Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(16):2825-2833. doi:10.7150/jca.25993 This issue Cite
Research Paper
Phosphorylation of the Cytoskeletal Protein CAP1 Regulates Non-Small Cell Lung Cancer Survival and Proliferation by GSK3β
Shuanshuan Xie1, Yang Liu1, Xuan Li1, Min Tan1, Changhui Wang1 ![]() , Jeffrey Field2, Guo-Lei Zhou3
, Jeffrey Field2, Guo-Lei Zhou3
1. Department of Respiratory Medicine, Shanghai Tenth People's Hospital, Tongji University, Shanghai 200072, China.
2. Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania, Philadelphia, PA, 19104, USA.
3. Department of Biological Sciences, Arkansas State University, State University, AR 72467, USA.
Received 2018-3-11; Accepted 2018-6-13; Published 2018-7-16
Abstract
Adenylate cyclase-associated protein 1 (CAP1) is an evolutionarily conserved protein that regulates actin dynamics. Our previous study indicates that CAP1 is overexpressed in NSCLC tissues and correlated with poor clinical outcomes. Further establishing the role and dissecting underlying mechanisms are imperative before targeting CAP1 can become a possibility for cancer treatment. Here we report our findings that knockdown of CAP1 inhibited cell proliferation and induced apoptosis in vitro and in vivo. Moreover, phosphor mutants of CAP1 at the S307/S309 regulatory site had compromised rescue effects for both the invasiveness and the proliferation in CAP1-knockdown cells and GSK3β kinase inhibitor LiCl inhibited cell phosphorylation site S307/S309 by up-regulating the expression of p53, BAK, BAD and cleaved PARP induced ROS production, decreased lung cancer cell viability, adhesion, proliferation, migration and invasion, and induction of apoptosis. These novel mechanistic insights may ultimately open up avenues for strategies targeting CAP1 in the treatment of lung cancer, tailored for specific types of the highly diverse disease.
Keywords: Adenylate cyclase-associated protein, Kinase, Phosphorylation, Lung cancer
Introduction
Lung cancer is the leading cause of cancer-related mortality, resulting in approximately 222,500 new cases and 155,870 deaths within the United States in 2017, the mortality related to lung cancer continues to rise [1]. Non-small cell lung cancer (NSCLC) represents approximately 85% of all lung cancers [2]. Despite extensive research and clinical efforts, the 5-year overall survival (OS) rate of NSCLC patients is about 17% [3]. Invasion and metastasis of cancer is a multi-step, multi-factor and multi-stage process [4], each step is regulated by many factors, such as 1) the activity of tumor cells; 2) the invasive ability of tumor cells;3) the movement ability of tumor cells; 4) the adhesion ability of tumor cells and so on [5]. Among those factors, the metastasis ability of tumor cells is the main reason that is different from other cell abilities [6]. The metastasis of NSCLC is the main cause of treatment failure and death [7].
As is known to all, actin cytoskeleton is essential for cancer metastasis [8]. CAP1 is cytoskeletal proteins that interact with cofilin and actin to regulate actin dynamics [9, 10]. In addition, recent studies, including those from our group, have established roles for mammalian CAP1 in regulating the actin cytoskeleton and cell migration [11-14].
Mounting evidence over the last decade suggests involvement of CAP1 in the invasiveness of a growing list of human cancers, including pancreatic, glioma, and ovarian cancer [15- 18]. However, Zhang et al found that in metastatic breast cancer cells, depletion of CAP1 stimulated both the invasiveness and the cell proliferation, while in non-metastatic breast cancer cells, it actually had opposite effects [19]. Furthermore, depletion of CAP1 in HeLa cells actually stimulated migration and invasion substantially. The role of CAP had a discrepancy in different cancer types [20]. Thus, we have evidence to confirm that CAP1 plays an important role in cell migration and invasion.
Our previous studies found that application and mutation were two most prominent alternations of CAP1 in cancers by cBioPortal and Tumorscape analysis [12]. Therefore, based on the above data, in the present study, we further investigated these application and mutation mechanisms in lung cancer by vivo and vitro experiment. Thus, further investigation of the molecular mechanisms underlying tumor metastasis is essential for developing new effective therapeutic targets and anti-lung cancer strategies.
Materials and Methods
Cell lines and cell culture reagents
Normal HP-AEpiC cells and lung cancer cell lines A549, 95 D, 95 C, H1299, H1975 and PC-9 were obtained from ATCC. HP-AEpiC cells were maintained in DME/F12 medium supplemented with 5% equine serum, 20 ng/mL EGF, 10 µg/mL insulin, 100 ng/mL cholera toxin and 500 ng/mL hydrocortisone. A549, H1299, H1975 and PC-9 were maintained in DMEM supplemented with 10% fetal bovine serum (FBS). 95 D and 95 C were maintained in RPMI-1640 medium supplemented with 10% FBS. Lithium chloride (LiCl, a conventional GSK3β inhibitor) was purchased from Sigma.
