Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(17):3177-3186. doi:10.7150/jca.24756 This issue Cite
Research Paper
Alternative Splicing of SMAD4 and Its Function in HaCaT Cells in Response to UVB Irradiation
Irfan Ullah1, Yi Liao2, Rongxue Wan1, Liling Tang1 ![]() , Jianguo Feng3
, Jianguo Feng3 ![]()
1. Key Laboratory of Biorheological Science and Technology, Ministry of Education, College of Bioengineering, Chongqing University, Chongqing 400044, China
2. Department of Cardiothoracic Surgery, Southwest Hospital, Third Military Medical University Chongqing, China
3. Department of Anesthesiology, The Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan Province, China
Received 2018-1-5; Accepted 2018-5-9; Published 2018-8-6
Abstract
Alternative splicing is one of the most common mechanisms of human gene regulation and plays a crucial role in increasing the diversity of functional proteins. Many diseases are linked to alternative splicing, especially cancer. SMAD4 is a member of the SMAD family and plays a critical role in mediating of TGF-β signal transduction and gene regulatory events. Smad4 is a tumour suppressor and acts as a shuttling protein between nucleus and cytoplasm. The splicing variants of Smad4 have been found in many cancers. The present study performed nested PCR to detect alternative splicing of Smad4 in HaCaT cells lines in response to UVB irradiation. The UVB induced a novel Smad4B isoform that led to decrease the Smad4 expression. The hnRNPA1 splicing factor is responsible for Smad4 alternative splicing in response to UVB. The UVB increased the expression of SF2 and hnRNPA1 Splicing factors. The hnRNPA1 overexpression induced Smad4B by regulating Smad4 alternative splicing. The Smad4B isoform supported the function of Smad4 full length in UVB resistance with certain limitation. The western blot analyses showed that the overexpressed Smad4 full length significantly increased N-cadherin expression while Smad4B overexpression decreased the expression the N-cadherin (P<0.05). Furthermore, overexpression of the isoform in HaCaT cells decreased cell invasion as compared to Smad4 full-length overexpression. These results will be helpful to understand the importance of Smad4 alternative splicing in skin tumorigenesis.
Keywords: Alternative splicing, Cancer, hnRNPA1, Proteins, Smad4, UVB
Introduction
The skin is the outermost organ of the body and can be damaged by environmental conditions such as sunlight and pollution [1]. It is continuously exposed to various environmental stress including ultraviolet irradiation. The ultraviolet radiation (UV) has both effects, beneficial and harmful. It increases pigmentation and vitamin D synthesis but also causes skin cancer and inflammation [2]. The ultraviolet radiation is the part of electromagnetic radiation having the wavelength range UVA 315-400nm, UVB 280-315nm and UVC 100-400nm. The oxygen and ozone layer are essential to absorb UVC radiation entirely and approximately 90 percent of UVB [3]. However, the excessive exposure of UVB and UVA cause various type of DNA damage. The DNA damage may have numerous harmful consequences like mutation, cell death, photoaging, and cancer. UVA and UVB are not only absorbed by DNA but also by the chromophore which is present in the skin. This process may result in DNA damage indirectly due to the formation of reactive oxygen species. However, the human cells have an elaborate system to control the harmful effects of reactive species by antioxidant and DNA damage repair mechanism [4]. UVB primary source of Vitamin D which maintain calcium hemostasis and another critical process [5]. Excessive exposure to UVB cause skin cancer, including basal cell carcinoma, squamous cell carcinoma, nonmelanoma and cutaneous malignant melanoma [6]. Literature study reveals that UVA and UVB wavelength triggers DNA damage and responsible for the majorities of this lesion [7]. The two primary DNA lesion appear due to UV irradiation, cyclobutane pyrimidine dimmer (CPDs) and 6-4 pyrimidine photoproducts (6-4 pps) [8,9]. The removal of CPDs is carried out by a complex and highly conserved mechanism called nucleotide excision repair (NER) in placental mammals. The loss or mutations in the NER regulatory pathway has been shown to result in a dramatic increase in cutaneous carcinogenesis in patients with xeroderma pigmentosa patients [10]. Keratinocytes have a defensive mechanism known as DNA damage response (DDR) against cutaneous carcinogenesis, which may have different physiological results such as cell cycle arrest, activation of DNA, apoptosis or senescence: Depending on the degree of DNA damage and the ability of keratinocytes to repair the damage [11,12].
Alternative splicing of the pre- mRNA is a crucial mechanism by which a gene can generate different mRNA transcripts and contribute to protein diversity, isoforms of proteins with different functions. Alternative splicing occurs in almost all human genes [13]. Approximately 95 percent of the human multi-exon genes are spliced alternatively [14]. Cancer is a complex disorder which is associated with a variety of genetic and epigenetic aberrations. Cells acquire eight common characteristics during cancer development. They include maintenance of proliferative signalling, elimination of growth suppressor, resistance to cell death, the possibility of replicative immortality, induced angiogenesis, and activation of invasion and metastasis. Many of these functions may be related directly resulting in abnormal gene regulation, dysregulation of alternative splicing during tumour progression [15]. The destruction of TGF-β signalling network at Smad level is commonly found in human cancers. Several sporadic epithelial tumours show missense, nonsense, frameshift, and point mutations at SMAD4 locus. The four alternatively spliced tumours associated Smad4 isoform have been found in a thyroid tumour. Smad4 is often mutated and deregulated by abnormal splicing in thyroid cancer [16]. The transforming growth factor beta (TGF-β) is a member of a large family of structurally related proteins that normally function to regulate embryonic development and cell homeostasis, including the cell proliferation regulation, differentiation, matrix production and apoptosis in a cell [17]. Alteration in the transforming growth factor beta (TGF-β) signalling pathway can cause human diseases, for example, developmental disorders, vascular disease, and cancer. TGF-β acts as a tumour-suppressing in the early stages of epithelial carcinogenesis, but TGF-β induces tumour development in advanced phases by inducing tumour growth, epithelial-mesenchymal transition (EMT), invasion, and metastasis [18]. The surface of the cell contains two kinds of the transmembrane kinase (serine/threonine) type I and II receptors. The TGF-β ligand binds to TBRII receptor and transmitting the signal from the surface of the outer membrane to the interior of the cell, activating the signalling cascade.
