS2 ETS gene fusion event. Data from subsequent sequencing studies have thus far supported Barbeiri's findings (24-29). Taken together, these findings suggest that SPOP mutation may be an early event in prostate cancer tumorigenesis and is a potential driver mutation of prostate cancer. Indeed, this hypothesis has been supported by in vivo data by two investigations showing that mutation or ablation of SPOP protein can lead to mouse prostate neoplasia (30, 31).
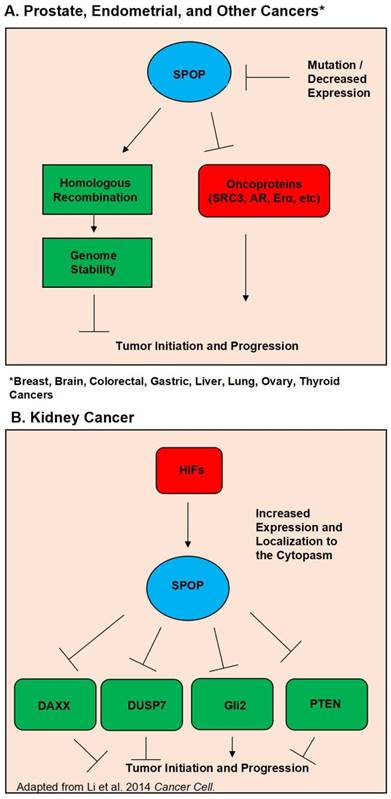
Further supporting the evidence of SPOP as a tumor suppressor is the steadily growing list of SPOP substrates, many of which are potent oncogenes. Perhaps foremost among these substrates in prostate cancer is the androgen receptor. In 2013 Geng et al first verified that SRC3 is a SPOP substrate, and that SPOP mutants lost the ability to regulate SRC-3 and thus AR activity (32). Further evidence of SPOP's regulation of AR was then discovered by An et al in 2014 and follow up study by Geng et al showing that SPOP can also directly regulate AR protein levels (33, 34). Another notable SPOP substrate is the ERG oncoprotein. Multiple studies have shown that SPOP regulates ERG protein levels, and that SPOP mutation led to ERG accumulation. This accumulation of ERG then promoted an invasive phenotype (35-37). Additionally, in 2015 An et al. showed that ERG gene fusion events protect ERG protein from regulation by SPOP (38). A study in 2014 by Theurillat et al demonstrated that SPOP, but not its mutants, ubiquitnates and promotes the degradation of a chromatin organizing protein, DEK. The authors also showed that DEK accumulates in mutated SPOP tumor samples (17). Wu et al have also shown that SPOP regulates CDC 20 (39). Trim 24, EgIN2, inF2, Senp7, DDIT3, SETD2, and C-myc have also been demonstrated to be SPOP substrates in prostate cancer (31, 40-45). Figure 1A outlines SPOP function in prostate cancer as well as other cancers where wild type SPOP is a tumor suppressor. Considering the vast collection of evidence, it is apparent that SPOP is a potent tumor suppressor in the prostate cancer setting.
Schematic diagram outlining the functional roles of SPOP. (A). Diagram of the normal functions of SPOP and how these are interrupted by mutations and/or loss of expression in Prostate, Endometrial, Breast, Brain, Colorectal, Gastric, Liver, Lung, Ovary, and Thyroid Cancers. (B). Diagram of how overexpression and localization to the cytoplasm alters SPOP function in Kidney Cancer.