Expression plasmids and establishment of CAP1 knockdown and re-expression stable cells
Invasive 95-D cells were seeded onto collagen I-coated 6-well plates at 5x104 cells/well. Following overnight culture, cells were transfected with CAP1 specific siRNA (siCAP1) or non-silencing control siRNA in serum-free RPMI-1640 using Oligofectamine (Invitrogen). The target sequence was AAACCGAGTCCTCAAAGAGTA. Both specific and control siRNA molecules were synthesized by GenePharma Co., Ltd. (Shanghai, China). Four hours after transfection, an equal volume of medium supplemented with 20% FBS was added. Transfectants were cultured until further analysis was conducted. The vector-based shRNA constructed S2 (nucleotides 519-537) and S3 (nucleotides 1074-1092) that target human CAP1 has been described previously. Plasmids that harbor introduced mismatches for re-expressing mouse CAP1 in the CAP1 knockdown A549 cells were described previously.
Cell viability assay
To determine the cell viability, cells were plated onto 24-well plates (5 × 104 cells/well). After 24 h incubation with different concentrations (0 mM, 10 mM, 20 mM, 40 mM, and 60 mM) of LiCl and incubation with 40mM LiCl for different time (24h, and 48h), 0.5 mg/mL3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide (MTT, Sigma) was added to the cell suspension for further 24 h, followed by addition of dimethyl sulfoxide (DMSO, Sigma) at 100 uL/well for cell lysis. Then, absorbance was measured at 562 nm. Each assay was carried out in triplicate.
Measurements of ROS generation
Lung cancer cells were cultured in 24-well plates, and after stimulation with different concentrations (0 mM, 20 mM, 40 mM, 60 mM, 80 mM, 100 mM) of LiCl for 24 h, cells were loaded with the fluorescent dye H2DCF-DA (10uM, Beyotime Institute of Biotechnology, Haimen, China) for 20 minutes at 37℃. H2DCF-DA fluorescence was detected at excitation and emission wavelengths of 488nm and 520nm, respectively. ROS formation was measured using a multi-well fluorescence scanner (EnSpire 2300, Perkin Elmer, USA).
Antibodies and Western blotting
The monoclonal human CAP1 antibody was previously described. Antibodies against GAPDH and GSK3β antibodies were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Antibodies against ERK1/2, phosphor-ERK1/2 (Thr202/Tyr204), phosphor-cofilin, and phospho-GSK3α/β (Ser21/Ser9) were from Cell Signaling Technology Inc. (Danvers, MA). Cofilin antibody was from Cytoskeleton Inc. (Denver, CO). Cell lysates were prepared for Western blotting similarly as previously described.
Cell migration and Matrigel invasion assays
For wound healing assays, at the designated time points, the images of the wound were captured under a Zeiss Axiovert 200M microscope. The relative migration rate was calculated by the following formula: (wound width at 0 hr-wound width at n hr)/wound width at 0 hr. Transwell migration was conducted similarly as we did previously. For Transwell assays, cells were starved for 4 hrs, harvested and re-suspended in the serum- free medium containing 0.1% bovine serum albumin before seeded onto Transwell chambers. The insert membranes in the invasion assays were coated with 0.25 µg/ml Matrigel. The experiments were repeated three times, and the results were analyzed using Student's t-test.
Immunofluorescence, phase imaging and cell spreading assays
Immunofluorescence was conducted similarly as described previously. Focal adhesions were stained with a vinculin antibody and visualized with Alexa Fluor 594-conjugated goat anti-mouse IgG (H). Confocal images were acquired with the BD Pathway 855 imaging station. For phase imaging, cells were cultured in a 6-well plate overnight, or for 24 hrs (MCF-7), and images were taken with a Zeiss Axiovert 200M microscope. For spreading assays, cells were plated onto fibronec- tin coated dishes, phase contrast images were taken with a Nikon Eclipse TE2000 microscope. For quantification of focal adhesions, 25 cells each field and a total of three fields were counted using the ImageJ software (rsb.info. nih.gov/ij) and the data were analyzed using Student's t-test.
In vivo tumor assay
The animal experiment was approved by the Animal Care and Use Committee at Shanghai Tenth People's Hospital (Shanghai, China). Nude mice were purchased from Shanghai Laboratory Animal Center of China. Stably expressing CAP1 cells were used for generating the xenograft model. For the subcutaneous tumor growth assay, 4 × 106 cells in 100 ml PBS were subcutaneously injected into nude mice (4 mice/group). All mice were sacrificed in 6 weeks, and the tumor weights were measured.