The heterogeneous nuclear ribonucleoprotein (hnRNP) and pre-mRNA splicing factor (SF2) regulate pre-mRNA splicing excellently. Previously it has been reported that hnRNPA1 and SF2 regulate gene alternative splicing. The overexpression of hnRNPA1 and SF2 in many cancer cells denote its relation with the motility and proliferation of cancer cells [19,20]. The present study determined Smad4 alternative splicing in HaCaT cell in response to UVB irradiation. We also studied whether hnRNAP A1 or SF2 splicing factor is involved in UVB induce alternative splicing of Smad4.
Materials and Methods
Cell culture
HaCaT cells is a spontaneously immortalized human keratinocyte cell line that has been widely used in scientific research for skin biology and differentiation studies. The HaCaT cells were maintained in RPMI-1640 (Hyclone) supplemented with 10% fetal bovine serum (FBS, Hyclone), 1% antibiotics penicillin-streptomycin (P/S, Hyclone) and were incubated at standard condition (37oC, 5% CO2 humidified incubator). Cell physiology and behaviour was progressively examined with the help of an inverted microscope.
UVB Irradiation
HaCaT cells were subcultured for 48 hours (80% confluence) in 60mm cell culture dishes in standard medium. UVB irradiation was carried out by using two 8-watts UVB lamps (SANKEY, Japan). The UVB intensity was measured by UV meter (photoelectric Instrument Factory of Beijing Normal University, China). The cells were washed with phosphate buffer saline (PBS) two-time and then irradiated at a different dose of UVB 30,50,100 and 400mj/cm2 without the plastic lid of the dishes. After UVB irradiation, the cells were incubated in a humidified incubator for six hours and RNA were extracted after incubation. The low dose of UVB (3,5,7 mj/cm2) at 24 hours of incubation was also studied.
The HaCaT stable cell lines of hnRNPA1 and SF2 splicing factor were donated by Xichao Xu and were maintained in RPMI-1640 (Hyclon) supplemented with 10% fetal bovine serum (FBS) 1% penicillin-streptomycin and 1.0 ug/ml puromycin. The culture cell was incubated at 37oC in 5% CO2.
RNA extraction and RT- PCR
Total RNA was isolated using TRIzol reagent (Tiangen) according to manufacturer instructions. The RNA was reverse transcribed by using Prime ScriptTM RT reagent with gDNA Eraser (Takara, Japan). The cDNA was used in nested PCR.
Nested PCR
The nested PCR was used to ensure the specificity. Two pairs of primers were used for a single locus. The second pairs (nested primers) were used to bind to the first step product. The total 20ul volume reaction has been amplified for 28 cycles by using the primer pair:
5/ AAGGATCAAAATTGCTTCAGA 3/ (Sense)
5/ CAGTCTAAAGGTTGTGGGTC 3/ (Antisense)
With denaturation 94oC for 30 secs, annealing at 56oC for 30sec and elongation at 72oC for 90 secs in nested first step. The following primers were used in nested second step with the denaturation at 94oC for 30 secs, annealing at 56oC for 30 sec and elongation was reduce at 72oC for 1 minute.
5/ GGTCGGAAAGGATTTCC 3/ (Sense)
5 /CCAACTGCACACCTTTGCCTA 3/ (Antisense)
The GAPDH was used as an internal control. The PCR products were loaded on 1% agarose gel.
Plasmid Construction and Sequencing Analysis
The cDNA was re-amplified to obtain the full length of Smad4 and Smad4B. 0.5% agarose gel was used to resolve the amplified product: The Smad4F and Smad4B bands were censored and purified by using gel extraction and purification kit (Omega). Dr Liao Yi donated the plasmid (Plenti-CMV). Sal1 and BamH1 restriction enzymes were used to censor the Plenti-CMV and BamH1, and Xho1 restriction enzymes were used for full length Smad4 and Smad4B isoform. The entire products were run on a 0.5% gel and purified by using gel extraction kit (Omega). The purification product was used in plasmid construction of Plenti-CMV-Smad4F, Plenti-CMV-Smad4B, and Plenti-CMV-GFP by a T4 ligase enzyme. The transformation was done by using E. Coli Top 10 competent cells through heat shock at 42oC, and the bacteria were grown on LB medium plate for one night. Overnight, some colonies were picked to confirm the positive colonies by PCR. The amplified bands corresponding to the correct size of the colonies was added to 10ml Amp+LB media culture flask. The flask was placed in transient thermoshaker or incubator at 37oC and 150 RPM shaking for one night. Overnight extract the bacteria by centrifugation and send to thermos fisher scientific company for BGI sequencing. The required plasmid was extracted by plasmid extraction kits (Omega) using company protocol. The plasmid concentration was checked by nano-dropper three times and takes the average. The plasmid concentrations were Plenti-CMV-Smad4F (647.9ng/ul) and Plenti-CMV-Smad4B (597.85ng/ul). The formula diluted the plasmid = total volume x desired concentration÷ Start concentration.
