Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(16):2973-2980. doi:10.7150/jca.25506 This issue Cite
Research Paper
Molecular analysis of gastric cancer identifies genomic markers of drug sensitivity in Asian gastric cancer
Qin Yang1,*, Chunchao Zhu2,*, Yanli Zhang1,*, Yangyang Wang2,*, Yahui Wang1, Lei Zhu1, Xiaomei Yang1, Jun Li1, Huizhen Nie1, Shuheng Jiang1, Xiaoxin Zhang1, Xiaoyan Cao1, Qing Li1, Xueli Zhang1, Guangang Tian1, Lipeng Hu1, Lili Zhu1, Gang Zhao2, ![]() , Zhigang Zhang1,
, Zhigang Zhang1, ![]()
1. State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, P.R. China
2. Department of Gastrointestinal Surgery, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, P.R. China
* These authors contributed equally to this work
Received 2018-2-12; Accepted 2018-6-16; Published 2018-7-30
Abstract
Background: Gastric cancer (GC) is one of the leading causes of lethal malignancies worldwide, especially in Eastern Asia. Clinical responses to antitumor therapies are often limited to a subset of patients.
Methods: To uncover new biomarkers of sensitivity and resistance to cancer therapeutics, we performed ultra-deep targeted sequencing in a cohort with 72 patients (41 with chemotherapy sensitivity and 31 with chemotherapy resistance).
Results: We found that sixteen mutated cancer genes were associated with widely used agent in chemotherapy of gastric cancer. Genes identified in these study are mainly involved in activation and inactivation of cancer chemotherapeutic agents, changes of apoptosis and proliferation, drug efflux, DNA damage repair, and the tumor microenvironment.
Discussion: A novel group of chemo-sensitivity related genes provided new therapeutic strategies to overcome the development and evolution of resistance to cancer chemotherapy.
Keywords: Gastric cancer, Chemotherapy response, Mutation
Introduction
Gastric carcinoma is one of the most common cancers and the second leading cause of cancer-related death in the world [1]. Despite a decline in morbidity and mortality, the burden remains high [2]. In gastric cancer patients, the treatment outcomes depend on the stage of the tumor at presentation and the condition of the patients. Surgical resection is the only possible curative treatment for gastric cancer. Moreover, more than two-thirds of patients have metastatic or surgically unresectable disease [3]. Therefore, Chemotherapy is used to relieve the symptoms of unresectable tumors and reduce the risk of recurrence and metastasis after surgery [4]. Although chemotherapy plays an important role in the treatment of recurrent and metastatic gastric cancer, multiple drug resistance (MDR) to chemotherapy is known as the major cause of treatment failure for gastric carcinomas [5]. Recently, numerous studies have reported the mechanisms of drug resistance in gastric carcinomas [6, 7]. However, the mechanisms of drug resistance in gastric cancer have not been fully elucidated.
To find mutations predicting chemotherapy benefit in gastric carcinomas, we performed ultra-deep targeted sequencing in a cohort with 72 patients (41 with a good response and 31 with a poor response). We sequenced 1,574 genes involved in tumor progression and development. The purpose of this study was twofold, firstly to find therapeutic response predictive biomarkers, secondly to identify new targets in gastric carcinomas treatment.
Material and Methods
Patients and Tissue Samples
72 pairs of tumor and normal samples were collected from patients diagnosed with gastric carcinomas; all patients had received radical surgery (total or partial gastrectomy). After surgery, all patients were received chemotherapy (fluorouracil, platinum or paclitaxel). All patients were Chinese. The original clinical and survival data of patients were provided in Table 1. The ethical committee of Ren Ji Hospital, Shanghai Jiao Tong University School of Medicine approved the studies and all patients signed informed consent before recruitment to the study.