Western blot analysis
Tissue samples were immediately homogenized in buffer containing 1 M Tris-HCl (pH 7.5), 1% Triton X-100, 1% NP-40 (Nonidet P-40), 10% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate, 0.5 M EDTA, 10 mg/ml leupeptin, 10 mg/ml aprotinin and 1 mM PMSF. The samples were centrifuged at 10,000 x g for 30 min to collect the supernatant. Proteins were mixed with loading and DTT (4:5:1) twice, boiled in water for 5-10 min and cooled in ice. Protein concentrations were determined using a Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA), and following total protein quantification, the lysates were loaded onto 10% polyacryl- amide-SDS gels, separated by electrophoresis and blotted onto NC membrane blots using a semi-dry transfer system. The blots were incubated with a mouse anti-human CAP1 antibody and a mouse anti-human actin antibody (both from Sigma, St. Louis, MO, USA) at 4℃ overnight. After washing, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies at room temperature for 45 min. The immunore-active signals for CAP1 and actin were visualized using the ECL system from GE Healthcare UK, Ltd. (Little Chalfont, Buckinghamshire, UK) and subjected to densitometric analyses using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Relative levels of CAP1 (after adjustment against actin) were determined based on the densitometric data.
H&E and immunohistochemical (IHC) SP assay
H&E sections were examined under a microscope to identify and mark the cancer nests. The formalin-fixed and paraffin-embedded (FFPE) sections were dewaxed in xylene and rehydrated in graded ethanol. Endogenous peroxidase activity was blocked by soaking in 0.3% hydrogen peroxide. The sections were then processed in 10 mmol/l citrate buffers (pH 6.0) and heated to 121℃ in an autoclave for 20 min to retrieve the antigen. After rinsing in phosphate-buffered saline (PBS) (pH 7.2), 10% goat serum was applied for 1 h at room temperature to block any non-specific reactions. The sections were then incubated overnight at 4℃ with anti-CAP1 (diluted at 1:500); mouse anti-human monoclonal antibodies against CAP1 were provided by Dr Zhou, University of Pennsylvania School of Medicine (Philadelphia, PA, USA). All the slides were processed using the peroxidase-antiperoxidase method (Dako, Hamburg, Germany). After rinsing with PBS, the peroxidase reaction was visualized by incubating the sections with diaminoben-zidine tetrahydrochloride in 0.05 mol/l Tris buffer (pH 7.6) containing 0.03% H2O2. After rinsing in water, the sections were counterstained with hematoxylin, dehydrated and cover-slipped. Stained sections were observed under a microscope. At least 10 high-power fields were randomly chosen, and ≥400 cells/field were counted.
Quantitative PCR
Total RNA was isolated from cells and tumor specimens using an RNA extraction kit from Isogen (Nippon Gene Co., Ltd., Toyama, Japan). RNA samples were treated with DNase I (Promega Corp., Madison, WI, USA) to remove genomic DNA. First Strand cDNAs were synthesized using a commercial First Strand cDNA Synthesis kit as per the manufacturer's instructions. PCR amplifications of the test gene CAP1 and the reference gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were performed using the primers, forward: 5'-ACT CGC TGC TTG CTG GTC-3' and reverse: 5'-ATG GGT GCC AAC AAA TCG-3', designed based on the human CAP1 mRNA sequence (GenBank accession No. BT007152) and the primers, forward: 5'-GAA GGT GA A GGT CGG AGTC-3' and reverse: 5'-CCC GA A TCA CAT TCT CCA AGA A-3', designed based on the human GAPDH cDNA sequence (GenBank access No. X01677). The reactions were carried out with the SYBR-Green PCR Core Reagents kit (Perkin-Elmer Applied Biosystems, Foster City, CA, USA). The PCR amplification parameters were: 50℃ for 2 min (one cycle), 95℃ for 10 min (one cycle), 95℃ for 15 sec and 60℃ for 1 min (40 cycles). The emission intensity of the SYBR-Green fluorescence was measured as real-time using the ABI PRISM 7700 Sequence Detector from Perkin-Elmer Applied Biosystems. Relative quantification of CAP1 mRNA abundance was performed using the DataAssist software (Life Technologies, Grand Island, NY, USA).
Statistical analysis
Statistical analysis was performed using SPSS version 20.0. Categorical data expressed as a percentage were analyzed using the χ2 test. Differences were considered significant when P<0.05. The results are expressed as the mean ± standard error (SE).
Results
CAP1 is associated with NSCLC
In our previous study [12], we analyzed the structure and function of CAP1 in lung cancer and found that the expression of CAP1 genes was elevated in lung cancer using the database TCGA, ONCOMINE. In addition, these data provide evidence that CAP1 expression plays an important role in lung cancer, and acts as a biomarker for cancer studies and a target for cancer diagnosis.
CAP1 is overexpressed in NSCLC tissues and correlated with poor clinical outcomes
Our previous study [13, 14] demonstrated that the expression of CAP1 was significantly higher in NSCLC tissues compared with their matched normal lung tissues. In addition, the expression of CAP1 in tumor tissues was significantly associated with tumor, lymph node metastasis and TNM stage in NSCLC patients, CAP1 was highly expressed in lung cancer with brain metastasis as compared to other metastatic groups (Bone metastasis, Visceral metastasis). Kaplan-Meier analysis showed that the OS rate in NSCLC patients with high CAP1 levels was significantly lower than that in those with low CAP1 levels. These results suggest that CAP1 may be closely related to the progression and metastasis of NSCLC.