SPOP in other cancers
Although a majority of research in SPOP has been in the context of prostate cancer, there are multiple reports of SPOP's anti-tumor effect in other cancer subtypes. Table 2 summarizes the different SPOP alterations that have been published and the tissue the studies were conducted in. Sequencing studies show that SPOP also has missense mutations in endometrial cancer, similarly to prostate cancer (46-49). However, the residues that are mutated in endometrial cancer are different from prostate cancer. Additionally, two studies have shown that SPOP has variants in ovarian cancer as well as liver cancer (50, 51). Interestingly, liver cancer had the S119N mutant which is also seen in prostate cancer (50). Sequencing studies in thyroid cancer have also found SPOP mutations in (52-54). In 2016 Yoo et al. showed that mutations were present in the MATH domain as in prostate (52). A different investigation by Ye et al in 2017 showed that SPOP mutations were mutually exclusive with alterations in EZH1 and ZNF148 (54). It is interesting to note that although the proteins are different SPOP mutation has mutual exclusivity with aberrations in other proteins as in prostate cancer.
List of SPOP alterations
| Description of SPOP Alterations in Different Cancer Subtypes | |
|---|---|
| Organ | Type(s) of Alteration(s) |
| Prostate | Missense Mutations, Loss of Expression |
| Endometrium | Missense Mutations, Loss of Expression |
| Breast | Loss of Expression |
| Brain | Loss of Expression |
| Colorectal | Loss of Expression |
| Gastric | Loss of Expression |
| Kidney | Overexpression, Cytoplasmic Localization |
| Liver | Missense Mutations |
| Ovary | Amplification, Deletion |
| Thyroid | Missense Mutations |
| Lung | Loss of Expression |
Along with missense mutations, multiple cancers have shown loss of SPOP genomic DNA or protein expression. In 2011 Li et al showed that SPOP can have loss of heterozygosity in breast cancer (8). Gao et al found that SPOP is crucial for the regulation of progesterone protein levels in breast cancer (55). This data combined with the data from prostate and endometrial suggest a pattern of SPOP being involved in hormonal regulation. SPOP expression has been found to be lost in colorectal, gastric, lung, and brain tumors (56-61). The researchers from all of these studies reported that loss of SPOP expression has a poor prognosis. Additional SPOP substrates have been discovered using gastric and lung cancer as a model. HDAC6, and MMP2 were found to be regulated by SPOP in the context of colorectal cancer (62, 63), and SIRT2 was found to be regulated by SPOP in non small cell lung cancer (64). Together, it appears that SPOP is a powerful tumor suppressor in solid tumors of a diverse tissue background.
Despite SPOP being almost universally hailed as a tumor suppressor across multiple types of solid cancers, there is evidence that in the context of kidney cancer SPOP acts as a tumor promoter. Liu et al published in 2009 that SPOP is involved in drosophila body segmentation and mediates the phosphorylation of JNK. Additionally they published that SPOP has increased expression in kidney cancer (12). In 2014 the same group released a second study showing that in kidney cancer Hypoxia Inducible Factors (HIF) induce SPOP localization to the cytoplasm instead of the nucleus. In this SPOP promotes tumorigenesis by mediating the degradation of Daxx, Gli2, PTEN and DUSP7 (65). In a follow up study they utilized a small molecule inhibitor targeted to SPOP. Their inhibitor was able to disrupt SPOP binding of substrate and promoted the killing of renal cancer cells. The inhibitor also showed in vivo efficacy in lowering tumor burden (66). A study by a separate group showed that depleting SPOP protein levels via siRNA was able to increase apoptosis in kidney cancer cell lines (67). SPOP's oncogenic function in kidney cancer is shown in Figure 1B.
SPOP Clinical Impact
Given the evidence reviewed here it is apparent that aberrations in the SPOP gene and / or protein will have a profound clinical impact. As discussed loss of SPOP or SPOP mutation corresponds with a poor prognosis in most solid tumor subtypes. However, the impact SPOP has on the efficacy of different treatments is still being investigated.