Clinicopathological Characteristics of 72 Primary Gastric Tumor Samples
| Parameter | Total | chemo-sensitive | chemo-resistant | p Value |
|---|---|---|---|---|
| (N=72) | (N=41) | (N=31) | ||
| Mean age (range) | 61(33-80) | 60(47-80) | 62(33-80) | 0.36 |
| Gender | ||||
| Male | 57 | 33 | 24 | 0.98 |
| Female | 15 | 8 | 7 | |
| Lauren type | ||||
| Intestinal | 23 | 15 | 8 | 0.64 |
| Diffuse | 42 | 22 | 20 | |
| Mix | 7 | 4 | 3 | |
| TNM(AJCC) | ||||
| Stage IIIA | 14 | 9 | 5 | 0.83 |
| Stage IIIB | 36 | 20 | 16 | |
| Stage IIIC | 22 | 12 | 10 | |
| Vascular invasion | ||||
| Absent | 46 | 30 | 16 | 0.1 |
| Present | 26 | 11 | 15 | |
| Perineuronal invasion | ||||
| Absent | 47 | 31 | 16 | 0.1 |
| Present | 25 | 10 | 15 |
Tumor cell purity was assessed in hematoxylin and eosin (H&E) sections. At least 5 slices of 10 μm of thickness were cut from the paraffin block and tumor regions were scraped according to the assessment of tumor enriched area.
Ultra-deep targeted sequencing and reads alignment
Genomic DNA was extracted using the GenReadTM DNA FFPE Kit (Qiagen; Germany) from formalin-fixed paraffin-embedded tissues, as well as matched normal DNA, and was fragmented and hybridized according to the manufacturer's protocol. For ultra-deep targeted sequencing, we used SeqCap EZ Choice kit (Roche). Raw fastq files were checked quality control using FastQC, and were removed low quality reads to clean reads. Burrows Wheller aligner (BWA) was used to align the sequencing reads onto the hg19 reference genome with default options [8]. Samtools was used to convert the SAM file into BAM; Picard was used to remove duplicate reads [9].
Detection of somatic mutations and indels
Mutect was used to detect somatic mutations, single-nucleotide variants (SNVs), small insertion and deletion (INDELs) [10]. To obtain a list of confident variants, we selected variants whose coverage was more than 20X. Vcf2maf was used to convert a vcf file into a maf file and annotate the somatic mutations [11].
Mutant-allele tumor heterogeneity
Mutant-allele tumor heterogeneity (MATH) analysis was initially developed to measure intratumor heterogeneity in gastric cancer samples [12]. Mutation allele frequencies (MAF) of mutated loci in each tumor were determined.
MATH = (absolute MAF - median MAF)* 1.4826 / median MAF
Statistical Analyses
Statistical analyses to test for correlation between mutations and clinicohistological variables such as age, sex and lymph node metastasis were performed using R/Bioconductor packages. We used the Fisher's exact test for difference between groups[13]. Using the clusterProfiler package, KEGG pathway enrichment analyses were performed with the significantly genes. The P value of 0.05 or less was considered statistically significant.
Results
Tissue Samples and Clinicopathological Data
Seventy-two cases of gastric carcinomas patients (forty-one chemo-sensitive and thirty-one chemo-resistant) were analyzed (Table 1). In our cohort, the mean age was 61 years (range 33-80; SD 9.4), 57 patients were male (79%). With a median follow up of 14.5 months (SD 27.3), 34 patients had a dead status (47%). Based on response to chemotherapy drug, 41 of patients have good prognosis which overall survival beyond 36 months, other 31 patients have poor prognosis, which had been dead in 18 months.
Identification of somatic mutations in Gastric Carcinomas
In all of the 72 primary tumor samples an adequate library for sequencing was obtained. After read quality filtering, mapping, and alignment to the reference genome (hg19), the median sequencing depth was 277×. A total of 17993 genetic variations (median 207, range 14-959, SD 107.7) were identified in exonic (including nonsense, missense, nonstop, frame_shift_del, frame_shift_ins, in_frame_del and in_frame_ins) (Figure 1A). The predominant nucleotide changes were cytosine to thymine transitions, which is consistent with previous gastric carcinomas genomics data (Figure 1B).
The mutation type and mutation spectrum in seventy-two gastric cancers (GC). (a) Somatic mutation type in each sample. (b) The six substitution subtypes in each sample: C>A, C>G, C>T, T>A, T>C, and T>G.