Formation of Stable Cell Lines
Stable cells formation
HEK 293T packaging cells were suspended in three 60mm dishes. After 24 h the cells were 80% confluent. HEK 293T cells were co-transfected with lentiviral Plenti-CMV-Gene of interest (Smad4F, Smad4B Isoform, and GFP) as well as lentiviral helper vectors pCMV-VSV-G plasmid and pCMV-dR8.2 dvpr plasmid. After 48h incubation, the viral supernatant fraction was collected. The 10ml collected medium was centrifuged 2000RPM for 5 minutes at 4Co. HaCaT cells were suspended in three 60mm dishes. After 24 h, the HaCaT cells were treated with 1.5ml collected lentiviral particles and add 6ul polybrene as transfection reagent. The medium was changed after 24 h and selected with the puromycin-containing medium over one week.
Cell Proliferation
Cell proliferation was analyzed by using CCK-8 assay (Beyotime, China). Briefly, same numbers of cells were seeded in forty-eight well plate. After 24 hours of incubation, the cells were irradiated with UVB. The cell viability was measured at 24h, 48h Pre-and post UVB by CCK-8 Assay.
Cell migration and invasion
The wound healing assay was used to study cell direction and migration rate. The HaCaT stable cell line was seeded in six-well plates overnight to make a monolayer. After 24h a scratch was made by pipet tip and wash with PBS twice to remove the dead cells. After 6, 12 and 24h the cell direction and migration rate was studied through inverted inflorescence microscope.
Western blotting
HaCaT stable cells were seeded in 60mm dishes. After 24h, the cells were treated with UVB 50mj/cm2. After 6 hours of UVB irradiation, cells were extracted a lysed by using RIPA lysis buffer (50 mMTris-HCl pH7.5, 1% NP-40, 0.5 mM EDTA, 0.1% SDS, 150 mMNaCl, 0.5% Sodium deoxycholate) with PMSF (Beyotime, China). The cell lysate was centrifuged at 13,000 RPM for 15 min at 4°C. BCA assay (Beyotime, China) was used to measure the protein concentration. The protein samples were separated by 15% SDS-PAGE and then transferred to polyvinylidene fluoride (PVDF) membranes (PALL, USA). PVDF membrane was blocked in 1XTBST with 5% nonfat milk for one hour and then incubated with primary antibodies at 4ºC overnight. After overnight incubation, the membrane was washed three times with 1xTBST and incubated with corresponding secondary antibodies for 1h at room temperature. Following a triple washing step with 1XTBST, the bands were visualized with the chemiluminescence method (Millipore, USA). Antibodies including GAPDH, Smad4, Bcl-2, Bax, and Caspases anti-rabbit or anti-mouse secondary antibodies were purchased from santa cruz and cell signalling (USA).
Results
UVB Irradiation leads to Smad4 splicing into Smad4-B in Keratinocyte cell
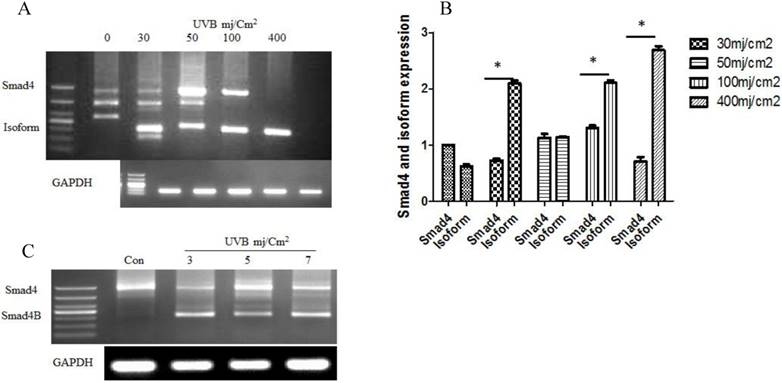
The given study determined whether UVB could induce alternative splicing of Smad4 in HaCaT cell line. Nested PCR was used to detect the Smad4 splicing after UVB treatment according to the previous protocol (2). The results showed that UVB induced only one isoform of Smad4 in HaCaT cells. DNA sequencing confirmed the isoform sequence. The translation capability was detected by PPM5 software. The expression of Smad4 decreases led to increases of Smad4B (Isoform) ultimately Smad4F (Full length) expression was entirely loss with a high dose of UVB. We also studied low dose of UVB at 24 hours. The same isoform Samd4B was obtained with low dose of UVB (Figure 1).