DNA damaging therapies such as radiation and chemotherapeutics have long been standards of cancer treatment. There are currently two studies that suggest SPOP has a role in the DNA damage response, the signaling pathway crucial for maintaining the genome. We have showed that after DNA damage SPOP interacted with ATM, a critical DDR protein, and appeared to co localize with yh2AX. Additionally, depletion of SPOP induced a radiosensitive phenotype (68). A different study by Boysen et al demonstrated that SPOP mutants favor using the relatively error prone non-homologous end joining (NHEJ) DNA damage pathway opposed to the higher fidelity homologous recombination (HR) pathway (Figure 1A) (69). Together, these findings suggest SPOP is involved in the DNA damage response although the exact mechanism is not yet understood. These studies also suggest that DNA damaging therapies could potentially have increased efficacy in patients with mutated or depleted SPOP.
BET inhibitors such as JQ1 are another favored treatment modality. As the name implies BET inhibitors impede BET containing proteins, which are epigenetic regulators that promote cell division (70). A trio of studies showed the impact of SPOP mutation on BET inhibitor efficacy. Two of which, using prostate cancer as a model found that Brd4 is substrate of SPOP. As such SPOP mutants had elevated Brd4. The elevated Brd4 resulted in a resistance to BET inhibitors in cells and tumors containing mutations in SPOP (71, 72). Interestingly, the third study by Janouskova et al published in 2017, which instead used endometrial cancer as a model found that SPOP mutation sensitized cells to BET inhibitor treatment (73). Another group investigated the efficacy of epigenetic related drugs on SPOP mutant tumors. In 2018 Yan et al publish that HDAC3 inhibitors blocked mTOR/AKT and AR signaling in tumors harboring SPOP mutations (74).
Immunotherapy has recently become a heavily investigated cancer treatment method. Among the types of immunotherapies used are immune checkpoint inhibitors. Checkpoint inhibitors block the apoptotic signaling proteins on tumor cells and / or immune cells to prevent tumor cells from inducing cell death in immune cells. PDL1 inhibitors are a well-studied treatment gaining use in combination with current standard therapy. In 2017 Zhang et al published that PDL1 is a SPOP substrate. They also showed that SPOP mutants did not bind to and mediate the ubiquitination of PDL1. Tumors with mutant SPOP had higher PDL1 levels, and a reduction in the number of CD8 tumor infiltrating T-cells (75).
Conclusions and Perspective
As an adapter for CUL3-based ligases, SPOP mediates the degradation of multiple proteins. SPOP mutations have been shown to affect several signaling pathways, such as SRC-3/AR, TNF/JNK and ERG pathways. Studies have provided evidence that SPOP is a tumor suppressor in prostate, endometrial, as well as other solid tumor forming cancers. However, there are reports showing that SPOP promotes tumorigenesis in clear cell renal cell carcinoma. SPOP mutation and loss of expression both can contribute to SPOP losing its tumor suppressor function. Aside from involvement in mediating ubiquitination, there is convincing evidence that SPOP plays a critical role in the DNA damage response, epigenetic regulation, and the immune response against tumors. Further evidence is needed to understand how current cancer therapies affect mutant SPOP tumors versus wild type SPOP tumors. It is clear that SPOP will play an important role in prostate cancer diagnosis, prognosis, and therapy in the future. It is clear SPOP is a critical protein for suppressing tumorigenesis and we are only beginning to understand its clinical impact.