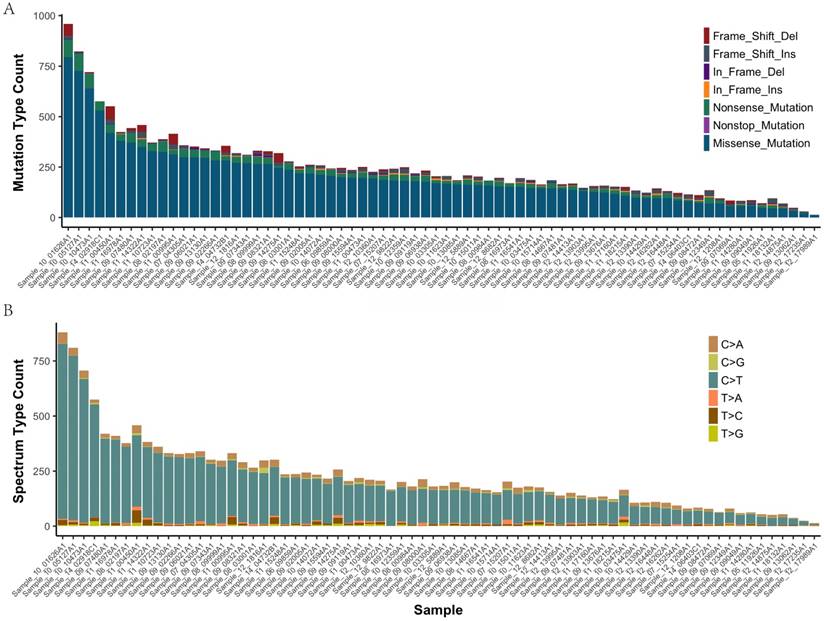
The mean number of mutations was 150, range 14-959, SD 107.7. These variants were distributed across 1495 different genes and were found in 72 samples (92% with at least two mutations). 4985 of the missense mutations identified are predicted to be possibly or probably damaging by Sift and Polyphen2, as these are more likely to play a causative role in tumorigenesis and represent therapeutic vulnerabilities. The number of damaging mutations considered to be chemo_sensitive samples was 71 (range 7-292), and 67 in chemo_resistant samples (range 6-233). The somatic mutation rates were similar for chemo-sensitive and chemo-resistant (average mutation rate of 243 versus 254 mutations per tumor, p = 0.79). Mutations in TTN, MUC16 and FSIP2 were the most frequently observed variants in this study (93%, 71%, and 69%, respectively). No significant difference between groups in Lauren type, vascular invasion and perineuronal invasion (Supplemental Figure 1).
The mutation burden and the Clonality do not correlate with chemotherapy
It has been reported that tumour mutational load or tumour mutational burden was associated with response to immunotherapy in multiple cancer types. To examine whether it also correlated with the chemotherapy in patients with GC, we investigated the number of missense mutations as tumor mutation burden in this tumor type. The number of missense mutations in these patients ranged from 13 to 794 (median value, 203). There were no significant differences between groups in chemotherapy (p = 0.34). We next investigated whether there was an association between clonality and chemotherapy. Clonality was positively correlated with tumor heterogeneity, as determined by MATH analysis. There had no significant statistical differences between groups in chemotherapy (p = 0.92). We next performed a survival analysis to evaluate the clinical relevance of clonality, and found no significant association between OS and mutation burden or clonality (Supplemental Figure 2). These results revealed that the number of missense mutations and clonality failed to correlate significantly with chemotherapy survival in GC.
Differences between chemo-resistant and chemo_sensitive
TP53 was the most predominantly mutated driver gene in gastric carcinomas, and there had no significant difference between the chemotherapy subgroups, with mutation rate was 27% vs. 35%. No statistically significant differences between the chemotherapy groups with the other commonly recurring genomic aberrations involve ARID1A, RHOA, RNF43, MUC6, KRAS, PTEN, CDH1, SMAD4, GLI3, ZIC4, ERBB2/3/4, NRG1, DCLK1, and PIK3CA (Supplemental Figure 3). However, cell adhesion gene CTNNA2 mutations were observed in 10 of 72 samples (9 in chemo_sensitive and 1 in chemp_resistant, p = 0.036) (Figure 2). CTNNA2 encoding catenin-a2 plays an important role in the regulation of B-catenin signaling. Six of the ten CTNNA2 single-nucleotide variants were probably damaging by Sift and Polyphen2 (R760H, T574K, T21M, A315V, R229C and S437C).
Oncoprint of mutations for the 31 significantly mutated genes in gastric cancer (GC) by deep sequencing.