CAP1 overexpression promotes NSCLC cell proliferation and invasion both in vitro and in vivo
Higher expression of CAP1 was detected in six NSCLC cell lines (A549, 95 D, 95 C, H1299, H1975 and PC-9) compared with human bronchial epithelial cells (HP-AEpiC) [Figure S1A, B]. To investigate the potential role of CAP1 in regulating NSCLC cell proliferation, we overexpressed or inhibited CAP1 in several NSCLC cell lines. GFP- CAP1 knockdown and RFP-CAP1 overexpression of lentivirus were transfected into lung cancer cell lines (A549, H1299 and 95D). The cell transfection efficiency was determined by Western Blot [Figure S2A, B].
To assess the function of CAP1, stable transfected cells were established, and functional analyses were performed, knockdown of CAP1 significantly inhibited NSCLC cell invasion [Figure 1A]. In contrast, overexpression of CAP1 significantly promoted cell invasion [Figure 1B].
CAP1 inhibits NSCLC metastasis in vitro and in vivo. (A) Transwell migration assay results show inhibition of CAP1 decreases A549 and 95-D cell invasion, (B) while CAP1 increases H1299 cell invasion. (C) The CAP1 inhibitors stably and (D) the CAP1 expressed cells were injected into the left flanks of nude mice. All mice were killed after six weeks. CAP1 was increased or decreased by overexpression or inhibition of CAP1 in tumor samples by IHC and HE.
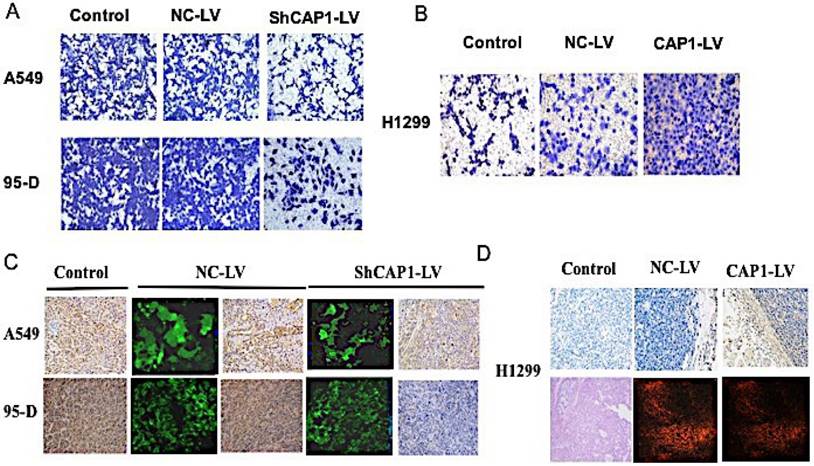
To explore the influence of CAP1 on regulation of oncogenicity of the lung cancer cell in vivo, tumor was implanted by subcutaneous injection of 1.0×107 cells/mouse in 200ul of a 50/50 dilution of DMEM in PBS into the left flanks of the nude mice. All mice were executed in 6 weeks after the inoculation, stripped and put out the tumor which to be calculated the tumor volume according to the formula. All the specimens were fixed with 10% formaldehyde solution, and the expression of CAP1 protein and nucleic acid in tumor tissue was detected by Western Blot, PCR [Figure S3B, C] and IHC [Figure 1C, D]. The tumor volume was significantly lower in CAP1 gene knockout tumor tissue than that in empty virus control group and blank control group [Figure S3A].
CAP1 is a phosphorylatable protein
Our previous studies found that application and mutation were two most prominent alternations of CAP1 in cancers by database analysis [12]. The results showed the application alternation of CAP1 in lung cancer. The following results will show the mutation alternation of CAP1 in lung cancer. A total of nine phosphorylation sites were identified in those mutation sites, CAP1 phosphorylation site S307/S309 was associated with lung cancer [20].