(A) Alternative splicing of Smad4 in response to a high dose of UVB at six hours. The UVB induced Smad4 B isoform that led to decrease Smad4F expression (B) Smad4F expression decreased with the increase of Smaad4B isoform (C) The alternative splicing of Smad4 at a low dose of UVB irradiation after 24h. The isoform significantly increased with the increasing of UVB dose. n=3 *P<0.05.
hnRNPA1 splicing factor is involved in Smad4 alternative splicing
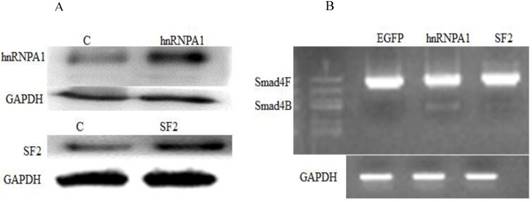
Next, we investigated whether a change in expression of splicing factors (hnRNPA1 and SF2) will affect the alternative splicing of Smad4. Western blot confirmed the overexpression of hnRNPA1 and SF2 in the HaCaT stable cell line (Figure 2A). It was determined that hnRNPA1 is responsible for alternative splicing of Smad4 to Smad4B (Figure 2B). The hnRNPA1 and SF2 overexpressed cells were subjected to nested PCR to detect the UVB induced alternative splicing of Smad4. The given results showed that overexpression of hnRNPA1 increases the expression of Smad4B isoform as compared to control and SF2 splicing factor (Figure 2).
Involvement of Splicing factors (hnRNPA1 and SF2) in alternative splicing of Smad4. (A) Overexpression of splicing factors hnRNPA1 and SF2. (B) Nested PCR results showed the involvement of hnRNPA1 in alternative splicing of Smad4 into Smad4B
UVB regulates the expression of hnRNPA1 and SF2
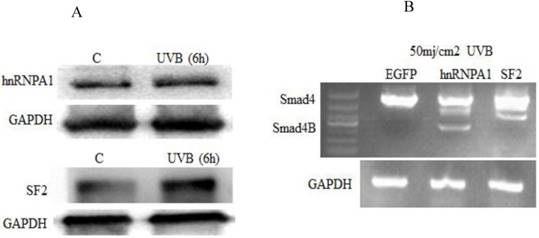
We further investigated whether UVB has an effect on hnRNPA1 and SF2 splicing factors and also studied whether hnRNPA1 or SF2 involved in UVB induced alternative splicing of Smad4. It was determined that UVB increases hnRNPA1 and SF2 splicing factors. The expression of hnRNPA1 and SF2 splicing factors increase at 6h and decrease after 24h post UVB irradiation. These two protein expressions are not correlated with ROS level in the cell. The given study reported that hnRNPA1 involved in UVB induced alternative splicing of Smad4.The results showed that UVB increases the expression of hnRNPA1 and SF2 splicing factors (Figure 3A). Which indirectly resulted the alternative splicing of Smad4. The overexpression of hnRNPA1 resulted smad4 alternative splicing (Figure 3B).
Effect of UVB on splicing factors (hnRNPA1 and SF2). (A) UVB increased the expression of hnRNPA1 and SF2 splicing factors. The UVB significantly increase the expression of hnRNPA1 and SF2 after six hours. (B) The hnRNPA1 splicing factor overexpression due to UVB irradiation resulted in alternative splicing of Smad4.
The Smad4B acts as a subsidiary of full length Smad4 during UVB irradiation
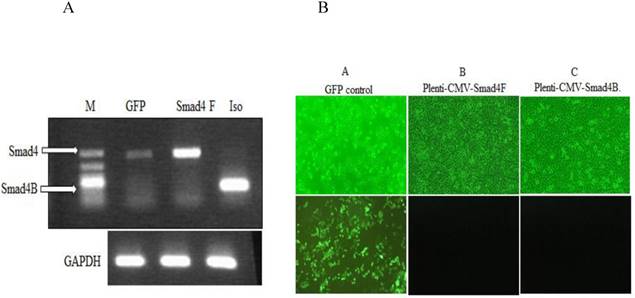
To investigate the function of Smad4B, the lentiviral Plenti-CMV-Gene of interest (Smad4F, Smad4B, and GFP), as well as lentiviral helper vectors pCMV-VSV-G plasmid and pCMV-dR8.2 dvpr plasmid, were constructed and transfected into HaCaT cell line by using polybrene transfection reagent. The Smad4F and Smad4B were transiently transfected into HaCaT cell. RT-PCR and inflorescence microscope was used detect the successful transfection the required plasmid (Figure 4).
(A) RT-PCR was used to detect the expression of Smad4 in GFP control, Overexpression of Smad4 and Smad4B isoform in HaCaT stable cell. (B) Inflorescence microscope was used to detect the transfection efficiency of Lentivirus. A GFP control B. Plenti-CMV-Smad4F C. Plenti-CMV-Smad4B. The transfection efficiency was more than 90 percent.