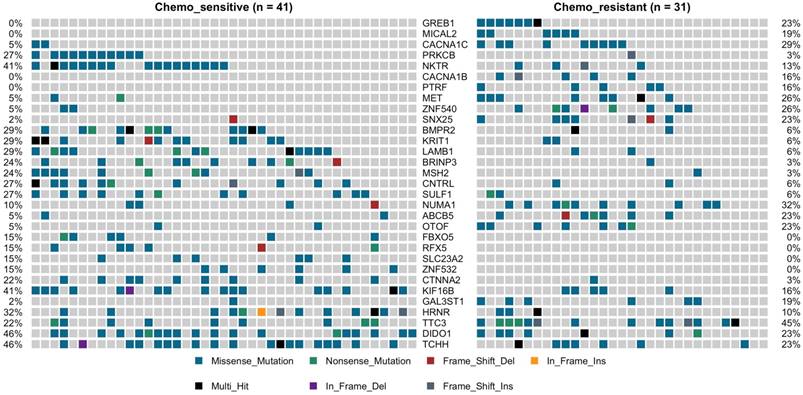
Applying the same procedure on the chemo-sensitive patients (n = 41) and chemo-resistant patients (n = 31), we also identified other 30 mutated genes, which were significant differences compared to the chemo-resistant mutation rate (fisher test, P < 0.05) (Figure 2, Supplemental Figure 4). This set of significant genes had two subgroups, one was higher mutation ratio in chemo_sensitive samples (gene 13), and another was lower (gene 18). The higher mutation ratio subgroup included a common driver cancer gene MSH2 (tumor suppressor gene). MutS Protein Homolog 2 (MSH2), which plays an important role in DNA mismatch repair, was identified 11 variants showing trends towards lower mutation rate compared to the chemo-sensitive mutation rate (3% vs. 24%, P= 0.019). The higher mutation ratio subgroup included a common driver cancer gene MET (oncogene). Hepatocyte Growth Factor Receptor (HGFR), which could regulate the receptor tyrosine kinase, and promote cell survival, motility and proliferation, was identified 10 variants showing trends towards higher mutation rate compared to the chemo-sensitive mutation rate (26% vs. 5%, P= 0.016). The lower mutation rate subgroup compared to the chemo-sensitive also included PRKCB (3% vs. 27%, P= 0.009), LAMB1 (6% vs. 29%, P= 0.018), BMPR2 (6% vs. 25%, P= 0.018), CTNNA2 (3% vs. 22%, P= 0.036), FBXO5 (0% vs. 15%, P= 0.033), SULF1 (6% vs. 27%, P= 0.032) and KRIT1 (6% vs. 29%, P= 0.018). The higher mutation ratio subgroup compared to the chemo-sensitive also included CACNA1B (16% vs. 0%, P= 0.012), CACNA1C (29% vs. 5%, P= 0.007), MICAL2 (19% vs. 0%, P= 0.005), ABCB5 (5% vs. 23%, p=0.033), GAL3ST1 (19% vs. 2%, P= 0.038) and PTRF (16% vs. 0%, P= 0.012) (Figure 2).
We next investigated whether mutations in different genes would cluster in some known signaling pathways. Using KEGG database, Genes which mutations have been identified significantly differences in our cohort was mapped. Hippo signaling pathway (BMPR2 and CTNNA2) and Rap1 signaling pathway (KRIT1 and PRKCB) were associated with the higher mutation ratio in chemo_sensitive group (P value 0.011 and 0.021). MAPK signaling pathway (CACNA1B and CACNA1C) was associated with the higher mutation ratio in chemo_resistant group (P value 0.011).