Acknowledgements
We thank contributions of all of authors in this study. This work was supported by grants from the National Natural Science Foundation of China (Grant No.81771852, 81672743), Research Fund for Tianjin Cancer Hospital Translational Oncology (No.1515), the fund (2016YFC0904601) from the Chinese Ministry of Science and Technology, NIH R01CA133093, and the Alabama Innovation Fund.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-74
2. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP. et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nature genetics. 2012;44(6):685-9
3. Nagai Y, Kojima T, Muro Y, Hachiya T, Nishizawa Y, Wakabayashi T. et al. Identification of a novel nuclear speckle-type protein, SPOP. FEBS letters. 1997;418(1-2):23-6
4. Hernandez-Munoz I, Lund AH, van der Stoop P, Boutsma E, Muijrers I, Verhoeven E. et al. Stable X chromosome inactivation involves the PRC1 Polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(21):7635-40
5. Kwon JE, La M, Oh KH, Oh YM, Kim GR, Seol JH. et al. BTB domain-containing speckle-type POZ protein (SPOP) serves as an adaptor of Daxx for ubiquitination by Cul3-based ubiquitin ligase. The Journal of biological chemistry. 2006;281(18):12664-72
6. Byun B, Tak H, Joe CO. BTB/POZ domain of speckle-type POZ protein (SPOP) confers proapoptotic function in HeLa cells. BioFactors. 2007;31(3-4):165-9
7. Byun B, Jung Y. Repression of transcriptional activity of estrogen receptor alpha by a Cullin3/SPOP ubiquitin E3 ligase complex. Molecules and cells. 2008;25(2):289-93
8. Li C, Ao J, Fu J, Lee DF, Xu J, Lonard D. et al. Tumor-suppressor role for the SPOP ubiquitin ligase in signal-dependent proteolysis of the oncogenic co-activator SRC-3/AIB1. Oncogene. 2011;30(42):4350-64
9. Zhang Q, Zhang L, Wang B, Ou CY, Chien CT, Jiang J. A hedgehog-induced BTB protein modulates hedgehog signaling by degrading Ci/Gli transcription factor. Developmental cell. 2006;10(6):719-29
10. Chen MH, Wilson CW, Li YJ, Law KK, Lu CS, Gacayan R. et al. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes & development. 2009;23(16):1910-28
11. Wen X, Lai CK, Evangelista M, Hongo JA, de Sauvage FJ, Scales SJ. Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Molecular and cellular biology. 2010;30(8):1910-22
12. Liu J, Ghanim M, Xue L, Brown CD, Iossifov I, Angeletti C. et al. Analysis of Drosophila segmentation network identifies a JNK pathway factor overexpressed in kidney cancer. Science. 2009;323(5918):1218-22
13. Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M. et al. Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Molecular cell. 2009;36(1):39-50
14. Takahashi I, Kameoka Y, Hashimoto K. MacroH2A1.2 binds the nuclear protein Spop. Biochimica et biophysica acta. 2002;1591(1-3):63-8
15. Liu A, Desai BM, Stoffers DA. Identification of PCIF1, a POZ domain protein that inhibits PDX-1 (MODY4) transcriptional activity. Molecular and cellular biology. 2004;24(10):4372-83
16. Choo KB, Chuang TJ, Lin WY, Chang CM, Tsai YH, Huang CJ. Evolutionary expansion of SPOP and associated TD/POZ gene family: impact of evolutionary route on gene expression pattern. Gene. 2010;460(1-2):39-47
17. Theurillat JP, Udeshi ND, Errington WJ, Svinkina T, Baca SC, Pop M. et al. Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science. 2014;346(6205):85-9
18. Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458(7237):438-44
19. Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nature reviews Cancer. 2006;6(5):369-81