CAP1 phosphorylation site S307/S309 affects NSCLC cell migration and invasion
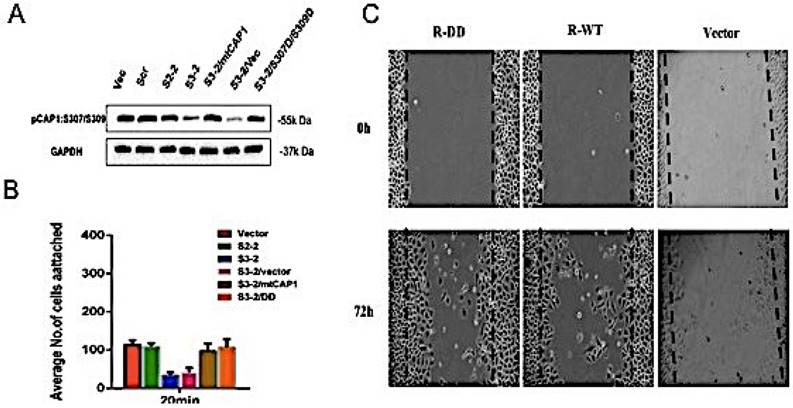
To investigate the role of CAP1 phosphorylation site S307/S309 in the biological characteristics of lung cancer cells, the mutant cells of CAP1 plasmids were transfected into CAP1KO lung cancer cell lines. The transfection efficiency was confirmed by Western Blot [Figure 2A]. Cell adhesion assay was used to detect the ability of CAP1 phosphorylation sites to adhere to lung cancer cells [Figure 2B]. The migration ability of CAP1 phosphorylation sites to lung cancer cells was detected by scratches [Figure 2C]. CAP1 phosphorylation site S307/S309 promoted the migration and invasion of lung cancer cells.
CAP1 phosphorylation site S307/S309 affects the biological characteristics of lung cancer cells. (A) Western blot with antibody confirmed re-expression of the exogenous WT CAP1 and the DD mutants in multiples CAP1 KO stable clones, with GAPDH serving as a loading control. (B) Statistic analysis of the number of focal adhesions per cell. The number of focal adhesions per cell. (C) Wound healing assays showed both wild type and mutated mouse CAP1 rescued the phenotype of elevated cell migration, while expression of the DD mutants further compromised cell migration.
GSK3β is a potential kinase of the CAP1 phosphorylation site S307/S309
To identify the potential kinases that may phosphorylate CAP1 in lung cancer, we also tested several other inhibitors and activators (several kinases that regulate the actin cytoskeleton and/or cell migration), including the ERK inhibitor U0126, the MEK inhibitor PD98059, the PKA inhibitor H89, the PKA activator Forskolin, the MAPK inhibitor PH797804, the MAPK activator Apocynin, PI3K inhibitor LY294002, the AKT inhibitor, the PAK4 inhibitor PF3758309, the JNK inhibitor XVI and the Raf-1 inhibitor BAY 439006, and none of them reduced phosphorylation at S307/S309 [Figure S4A-C]. Those results were further confirmed by the MTT in DD mutation lung cancer cell, however, none of these inhibitors and activators influence proliferation of phosphorylated CAP1 except GSK3β inhibitor LiCl [Figure S4D]. Western Blot test showed that GSK3β was a potential kinase of CAP1 phosphorylation site S307/S309 [Figure 3].
GSK3β inhibitor LiCl reduced CAP1 phosphorylation. Treatment of lung cells with GSK3β inhibitors LiCl reduced CAP1 phosphorylation at S307/S309.
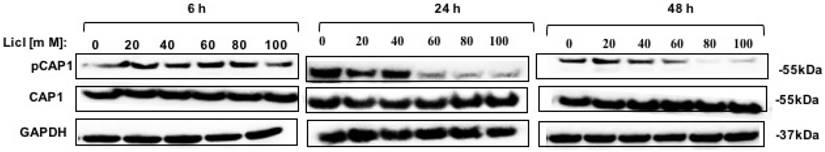
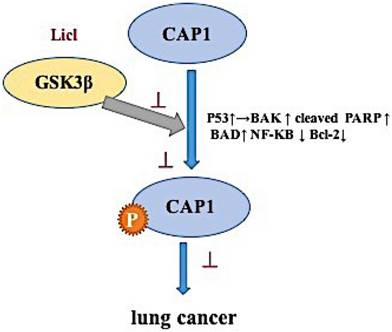
Thus, the inhibitor data suggest that GSK3 is a kinase targeting S307/S309. GSK3β may be an upstream target gene that regulates the phosphorylation site of CAP1, which affects the biological characteristics of lung cancer.
GSK3β inhibitor LiCl affects CAP1 phosphorylation to regulate the biological characteristics of lung cancer cells
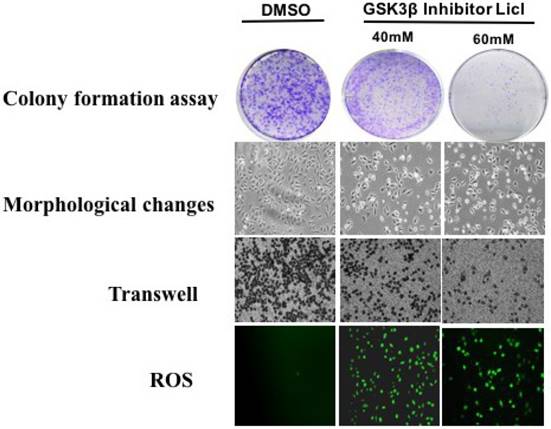
Plate cloning assay, Morphological changes and Transwell method were used to detect the effect of GSK3β inhibitor LiCl on CAP1 phosphorylation of lung cancer cells. GSK3β inhibitor LiCl affects CAP1 phosphorylation to regulate the biological characteristics of lung cancer cells. GSK3β inhibitors LiCl decreased survival and proliferation of lung cancer cell [Figure 4].