Mutational status and overall survival
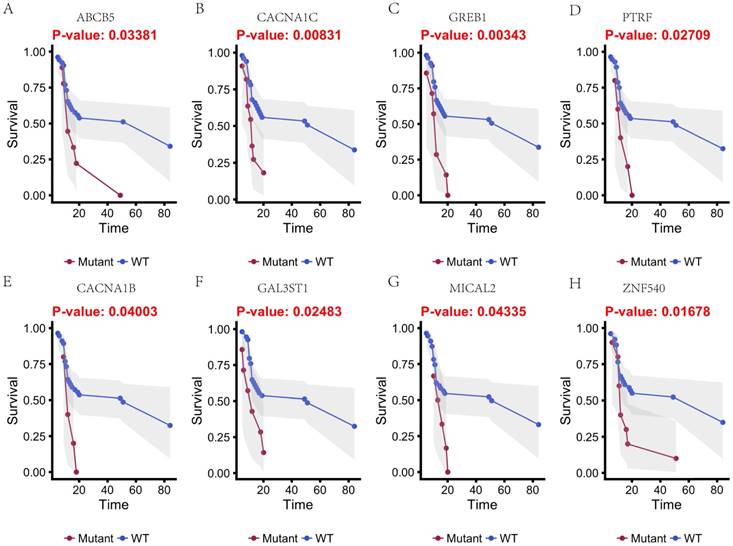
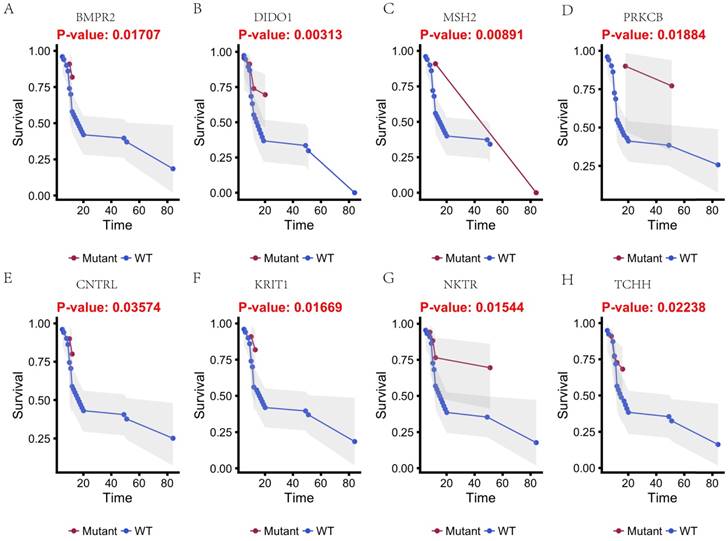
In this study, 31 genes had associated with chemotherapy. Then Univariate Kaplan-Meier analysis was used to assess associations between mutations in chemotherapy genes and overall survival from diagnosis. We found a significant association between OS and 16 genes (p < 0.05) (Figure 3 and Figure 4). This analysis revealed that the risk of death was significantly lower for patients with mutant type than for those with wild type, including ABCB5, CACNA1B, CACNA1C, GAL3ST1, GREB1, MICAL2, PTRF and ZNF540 (Figure 3). The other eight genes had high risk of death in mutant type than wild type, including BMPR2, CNTRL, DIDO1, KRIT1, MSH2, NKTR, PRKCB and TCHH (Figure 4).
Kaplan-Meier estimates of the overall survival of patients in each subgroup of gastric cancers based on significant genes (A-H). The wild-type cases (blue line) have better prognosis than mutated type cases (red line).
Kaplan-Meier estimates of the overall survival of patients in each subgroup of gastric cancers based on significant genes (A-H). The mutated type cases (blue line) have better prognosis than wild-type cases (red line).
Discussion
Multiple molecular mechanisms have been reported in drug resistance, such as drug inactivation, drug target alteration, drug efflux, DNA damage repair, cell death inhibition and the epithelial-mesenchymal transition (EMT) [14]. However, the specific mechanisms mediating escape from chemotherapy and gastric cancer recurrence remain largely unknown. To address this issue, we target-sequenced a cohort of 72 gastric cancer tissues to find chemotherapy predictive biomarkers and new treatment targets. We found that the commonly recurring genomic aberrations involved TP53, ARID1A, RHOA, RNF43, MUC6, KRAS, PTEN, CDH1, SMAD4, GLI3, ZIC4, ERBB2/3/4, NRG1, DCLK1, and PIK3CA had similar mutation rate between the chemo sensitive subgroup and the chemo resistant subgroup. As a result, sixteen genes including ABCB5, CACNA1B, CACNA1C, GAL3ST1, GREB1, MICAL2, PTRF, ZNF540, BMPR2, CNTRL, DIDO1, KRIT1, MSH2, NKTR, PRKCB and TCHH had significantly difference between the two groups (Figure 5).