20. Laney JD, Hochstrasser M. Analysis of protein ubiquitination. Current protocols in protein science. 2011 Chapter 14:Unit14 5
21. Pintard L, Willems A, Peter M. Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. The EMBO journal. 2004;23(8):1681-7
22. Stogios PJ, Downs GS, Jauhal JJ, Nandra SK, Prive GG. Sequence and structural analysis of BTB domain proteins. Genome biology. 2005;6(10):R82
23. Brenner JC, Chinnaiyan AM. Disruptive events in the life of prostate cancer. Cancer cell. 2011;19(3):301-3
24. Haffner MC, Mosbruger T, Esopi DM, Fedor H, Heaphy CM, Walker DA. et al. Tracking the clonal origin of lethal prostate cancer. The Journal of clinical investigation. 2013;123(11):4918-22
25. Blattner M, Lee DJ, O'Reilly C, Park K, MacDonald TY, Khani F. et al. SPOP mutations in prostate cancer across demographically diverse patient cohorts. Neoplasia. 2014;16(1):14-20
26. Khani F, Mosquera JM, Park K, Blattner M, O'Reilly C, MacDonald TY. et al. Evidence for molecular differences in prostate cancer between African American and Caucasian men. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20(18):4925-34
27. Jung SH, Shin S, Kim MS, Baek IP, Lee JY, Lee SH. et al. Genetic Progression of High Grade Prostatic Intraepithelial Neoplasia to Prostate Cancer. European urology. 2016;69(5):823-30
28. Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163(4):1011-25
29. Vinceneux A, Bruyere F, Haillot O, Charles T, de la Taille A, Salomon L. et al. Ductal adenocarcinoma of the prostate: Clinical and biological profiles. The Prostate. 2017;77(12):1242-50
30. Blattner M, Liu D, Robinson BD, Huang D, Poliakov A, Gao D. et al. SPOP Mutation Drives Prostate Tumorigenesis In Vivo through Coordinate Regulation of PI3K/mTOR and AR Signaling. Cancer cell. 2017;31(3):436-51
31. Geng C, Kaochar S, Li M, Rajapakshe K, Fiskus W, Dong J. et al. SPOP regulates prostate epithelial cell proliferation and promotes ubiquitination and turnover of c-MYC oncoprotein. Oncogene. 2017;36(33):4767-77
32. Geng C, He B, Xu L, Barbieri CE, Eedunuri VK, Chew SA. et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(17):6997-7002
33. An J, Wang C, Deng Y, Yu L, Huang H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell reports. 2014;6(4):657-69
34. Geng C, Rajapakshe K, Shah SS, Shou J, Eedunuri VK, Foley C. et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer research. 2014;74(19):5631-43
35. Duan S, Pagano M. SPOP Mutations or ERG Rearrangements Result in Enhanced Levels of ERG to Promote Cell Invasion in Prostate Cancer. Molecular cell. 2015;59(6):883-4
36. Huang Y, Tan N, Jia D, Jing Y, Wang Q, Li Z. et al. Speckle-type POZ protein is negatively associated with malignancies and inhibits cell proliferation and migration in liver cancer. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015;36(12):9753-61
37. Gan W, Dai X, Lunardi A, Li Z, Inuzuka H, Liu P. et al. SPOP Promotes Ubiquitination and Degradation of the ERG Oncoprotein to Suppress Prostate Cancer Progression. Molecular cell. 2015;59(6):917-30
38. An J, Ren S, Murphy SJ, Dalangood S, Chang C, Pang X. et al. Truncated ERG Oncoproteins from TMPRSS2-ERG Fusions Are Resistant to SPOP-Mediated Proteasome Degradation. Molecular cell. 2015;59(6):904-16
39. Wu F, Dai X, Gan W, Wan L, Li M, Mitsiades N. et al. Prostate cancer-associated mutation in SPOP impairs its ability to target Cdc20 for poly-ubiquitination and degradation. Cancer letters. 2017;385:207-14