GSK3β inhibitor LiCl affects CAP1 phosphorylation to regulate the biological characteristics of lung cancer cells.
GSK3β inhibitor LiCl influences CAP1 phosphorylation inducing ROS-dependent lung cancer cell apoptosis
Reactive oxygen species (ROS) may participate in the process of tumor cell apoptosis signal transduction. We monitored the level of ROS in lung cells. The expression of phosphorylated CAP1, pGSK3/GSK3, NF-κB, p53, cleaved PARP, BAD, BAK and Bcl-2 in lung cancer cell lines was detected by western blot. The western blot result showed that after treating lung cancer A549 cells with GSK3β inhibitor LiCl for 24h, it promotes the expression of ROS and apoptosis-related proteins such as p53, cleaved PARP, BAD, BAK and inactivation of Bcl-2, NF-κB by the re-expression of the DD mutants. [Figure 5]. These results suggested that GSK3β inhibitor LiCl affects CAP1 phosphorylation which induces ROS-dependent lung cancer cell apoptosis.
The western blots result showed that after treating lung cancer A549 cells with GSK3β inhibitor LiCl for 24h, it promotes the expression of ROS and apoptosis-related proteins such as p53, cleaved PARP, BAD, BAK and inactivation of Bcl-2, NF-κB by the re-expression of the DD mutants. The experiments were repeated at least three times with similar results.
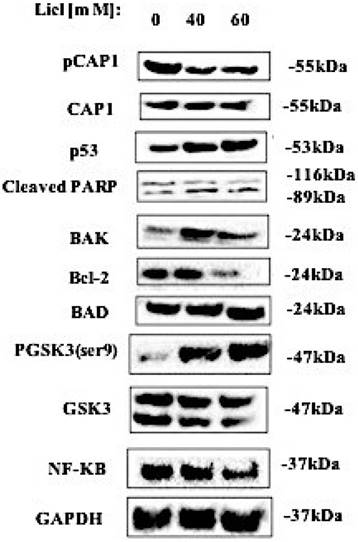
GSK3β kinase inhibitor LiCl inhibits cell phosphorylation site S307/S309 by up-regulating the expression of p53, BAK, BAD and cleaved PARP induced ROS production, decreased lung cancer cell viability, adhesion, proliferation, migration and invasion, and induction of apoptosis [Figure 6].
The mechanism of phosphorylation sites on cyclase-associated protein 1 in the metastasis of lung cancer
Discussion
Consistent with its cellular function as a key actin-regulating protein, mounting evidence supports involvement of CAP1 in the invasiveness of human cancers [8-10]. CAP1 regulation of the actin cytoskeleton is widely conserved [21-24]. Mutation or knockdown of CAPs leads to problems with the actin cytoskeleton, including aberrant cell morphology, polarity and motility [25,26]. Results from our well-controlled and in-depth studies support up-regulation of CAP1 in lung cancer cells, and further, we reveal cell context-dependent functions for CAP1 in the invasiveness, as well as proliferation of lung cancer cells, along with underlying mechanism where GSK3 signaling plays a pivotal role. Next, we found that the functions of CAP1 are regulated through a phosphorylation-dependent mechanism in lung cancer: phosphorylation sites-at S307 and S309 tandem site controls CAP1. We found that GSK3 phosphorylates S307 and S309 by using inhibitors LiCl. Inhibition of GSK3β can cause loss of cell polarity as well as accumulation of stress fibers. We propose that GSK3 regulates CAP1 through at least two mechanisms. First, GSK3 (and potentially other kinases) phosphorylate CAP1 at S309 and promote CAP1 localization to the cytosol. Second, phosphorylation at S309 affects protein-protein interactions with actin and cofilin. The loss of this phospho-regulation by GSK3 inhibition is expected to disrupt CAP1 function and actin dynamics. Additionally, GSK3 can also, as observed by us, affect actin dynamics through other mechanisms.
In recent years, there are more and more clinical targeted kinase inhibitor drugs. For example, Alectinib, a powerful chosen ALK inhibitors, can be effective against ALK kinase region of most mutations, studies have shown that Alectinib without a gram of patients treated for his objective response rate can reach 94%, grams of ALK of drug resistance for his positive NSCLC remission rate is 54%, and no disease progression of survival rate is 83% a year [27, 28]. Our study found CAP1 is mainly through phosphorylation site S307/S309 lung cancer cell invasion and migration function. In addition, GSK3 beta is CAP1 phosphorylation site S307/S309 kinase. Building inhibiting CAP1 phosphorylation sites kinase drug or changing the state of phosphorylation sites can inhibit lung cancer metastasis and revealing the molecular mechanism of interaction between them is very important.