Chemotherapy resistances can be affected by a number of possible determinants such as activation and inactivation of chemotherapeutic drugs, changes of apoptosis and proliferation, drug efflux, DNA damage repair, and the tumor microenvironment.
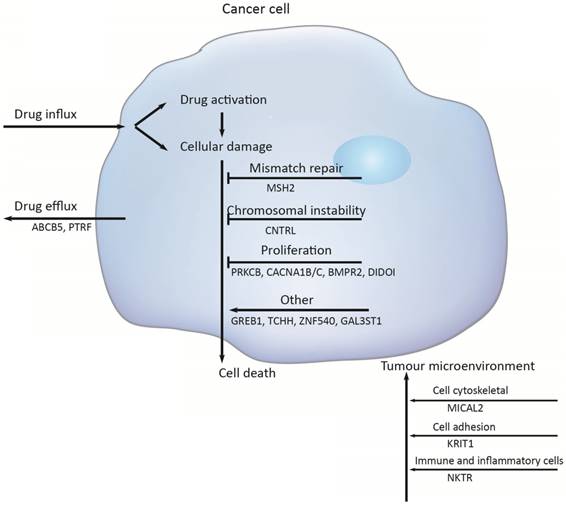
Firstly, activation and inactivation of cancer chemotherapeutic agents can play a major role in the development of resistance. One example of this is observed in the treatment of gastric cancer, capecitabine that is activated after phosphorylation that convert it to 5‑fluorouracil [15]. However, hypermethylation of the promoter of gene encoding thymidine phosphorylase is causing capecitabine resistance [16]. Secondly, the damaged DNA repair has also a clear role in anticancer drug resistance [17]. DNA mismatch repair (MMR) in the DNA damage response mediates the cytotoxicity of some DNAdamaging anticancer agents; for example, hypermethylation of MLH1 has been reported to cause resistance to cisplatin and carboplatin [18]; and the absence of MSH2 has been shown to increase resistance to thiopurines [19]. But in this study, the mutation of MSH2 was identified to increase sensitive to chemotherapy, with the mutation rate was 24%. Thirdly, Genomic instability that is called chromosomal instability (CIN) drives cancer development and increases chemotherapy resistance [20, 21]. The CIN gene (NEK2) mediated drug resistance was further demonstrated to be through AKT-mediated upregulation of ABC transporters [22]. Several studies have clearly implicated centrosome has a role in tumorigenesis and centrosome dysfunction can result in chromosome instability [23]. Centriolin is encoded by CNTRL gene and involves in maintaining centrosome structure and functions [24]. In our study, the higher mutation rates of gene CNTRL in chemo_sensitive subgroup ensure that they have a decreased risk of chromosomal instability in tumorigenesis.
Additionally, PRKCB, CACNA1A/B and BMP signaling pathway mainly function in regulating cell proliferation. The gene PRKCB may activate the MAPK pathway, regulating cell proliferation [25]. And a substantial alteration of Ca2+ could be responsible for cell proliferation and cell apoptosis through the cell cycle [26]. Bone morphogenetic protein (BMPs) is a large family of growth factors that belongs to tumor growth factor-β superfamily. They have crucial roles in regulating a wide range of developmental functions, such as pattern formation, differentiation, proliferation, migration and so on [27]. BMP protein binding to type 1 and 2 receptors (BMPR1 and BMPR2), activated BMP signaling, increased migration and invasion. In gastric cancer, bone morphogenetic protein-2 (BMP2) significantly inhibited cancer cell proliferation by suppressing the expression of β-catenin, c-Myc, and aurora kinases [28]. Death inducer-obliterator 1 (DIDOI) as a BMP-specific Smad-regulated target gene, promotes the attachment, migration, invasion and apoptosis resistance of melanoma cells [29]. In our study, there is a significant difference in the mutation rate of PRKCB, CACNA1A/B, BMPR2, and DIDOI in different groups.
The tumor microenvironment (TME) not only plays a vital role during cancer progression and metastasis but also has profound effects on therapeutic efficacy. In our study, mutation rates among genes related to cytoskeleton (MICAL2)[30], cell adhesion (KRIT1)[31, 32], were also significantly difference (MICAL2 p = 0.005, KRIT1 p = 0.018).