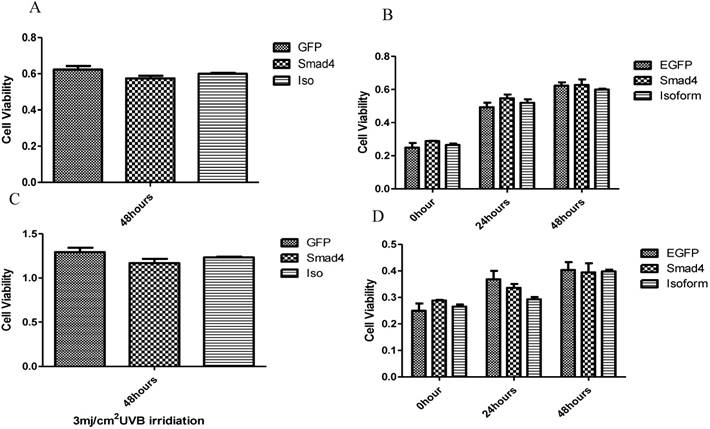
The effect of Smad4F and Smad4B was observed on cell proliferation pre-and post UVB treatment by using CCK-8 cell proliferation assay. The results showed that the Smad4B and Smad4F overexpression had no significant effect on cell proliferation (Figure 5 A, B). To detect the UVB effect on isoform, the cell was treated with 3mj/cm2 with UVB. After 24, 48 hours of UVB treatment there was no significant difference between isoform and Smad4 overexpression (Figure 5 C, D). The Smad4B acted as a subsidiary of Smad4F against UVB irradiation (Figure 5).
The influence of UVB on Smad4 and Smad4B overexpression. (A, B) Overexpression of Smad4 and Isoform had no significant effect on cell proliferation. (C, D) After 24, 48 hours of UVB treatment there was no significant difference between isoform and Smad4 overexpression on cell proliferation. The P value was P>0.05, therefore no significant difference between isoform and full length of Smad4.
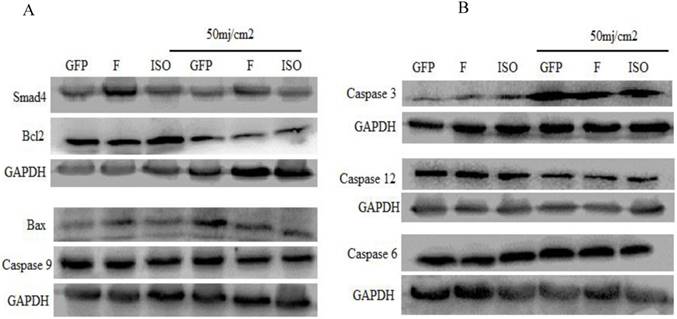
The pro-apoptotic and anti-apoptotic proteins (Bcl2 and BAX, caspase 9, Caspase-6, Caspase-12, and Cleaved Caspase-3 was detected by western blot. The western blot results indicated that overexpression of Smad4F and Smad4B had no significant difference on Bcl2, Bax, and other Caspases. The isoform showed little effect on Bax, Bcl2 and caspases but had no significant difference (Figure 6).
The western blot was used to determine the effect of Smad4F and Smad4B (Isoform) overexpression on caspases, Bcl2 and Bax protein levels. The results showed that there is no significant difference between Smad4F and Smad4B on Bcl2, Bax, Caspase 6, Caspase 3, 9,12.
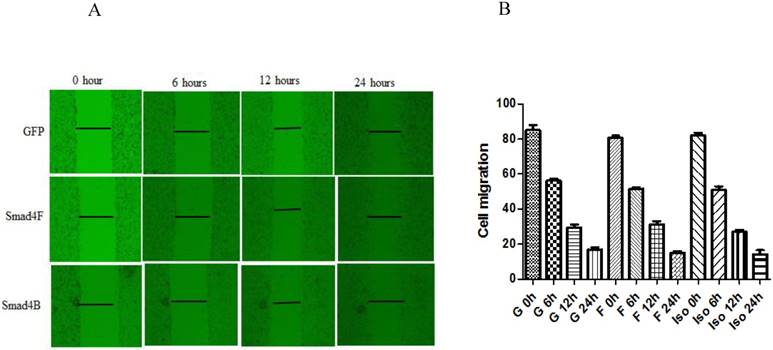
Smad4F and Smad4B have no significant difference in cell migration
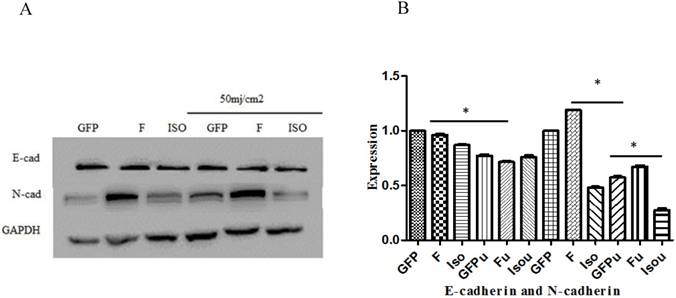
Wound healing assay was used to study the effect of Smad4B and Smad4F on cell migration. The results of wound healing assay showed that Smad4F and Smad4B had no significant difference in cell migration (Figure 7). The western blot results revealed that there was no significant differential effect on E-cad expression, but the expression of N-cad significantly decreased in Smad4B as compared to Smad4F overexpression and GFP control (Figure 8). The N-cad function in cell adhesion is more critical than E-cad for motility and invasion (21). The isoform decreases cell invasion while Smad4F overexpression increase (Figure 7) (Figure 8).
The wound healing assay was used to determine the effect of Isoform and Smad4 overexpression on HaCaT cell migration, but no significant difference was found in cell migration. The P-Value was P>0.05.
E-cadherin and N-cadherin expression. The Smad4 and Smad4 B isoform was no significant effect on expression of E-Cad, but the Smad4B decreased the expression of N-cad as compared to Smad4F and GFP control. The Smad4 overexpression significantly increases N-cad expression while the Isoform Smad4B decreases the N-cad expression. *P<0.05. The UVB significantly decrease the expression of E-cad and N-cad. *P<0.01.