Apoptosis that maintains the balance of cell proliferation and cell death is crucial [29]. Reactive oxygen molecules (ROMs) are of great significance to apoptosis [30]. Under normal circumstances, the body will produce a small number of ROMs, which play an important role in normal physiology. While under the influence of some factors, a large number of ROMs are involved in the occurrence and development of disease. Some anticancer drugs, such as cisplatin, paclitaxel and arsenic trioxide can produce ROMs and induce cancer cell apoptosis [31-33]. Thus, oxidative stress is an overproduction of reactive oxygen species that overwhelms the cellular antioxidant capacity. Hence, it seems that increased ROMs generation and oxidative stress have the central role in lithium cytotoxic mechanism. In our study, we showed for the first time that LiCl exerted the prooxidant effects in lung cancer cells.
GSK-3 is a pluripotent serine-threonine kinase with numerous intracellular target proteins. GSK-3 isoforms are encoded by distinct genes. LiCl has been shown to induce cell growth arrest, apoptosis, and terminal differentiation in various human malignant tumors by targeting GSK-3[34]. Therefore, we used LiCl to inhibit the activity of GSK-3 pharmacologically and found the increased generation of ROS, which implied the involvement of ROS inactivation of GSK3β in the regulation of cancer development. NF-κB is an important transcription factor involved in growth arrest and apoptosis by regulating the expression of numerous target genes such as Bcl-2[35]. Our study found that GSK3β inhibitor LiCl active transcription factor by NF-κB, p53, Bax, Bcl-2 gene expression, such as induction of ROMs generation, thus inhibiting cell proliferation, inducing cell apoptosis.
GSK3β inhibitor LiCl promotes cell apoptosis by decreasing phosphorylation CAP1. LiCl-induced cell death is dependent on p53, GSK-3 may represent a promising target for cancer therapy. The results of comparing the single point mutations of S309 and S307 with the AA and DD double mutations in biochemical and cell biological analyses support the notion of the two residues function in tandem. Of note, there are two actin binding sites on CAP1, the Cofilin-ADP-G-actin binding site at the N-terminus and the G-actin binding/sequestering site at the C-terminus. However, it remains to be determined whether the differences in binding Cofilin and actin, as well as the subcellular localization of CAP1, are solely responsible for the cellular phenotype caused by these mutants. Next, we will further study how mutant sites infect CAP1-terminal domain in lung cancer.
Conclusions
In summary, our work reveals a phosphorylation-dependent regulatory mechanism of CAP1, by identifying a direct phosphorylation of CAP1 by GSK3 at S309, which, together with phosphorylation of S307, collaboratively regulates CAP1 function. These findings contribute mechanistic insights into how cell signaling controls the actin cytoskeleton through molecules that regulate actin dynamics.
Supplementary Material
Supplementary figures.
Acknowledgements
The authors thank Jeffrey Field (University of Pennsylvania, Philadelphia, PA) for helpful discussions, Jeffrey Field (University of Pennsylvania, Philadelphia, PA), Guolei Zhou (Arkansas State University, State University, AR 72467, USA) for the gift of CAP1 siRNA, S307/S309DD plasmid and S307/S309 antibody.
Author contributions
SSX and J.Z conceived, designed and supervised most of the project, and wrote the manuscript. SS X, CH.W. and JF. conducted the experiments and analyzed the data. J.F. conceived and supervised the project at the early stage. MT and ZGL organized the figures and tables.