Several the ATP-binding cassette (ABC) transporter family of transmembrane proteins such as ABCB1 (MDR1, P-glycoprotein), ABCC1 (MRP1, multidrug resistance-associated protein 1) and ABCG2 (BCRP, Breast Cancer Resistance Protein), has been linked to mediate multidrug resistance of human cancer by promoting anticancer drug efflux [33, 34]. And ABCB5 (ATP-binding cassette member B5) was also as a clinically relevant multidrug resistance mediator in colorectal cancer [35], hepatocellular carcinoma [36] and human malignant melanoma [37]. In this study we observed that 23 % of the tumors had an ABCB5 mutation in chemoresistant subgroup and 5 % in chemosensitive subgroup, indicating that the mutation of ABCB5 plays an important role in drug resistant in gastric cancer. And PTRF knockdown also reduced multidrug resistance in cancer cells via the fortification of lipid rafts [38]. In this study, the mutant PTRF increased the drug resistance in gastric cancer.
Conclusion
Two important conclusions can be inferred from these data. First, genes including TP53, CTNNB1, ARID1A, CDH1, PTEN, KRAS and so on, which were as drivers of gastric carcinogenesis, play an important role in tumor development and progression. However most of these genetic variants do not influence the drug responses. Second, drug resistance to chemotherapy in gastric carcinomas is a complex and integrated activation and inactivation, changes of apoptosis and proliferation, drug efflux, DNA damage repair, and the tumor microenvironment. To discover and prioritize recurrent mutations associated with response to chemotherapy agents, larger sample series are urgently needed.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by the Shanghai Natural Science Foundation (15ZR1439200) and the National Science Foundation of China (81502382; 81672358).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. The Lancet. 2016;388:2654-64
2. McLean MH, El-Omar EM. Genetics of gastric cancer. Nat Rev Gastroenterol Hepatol. 2014;11:664-74
3. Wohrer SS, Raderer M, Hejna M. Palliative chemotherapy for advanced gastric cancer. Ann Oncol. 2004;15:1585-95
4. Casaretto L, Sousa PL, Mari JJ. Chemotherapy versus support cancer treatment in advanced gastric cancer: a meta-analysis. Braz J Med Biol Res. 2006;39:431-40
5. Shi WJ, Gao JB. Molecular mechanisms of chemoresistance in gastric cancer. World J Gastrointest Oncol. 2016;8:673-81
6. Zhang DX, Fan DM. New insights into the mechanisms of gastric cancer multidrug resistance and future perspectives. Future Oncol. 2010;6:527-37
7. Marin JJG, Al-Abdulla R, Lozano E, Briz O, Bujanda L, Banales JM. et al. Mechanisms of Resistance to Chemotherapy in Gastric Cancer. Anti-Cancer Agent Me. 2016;16:318-34
8. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589-95
9. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078-9
10. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213-9
11. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A. et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122
12. Mroz EA, Tward AM, Hammon RJ, Ren Y, Rocco JW. Intra-tumor Genetic Heterogeneity and Mortality in Head and Neck Cancer: Analysis of Data from The Cancer Genome Atlas. Plos Medicine. 2015:12
13. Mayakonda A, Koeffler HP. Maftools: Efficient analysis, visualization and summarization of MAF files from large-scale cohort based cancer studies. BioRxiv. 2016
14. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714-26
15. Pietrantonio F, De Braud F, Da Prat V, Perrone F, Pierotti MA, Gariboldi M. et al. A Review on Biomarkers for Prediction of Treatment Outcome in Gastric Cancer. Anticancer Res. 2013;33:1257-66
16. Kosuri KV, Wu X, Wang L, Villalona-Calero MA, Otterson GA. An epigenetic mechanism for capecitabine resistance in mesothelioma. Biochem Biophys Res Commun. 2010;391:1465-70
17. Kerbel RS. Molecular and physiologic mechanisms of drug resistance in cancer: An overview. Cancer Metast Rev. 2001;20:1-2
18. Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573-84