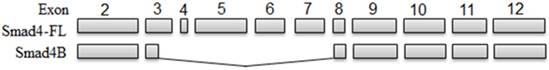
UVB-induced Smad4 splicing (Smad4) led to the decrease the full length of Smad4 and increase of Smad4B. The Smad4B lacks Exon4Δ3-7 and part of Exon 3 compared with Smad4 full length
Discussion
Alternative splicing plays a significant role in creating the diversity of the protein by including or excluding specific exons. Majorities of the transcript are degraded in the nucleus or encode for unstable protein [22,23]. Alternative splicing is a central process for gene regulation. Most of the multi-exon genes in humans are alternatively spliced, and the transcript displays different fantastic function in physiological and pathological condition [24]. It has been found that aberrant splicing is associated with various diseases, including cancer. Mutations in splicing regulatory elements in the nucleotide sequence and changes in the cellular splicing regulatory device both lead to changes in the splicing pattern of many cancer-associated genes [25].
The present study analysed for the first time the alternative splicing of Smad4 in HaCaT cells in response to UVB irradiation and found that UVB can induce only one isoform (Smad4B) of Smad4 in HaCaT cell at 6 hours of the incubation period (Figure 1). The given isoform (Smad4B) was more potent against UVB irradiation as compare to full length of Smad4. The same isoform was obtained at low dose (3,5, and 7) of UVB treatment and 24 hours incubation period. Both high and low dose UVB induced Smad4B isoform in HaCaT cell line. The UVB decreases the expression of Smad4F led to increases the expression of Smad4B isoform. The Smad4B acts as a subsidiary of Smad4F during UVB irradiation. Nested PCR was used to detect the alternative splicing of Smad4 and followed by sequencing. Smad4 plays a fundamental role in all transforming growth factor β (TGF-β) signalling pathways. It acts as a shuttling protein between cytoplasm and nucleus. The previous study determines that several mRNA splicing of Smad4 in different cells. It is investigated that Smad4 mRNA is alternatively spliced in the linker region to give six different mRNA isoforms (Smad4 Δ3, Δ4, Δ5-6, Δ6, Δ4-6, and Δ4-7 in HaCaT cell from both normal and tumour cells [26]. The Smad4B is a new isoform, no one reported it before. High dose of UVB cause complete loss of Smad4 but the Smad4B was still expressed. The results showed that this Smad4B shows more resistance to UVB irradiation as compared to Smad4F. The given Smad4 B isoform deleted in linker region from exon 3 to exon 7 (Smad4 Δ3-7).
The Smad4, a tumour suppressor gene, is inactive in human pancreatic ductal carcinoma with high frequency [27]. The inactivation is commonly due to homozygous deletion and may also by heterozygous mutation in the remaining allele. Inactivation of nonsense mutations can result in loss of protein expression by enhanced proteasome degradation. Even if expressed as protein, missense mutations may lead to loss of specific functions of the Madh4 protein, such as DNA binding or Smad protein interactions [28]. Many studies have demonstrated that some RNA-binding proteins can participate in inflammatory processes and the regulation of tumorigenesis by regulating the splicing or mRNA stability of inflammation and tumour-associated genes [29,30,31]. The processing of post-transcriptional miRNA involves in a sequence of reactions that convert a prior-miRNA precursor into a biologically active and mature miRNA. RNA binding proteins (RBP) are critical for determining the function of miRNAs that are required in all stages of the miRNA pathway. The given study was focused on two most common and vital splicing factors hnRNPA1 and SF2 and their role in alternative splicing of Smad4 in UVB response.
The two nuclear RNA binding proteins, the heterogeneous nuclear ribonucleoprotein family (hnRNP) and the serine/arginine-rich protein (SR) family play a key role in regulating alternative splicing and mRNA stability [32]. The heterogeneous nuclear ribonucleic acid (hnRNP) family of proteins forms a diverse RNA-binding protein group that deals with various functions of metazoan [33]. The two splicing factors (hnRNPA1 and SF2) were examined in the present study. Some previous study reported that hnRNPA1 and SF2 splicing factors involved in UVB regulated gene alternative splicing [1,34,35,36]. Some authors reported that hnRNPA1 and SF2 also upregulated in various human cancers. It can be related to cancer cell proliferation and metastasis [14,37]. The hdm2 alternative splicing under UVB in human keratinocyte has been reported [37]. The overexpression of hnRNPA1 and SF2 were detected by western blot (Figure 2A). It was found that hnRNPA1 is responsible for regulating the alternative splicing of Smad4. The production of Smad4B led to decrease the expression of Smad4F. The overexpression of hnRNPA1 stimulates the alternative splicing of Smad4 while SF2 overexpression does not affect (Figure 2B). The UVB significantly increase the expression of hnRNPA1 and SF2 splicing factors (Figure 3A). The overexpressed hnRNPA1 is directly proportional to alternative splicing of Smad4 in response to UVB (Figure 3B). There was no definite effect of SF2 splicing factor on alternative splicing of Smad4. It was determined that the overexpression of Smad4F and Smad4B isoform have no significant difference in cell proliferation, after UVB irradiation and without UVB (Figure 4). Alternative splicing increases the numbers and variety of proteins expressed in eukaryotic cells. Recently the relationship between human genetic diseases and cancer are associated with different abnormal alternative splicing enhanced [38]. There are four Smad4 isoform have been reported in a thyroid tumour. All the isoform loss different portion of the linker region such as ΔEx6, ΔEx5-6, ΔEx4-6, and ΔEx4-7. The Smad4 often mutates and deregulated abnormally in thyroid tumours, and these changes may contribute to the onset of thyroid neoplasms. ΔEx5-6 is previously reported in primary neuroblastoma [39] in the MDA-MB231 mammary cell [40] and HaCaT cell line [26]. The isoform ΔEx4-6 is also reported in neuroblastoma [39]. The present study reported lacking almost the entire linker region. The ΔEx4-7 variants are reported in Hacat cell [26] and papillary thyroid carcinoma [16].