40. Groner AC, Cato L, de Tribolet-Hardy J, Bernasocchi T, Janouskova H, Melchers D. et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer cell. 2016;29(6):846-58
41. Zhang L, Peng S, Dai X, Gan W, Nie X, Wei W. et al. Tumor suppressor SPOP ubiquitinates and degrades EglN2 to compromise growth of prostate cancer cells. Cancer letters. 2017;390:11-20
42. Jin X, Wang J, Gao K, Zhang P, Yao L, Tang Y. et al. Dysregulation of INF2-mediated mitochondrial fission in SPOP-mutated prostate cancer. PLoS genetics. 2017;13(4):e1006748
43. Zhu H, Ren S, Bitler BG, Aird KM, Tu Z, Skordalakes E. et al. SPOP E3 Ubiquitin Ligase Adaptor Promotes Cellular Senescence by Degrading the SENP7 deSUMOylase. Cell reports. 2015;13(6):1183-93
44. Zhang P, Gao K, Tang Y, Jin X, An J, Yu H. et al. Destruction of DDIT3/CHOP protein by wild-type SPOP but not prostate cancer-associated mutants. Human mutation. 2014;35(9):1142-51
45. Zhu K, Lei PJ, Ju LG, Wang X, Huang K, Yang B. et al. SPOP-containing complex regulates SETD2 stability and H3K36me3-coupled alternative splicing. Nucleic acids research. 2017;45(1):92-105
46. Le Gallo M, O'Hara AJ, Rudd ML, Urick ME, Hansen NF, O'Neil NJ. et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nature genetics. 2012;44(12):1310-5
47. Hu X, Yang Z, Zeng M, Liu YI, Yang X, Li Y. et al. Speckle-type POZ (pox virus and zinc finger protein) protein gene deletion in ovarian cancer: Fluorescence in situ hybridization analysis of a tissue microarray. Oncology letters. 2016;12(1):658-62
48. Le Gallo M, Rudd ML, Urick ME, Hansen NF, Zhang S, Program NCS. et al. Somatic mutation profiles of clear cell endometrial tumors revealed by whole exome and targeted gene sequencing. Cancer. 2017;123(17):3261-8
49. DeLair DF, Burke KA, Selenica P, Lim RS, Scott SN, Middha S. et al. The genetic landscape of endometrial clear cell carcinomas. The Journal of pathology. 2017;243(2):230-41
50. Jia D, Dong R, Jing Y, Xu D, Wang Q, Chen L. et al. Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology. 2014;60(5):1686-96
51. Arildsen NS, Jonsson JM, Bartuma K, Ebbesson A, Westbom-Fremer S, Masback A. et al. Involvement of Chromatin Remodeling Genes and the Rho GTPases RhoB and CDC42 in Ovarian Clear Cell Carcinoma. Frontiers in oncology. 2017;7:109
52. Yoo SK, Lee S, Kim SJ, Jee HG, Kim BA, Cho H. et al. Comprehensive Analysis of the Transcriptional and Mutational Landscape of Follicular and Papillary Thyroid Cancers. PLoS genetics. 2016;12(8):e1006239
53. Jung SH, Kim MS, Jung CK, Park HC, Kim SY, Liu J. et al. Mutational burdens and evolutionary ages of thyroid follicular adenoma are comparable to those of follicular carcinoma. Oncotarget. 2016;7(43):69638-48
54. Ye L, Zhou X, Huang F, Wang W, Qi Y, Xu H. et al. The genetic landscape of benign thyroid nodules revealed by whole exome and transcriptome sequencing. Nature communications. 2017;8:15533
55. Gao K, Jin X, Tang Y, Ma J, Peng J, Yu L. et al. Tumor suppressor SPOP mediates the proteasomal degradation of progesterone receptors (PRs) in breast cancer cells. American journal of cancer research. 2015;5(10):3210-20
56. Kim MS, Je EM, Oh JE, Yoo NJ, Lee SH. Mutational and expressional analyses of SPOP, a candidate tumor suppressor gene, in prostate, gastric and colorectal cancers. APMIS: acta pathologica, microbiologica, et immunologica Scandinavica. 2013;121(7):626-33
57. Zhi X, Tao J, Zhang L, Tao R, Ma L, Qin J. Silencing speckle-type POZ protein by promoter hypermethylation decreases cell apoptosis through upregulating Hedgehog signaling pathway in colorectal cancer. Cell death & disease. 2016;7(12):e2569