19. Diouf B, Cheng Q, Krynetskaia NF, Yang W, Cheok M, Pei D. et al. Somatic deletions of genes regulating MSH2 protein stability cause DNA mismatch repair deficiency and drug resistance in human leukemia cells. Nat Med. 2011;17:1298-303
20. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability-an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220-8
21. Kuznetsova AY, Seget K, Moeller GK, de Pagter MS, de Roos JA, Durrbaum M. et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle. 2015;14:2810-20
22. Zhou W, Yang Y, Xia JL, Wang H, Salama ME, Xiong W. et al. NEK2 Induces Drug Resistance Mainly through Activation of Efflux Drug Pumps and Is Associated with Poor Prognosis in Myeloma and Other Cancers. Cancer Cell. 2013;23:48-62
23. Nigg EA. Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer. 2002;2:815-25
24. Kumar A, Rajendran V, Sethumadhavan R, Purohit R. CEP proteins: the knights of centrosome dynasty. Protoplasma. 2013;250:965-83
25. Ueda Y, Hirai S, Osada S, Suzuki A, Mizuno K, Ohno S. Protein kinase C activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. J Biol Chem. 1996;271:23512-9
26. Schreiber R. Ca2+ signaling, intracellular pH and cell volume in cell proliferation. J Membr Biol. 2005;205:129-37
27. Jiao G, Guo W, Ren T, Lu Q, Sun Y, Liang W. et al. BMPR2 inhibition induced apoptosis and autophagy via destabilization of XIAP in human chondrosarcoma cells. Cell Death Dis. 2014;5:e1571
28. Lee KB, Jin H, Ye S, Park BH, Kim SM. Recombinant human bone morphogenetic protein-2 inhibits gastric cancer cell proliferation by inactivating Wnt signaling pathway via c-Myc with aurora kinases. Oncotarget. 2016;7:73473-85
29. Braig S, Bosserhoff AK. Death inducer-obliterator 1 (Dido1) is a BMP target gene and promotes BMP-induced melanoma progression. Oncogene. 2013;32:837-48
30. Giridharan SS, Caplan S. MICAL-Family Proteins: Complex Regulators of the Actin Cytoskeleton. Antioxid Redox Signal. 2014;20:2059-73
31. Fanjul-Fernandez M, Quesada V, Cabanillas R, Cadinanos J, Fontanil T, Obaya A. et al. Cell-cell adhesion genes CTNNA2 and CTNNA3 are tumour suppressors frequently mutated in laryngeal carcinomas. Nat Commun. 2013;4:2531
32. van den Berg MC, Burgering BM. CCM1 and the second life of proteins in adhesion complexes. Cell Adh Migr. 2014;8:146-57
33. Choi YH, Yu AM. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr Pharm Des. 2014;20:793-807
34. Shaffer BC, Gillet JP, Patel C, Baer MR, Bates SE, Gottesman MM. Drug resistance: still a daunting challenge to the successful treatment of AML. Drug Resist Updat. 2012;15:62-9
35. Wilson BJ, Schatton T, Zhan Q, Gasser M, Ma J, Saab KR. et al. ABCB5 identifies a therapy-refractory tumor cell population in colorectal cancer patients. Cancer Res. 2011;71:5307-16
36. Cheung ST, Cheung PF, Cheng CK, Wong NC, Fan ST. Granulin-epithelin precursor and ATP-dependent binding cassette (ABC)B5 regulate liver cancer cell chemoresistance. Gastroenterology. 2011;140:344-55
37. Wilson BJ, Saab KR, Ma J, Schatton T, Putz P, Zhan Q. et al. ABCB5 Maintains Melanoma-Initiating Cells through a Proinflammatory Cytokine Signaling Circuit. Cancer Res. 2014;74:4196-207
38. Yi JS, Mun DG, Lee H, Park JS, Lee JW, Lee JS. et al. PTRF/Cavin-1 is Essential for Multidrug Resistance in Cancer Cells. Journal of Proteome Research. 2013;12:605-14
Author contact
![]() Corresponding authors: Zhigang Zhang, Ph.D. 800 Dongchuan Road, Shanghai 200240, P.R. China. State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University. E-mail: zzhangcom and Gang Zhao, M.D & Ph.D. 160 Pujian Road, Shanghai 200127, P.R. China. Department of Gastrointestinal Surgery, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University. E-mail: zhaogang74313com
Corresponding authors: Zhigang Zhang, Ph.D. 800 Dongchuan Road, Shanghai 200240, P.R. China. State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University. E-mail: zzhangcom and Gang Zhao, M.D & Ph.D. 160 Pujian Road, Shanghai 200127, P.R. China. Department of Gastrointestinal Surgery, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University. E-mail: zhaogang74313com