The Wound healing assay was used to determine the effect of Smad4B and Smad4F overexpression on HaCaT cell migration, but no significant differences were found on cell migration (Figure 7). The Smad4B and Smad4 F have no significant difference E-cad expression but decrease the expression of N-cad. E-cadherin is identified to play a role in tumour progression and metastasis because the loss of E-cadherin expression has been found to be associated with aggressive and undifferentiated phenotypes of many cancers, including pancreatic cancer. Another adhesion molecule, N-cadherin, is correlated with an increased invasion of cancer [41]. The previous study has shown that overexpression of N-cadherin in breast cancer is associated with invasiveness as a result of N-cadherin-mediated interaction between cancer and the stromal Cell [42]. The transfection of N-cadherin promotes motility and invasion in breast cancer cells without a reduction in E-cadherin expression. It has been determined that N-cadherin, which acts as adhesion molecules, can be more critical than the E-cadherin for metastasis and invasion [21]. The Smad4F overexpression significantly increases the expression of N-cadherin while the Smad4B decrease the expression of N-cadherin (Figure 8).
Conclusion
Pathogenesis and treatment of skin cancer caused by UV radiation have been the focus of research recently, UV irradiation may cause activation of a series of intracellular signalling pathways, changes in protein levels as well as the variable genes Splicing, these stress changes have significant implications for the formation of skin cancer. Alternative splicing is a fundamental mechanism to yield many diverse proteins; multiple transcriptions can create numerous mRNA transcripts. The given study determined the alternative splicing of Smad4 and its function in HaCaT cell in response to UVB irradiation. It was investigated that UVB induced only one isoform of Smad4 under different doses of UVB irradiation. This isoform showed more resistance to UVB as compared the Smad4.The given isoform act as a supporter of Smad4 against UVB. At high dose of UVB irradiation, Smad4 suppress entirely while Smad4B still showing resistance. The overexpression of hnRNPA1 is responsible for UVB induced alternative splicing of Smad4 in HaCaT cells. The UVB increase the expression of hnRNPA1 and SF2 splicing factors. The overexpression of Smad4 full length increases N-cadherin expression while the isoform decreased N-cadherin expression. However, there was no effect on E-cadherin.
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (No 106112017CDJXSYY0001, 2018CQDYSG0021) and the Graduated Scientific Research and Innovation Foundation of Chongqing, China (No CYB16039).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Naylor EC, Watson REB, Sherratt MJ. Molecular aspects of skin ageing. Maturitas. 2011;69:249-56
2. Bertrand JU, Petit V, Hacker E, Berlin I, Hayward NK, Pouteaux M, Sage E, Whiteman DC, Larue L. UVB represses melanocyte cell migration and acts through beta-catenin. Exp Dermatol. 2017;26(10):875-882
3. Schuch AP, Garcia CC, Makita K, Menck CF. DNA damage as a biological sensor for environmental sunlight. Photochem.Photobiol.Sci. 2013;12(8):1259-72
4. Schuch AP, Moreno NC, Schuch NJ, Menck CF, Garcia CCM. Sunlight damage to cellular DNA: Focus on oxidatively generated lesions. Free Radic Biol Med. 2017;107:110-124
5. Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266-281
6. Lucas RM, Norval M, Neale RE, Young AR, de Gruijl FR, Takizawa Y, van der Leun JC. The consequences for human health of stratospheric ozone depletion in association with other environmental factors. Photochem.Photobiol.Sci. 2015;14(1):53-87
7. Markovitsi D. UV-induced DNA Damage: The Role of Electronic Excited States. Photochem Photobiol. 2016;92(1):45-51
8. Ikehata H, Ono T. The mechanisms of UV mutagenesis. J Radiat Res. 2011;52(2):115-25
9. Rastogi RP, Richa, Kumar A, Tyagi MB, Sinha RP. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids. 2010;2010:592980
10. DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. The Journal of investigative dermatology. 2012;132(3 Pt 2):785-96
11. Giglia-Mari G, Zotter A, Vermeulen W. DNA damage response. Cold Spring Harb Perspect Biol. 2011;3(1):a000745
12. Nakanishi M, Niida H, Murakami H, Shimada M. DNA damage responses in skin biology-implications in tumor prevention and aging acceleration. J Dermatol Sci. 2009;56(2):76-81
13. Liu S, Cheng C. Alternative RNA splicing and cancer. Wiley Interdiscip Rev RNA. 2013;4(5):547-66
14. Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40(12):1413-5
15. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-74
16. Lazzereschi D, Nardi F, Turco A, Ottini L, D'Amico C, Mariani-Costantini R, Gulino A, Coppa A. A complex pattern of mutations and abnormal splicing of Smad4 is present in thyroid tumours. Oncogene. 2005;24(34):5344-54
17. Asiedu MK, Ingle JN, Behrens MD, Radisky DC, Knutson KL. TGF-beta/TNF(alpha)-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011;71(13):4707-19
18. Huibin YGL, Wu JJ, Wang L, Uhler M, Simeone DM. Protein Kinase A Modulates Transforming Growth Factor-β Signaling through a Direct Interaction with Smad4 Protein. J Biol Chem. 2013;288(12):8737-8749
19. Feng J, Li L, Tong L, Tang L, Wu S. The Involvement of Splicing Factor hnRNP A1 in UVB-induced Alternative Splicing of hdm2. Photochem Photobiol. 2016;92(2):318-24
20. Bonomi S, Di Matteo A, Buratti E, Cabianca DS, Baralle FE, Ghigna C. HnRNP A1 controls a splicing regulatory circuit promoting mesenchymal-to-epithelial transition. Nucleic Acids Res. 2013;41(18):8665-79
21. Nakajima S, Doi R, Toyoda E, Tsuji S, Wada M, Koizumi M. N-Cadherin Expression and Epithelial-Mesenchymal Transition in Pancreatic Carcinoma. Clin Cancer Res. 2004;10:4125-33
22. Grellscheid S, Dalgliesh C, Storbeck M, Best A, Liu Y, Jakubik M. Identification of Evolutionarily Conserved Exons as Regulated Targets for the Splicing Activator Tra2β in Development. PLoS Genet. 2011;7:e1002390
23. Hao Y, Colak R, Teyra J, Corbi-Verge C, Ignatchenko A, Hahne H. Semi-supervised Learning Predicts Approximately One-Third of the Alternative Splicing Isoforms as Functional Proteins. Cell Rep. 2015;12:183-9
24. Bardot B, Toledo F. Targeting MDM4 splicing in cancers. Genes. 2017;8(2):E82
25. Pajares MJ, Ezponda T, Catena R, Calvo A, Pio R, Montuenga LM. Alternative splicing: an emerging topic in molecular and clinical oncology. Lancet Oncol. 2007;8(4):349-57
26. Pierreux CE, Nicola FJ, Hill CS, Nicolás FJ, Caroline S. Transforming Growth Factor b-Independent Shuttling of Smad4 between the Cytoplasm and Nucleus. Mol Cell Biol. 2000;20:9041-54
27. Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350-3
28. Maurice D, Pierreux CE, Howell M, Wilentz RE, Owen MJ, Hill CS. Loss of Smad4 Function in Pancreatic Tumors: C-terminal truncation leads to decreased stability. J Biol Chem. 2001;276:43175-81
29. Ten Dijke P, Hill CS. New insights into TGF-b-Smad signalling. Trends Biochem Sci. 2004;29:265-73
30. Guo R, Li Y, Ning J, Sun D, Lin L, Liu X. HnRNP A1/A2 and SF2/ASF Regulate Alternative Splicing of Interferon Regulatory Factor-3 and Affect Immunomodulatory Functions in Human Non-Small Cell Lung Cancer Cells. PLoS One. 2013;29:8 (4), e62729
31. David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010;24(21):2343-64
32. Martinez-Contreras R, Cloutier P, Shkreta L, Fisette JF, Revil T, Chabot B. hnRNP Proteins and Splicing Control. In: Adv Exp Med Biol. 2007;623:123-147
33. Caceres JF, Kornblihtt AR. Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet. 2002;18(4):186-93
34. Cavinato M, Waltenberger B, Baraldo G, Grade CVC, Stuppner H, Jansen-Dürr P. Plant extracts and natural compounds used against UVB-induced photoaging. Biogerontology. 2017;18:499-516
35. Liu Y, Wu B, Zhong H, Tian X, Fang W. Quantification of alternative splicing variants of human telomerase reverse transcriptase and correlations with telomerase activity in lung cancer. PLoS One. 2012;7(6):e38868
36. Tauler J, Mulshine JL. Lung cancer and inflammation: interaction of chemokines and hnRNPs. Curr Opin Pharmacol. 2009;9(4):384-8
37. He Y, Smith R. Nuclear functions of heterogeneous nuclear ribonucleoproteins A/B. Cell Mol Life Sci. 2009;66(7):1239-56
38. Cáceres JF, Kornblihtt AR. Alternative splicing: Multiple control mechanisms and involvement in human disease. Trends in Genet. 2002;18(4):186-93
39. Kageyama H, Seki N, Yamada S, Sakiyama S, Nakagawara A. DPC4 splice variants in neuroblastoma. Cancer Lett. 1998;122:187-93
40. De Winter JP, Roelen BA, ten Dijke P, van der Burg B, van den Eijnden-van Raaij AJ. DPC4 (SMAD4) mediates transforming growth factor-beta1 (TGF-beta1) induced growth inhibition and transcriptional response in breast tumour cells. Oncogene. 1997;14:1891-9
41. Bukholm IK, Nesland JM, Børresen-Dale AL. Re-expression of E-cadherin, alpha-catenin and beta-catenin, but not of gamma-catenin, in metastatic tissue from breast cancer patients. J Pathol. 2000;190:15-9
42. Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol. 2000;148:779-90
Author contact
![]() Corresponding authors: tanglilingedu.cn/ffengjianguocom
Corresponding authors: tanglilingedu.cn/ffengjianguocom