58. Zeng C, Wang Y, Lu Q, Chen J, Zhang J, Liu T. et al. SPOP suppresses tumorigenesis by regulating Hedgehog/Gli2 signaling pathway in gastric cancer. Journal of experimental & clinical cancer research: CR. 2014;33:75
59. Li JJ, Zhang JF, Yao SM, Huang H, Zhang S, Zhao M. et al. Decreased expression of speckle-type POZ protein for the prediction of poor prognosis in patients with non-small cell lung cancer. Oncology letters. 2017;14(3):2743-8
60. Ding D, Song T, Jun W, Tan Z, Fang J. Decreased expression of the SPOP gene is associated with poor prognosis in glioma. International journal of oncology. 2015;46(1):333-41
61. Hu Y, Yang L, Zhang M, Huang Z, Lin J, Zhang N. Expression and clinical relevance of SPOPL in medulloblastoma. Oncology letters. 2017;14(3):3051-6
62. Tan Y, Ci Y, Dai X, Wu F, Guo J, Liu D. et al. Cullin 3SPOP ubiquitin E3 ligase promotes the poly-ubiquitination and degradation of HDAC6. Oncotarget. 2017;8(29):47890-901
63. Zhang S, Xiao J, Chai Y, Hong Z, Liu Z, Yuan R. et al. Speckle-Type POZ Protein Down-Regulates Matrix Metalloproteinase 2 Expression via Sp1/PI3K/Akt Signaling Pathway in Colorectal Cancer. Digestive diseases and sciences. 2018;63(2):395-402
64. Luo J, Bao YC, Ji XX, Chen B, Deng QF, Zhou SW. SPOP promotes SIRT2 degradation and suppresses non-small cell lung cancer cell growth. Biochemical and biophysical research communications. 2017;483(2):880-4
65. Li G, Ci W, Karmakar S, Chen K, Dhar R, Fan Z. et al. SPOP promotes tumorigenesis by acting as a key regulatory hub in kidney cancer. Cancer cell. 2014;25(4):455-68
66. Guo ZQ, Zheng T, Chen B, Luo C, Ouyang S, Gong S. et al. Small-Molecule Targeting of E3 Ligase Adaptor SPOP in Kidney Cancer. Cancer cell. 2016;30(3):474-84
67. Liu X, Sun G, Sun X. RNA interference-mediated silencing of speckle-type POZ protein promotes apoptosis of renal cell cancer cells. OncoTargets and therapy. 2016;9:2393-402
68. Zhang D, Wang H, Sun M, Yang J, Zhang W, Han S. et al. Speckle-type POZ protein, SPOP, is involved in the DNA damage response. Carcinogenesis. 2014;35(8):1691-7
69. Boysen G, Barbieri CE, Prandi D, Blattner M, Chae SS, Dahija A. et al. SPOP mutation leads to genomic instability in prostate cancer. eLife. 2015:4
70. Doroshow DB, Eder JP, LoRusso PM. BET inhibitors: a novel epigenetic approach. Annals of oncology: official journal of the European Society for Medical Oncology. 2017;28(8):1776-87
71. Dai X, Gan W, Li X, Wang S, Zhang W, Huang L. et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nature medicine. 2017;23(9):1063-71
72. Zhang P, Wang D, Zhao Y, Ren S, Gao K, Ye Z. et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nature medicine. 2017;23(9):1055-62
73. Janouskova H, El Tekle G, Bellini E, Udeshi ND, Rinaldi A, Ulbricht A. et al. Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nature medicine. 2017;23(9):1046-54
74. Yan Y, An J, Yang Y, Wu D, Bai Y, Cao W. et al. Dual inhibition of AKT-mTOR and AR signaling by targeting HDAC3 in PTEN- or SPOP-mutated prostate cancer. EMBO molecular medicine. 2018:10 (4)
75. Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT. et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553(7686):91-5
Author contact
![]() Corresponding author: Bo Xu, MD, PhD, Department of Oncology, Drug Discovery Division, Southern Research Institute. Email: bxuorg
Corresponding author: Bo Xu, MD, PhD, Department of Oncology, Drug Discovery Division, Southern Research Institute. Email: bxuorg