Funding
This study was funded by the National Natural Science Foundation of China (No.81472180)
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
2. Ramalingam SS, Owonikoko TK, Khuri FR.Lung cancer. new biological insights and recent therapeutic advances. CA Cancer J Clin. 2011;61:91-112
3. American Cancer Society: Cancer Facts & Figures 2014, Atlanta, Georgia, 2014. http://www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2014/
4. Ono S. The role of cyclase-associated protein in regulating actin filament dynamics-more than a monomer-sequestration factor. J Cell Sci. 2013;126:3249-58
5. Pappa KI, Lygirou V, Kontostathi G. et al. Polyzos A, Anagnou NP. Proteomic Analysis of Normal and Cancer Cervical Cell Lines Reveals Deregulation of Cytoskeleton-associated Proteins. Cancer Genomics Proteomics. 2017;14:253-266
6. Hubberstey A. V. and Mottillo, E. P. Cyclase-associated proteins: CAPacity for linking signal transduction and actin polymerization. FASEB J. 2002;16:487-499
7. Ono S. The role of cyclase-associated protein in regulating actin filament dynamics - more than a monomer-sequestration factor. J. Cell Sci. 2003;126:3249-3258
8. Chang CY, Leu J, Lee YJ. The actin depolymerizing factor (adf)/cofilin signaling pathway and DNA damage responses in cancer. Int J Mol Sci. 2015;16:4095-4120
9. Nishida E, Maekawa S, Sakai H. Cofilin, a protein in porcine brain that binds to actin filaments and inhibits their interactions with myosin and tropomyosin. Biochemistry. 1984;23:5307-5313
10. Bravo-Cordero JJ, Magalhaes MA, Eddy RJ. et al. Functions of cofilin in cell locomotion and invasion. Nat Rev Mol Cell Biol. 2013;14:405-415
11. Zhou G. L, Zhang, H, Field, J. Mammalian CAP (Cyclase-associated protein) in the world of cell migration: Roles in actin filament dynamics and beyond. Cell Adhes Migr. 2014;8:55-59
12. Xie S, Shen C, Tan M. et al. Systematic analysis of gene expression alterations and clinical outcomes of adenylate cyclase-associated protein in cancer. Oncotarget. 2017;8:27216-27239
13. Xie SS, Tan M, Lin HY. et al. Overexpression of adenylate cyclase-associated protein 1 may predict brain metastasis in non-small cell lung cancer. Oncol Rep. 2015;33:363-71
14. Tan M, Song X, Zhang G. et al. Overexpression of adenylate cyclase-associated protein 1 is associated with metastasis of lung cancer. Oncol Rep. 2013;30:1639-44
15. Yamazaki K, Takamura M, Masugi Y. et al. Adenylate cyclase-associated protein 1 overexpressed in pancreatic cancers is involved in cancer cell motility. Lab Invest. 2009;89:425-432
16. Yu XF, Ni QC, Chen JP. et al. Knocking down the expression of adenylate cyclase-associated protein 1 inhibits the proliferation and migration of breast cancer cells. Exp Mol Pathol. 2014;96:188-194
17. Fan YC, Cui CC, Zhu YS. et al. Overexpression of CAP1 and its significance in tumor cell proliferation, migration and invasion in glioma. Oncol Rep. 2016;36:1619-25
18. Hua M, Yan S, Deng Y. et al. CAP1 is overexpressed in human epithelial ovarian cancer and promotes cell proliferation. Int J Mol Med. 2015;35:941-9
19. Zhang H, Zhou GL. CAP1 (Cyclase-Associated Protein 1) Exerts Distinct Functions in the Proliferation and Metastatic Potential of Breast Cancer Cells Mediated by ERK. Sci Rep. 2016;13:6 25933
20. Zhou GL, Zhang H, Wu H. et al. Phosphorylation of the cytoskeletal protein CAP1 controls its association with cofilin and actin. J Cell Sci. 2014;127:5052-65
21. Zhang H, Ghai P, Wu H. et al. Mammalian adenylyl cyclase-associated protein 1 (CAP1) regulates cofilin function, the actin cytoskeleton, and cell adhesion. J Biol Chem. 2013;288:20966-77
22. Kawamukai M Gerst J, Field J. et al. Genetic and biochemical analysis of the adenylyl cyclase-associated protein, cap, in Schizosaccharomyces pombe. Mol Biol Cell. 1992;3:167-80
23. Matviw H, Yu G. and Young, D. Identification of a human cDNA encoding a protein that is structurally and functionally related to the yeast adenylyl cyclase-associated CAP proteins. Mol. Cell. Biol. 1992;12:5033-40
24. Zelicof A, Gatica J. and Gerst, J. E. Molecular cloning and characterization of a rat homolog of CAP, the adenylyl cyclase-associated protein from Saccharomyces cerevisiae. J Cell Sci. 2005;118:19-26
25. DesMarais V, Ghosh M, Eddy R, Condeelis J.Cofilin takes the lead. J. Cell Sci. 2005;118:19-26
26. Lappalaien P. nd Drubin, D. G. (1997). Cofilin promotes rapid actin filament turnover in vivo. Nature 388, 78-82.
27. Soda M, Choi YL, Enomoto M. et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561-566
28. Soda M, Takada S, Takeuchi K. et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci U S A. 2008;105:19893-19897
29. ML Circu, TY Aw. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radical Biology & Medicine. 2010;48:749-762
30. Cai XB, Jing XB, Hu H. et al. The role of reactive oxygen species in cisplatin-induced apoptosis of esophageal cancer cell line EC-109. Ai Zheng. 2006;25:427-31
31. Woo SH, Park IC, Park MJ. et al. Arsenic trioxide sensitizes CD95/Fas-induced apoptosis through ROS-mediated upregulation of CD95/Fas by NF-kappaB activation. Int J Cancer. 2004;112:596-606
32. Xiao D, Herman-Antosiewicz A, Antosiewicz J. et al. Diallyl trisulfide-induced G(2)-M phase cell cycle arrest in human prostate cancer cells is caused by reactive oxygen species-dependent destruction and hyperphosphorylation of Cdc 25 C. Oncogene. 2005;15:24 6256-68
33. Hochman A, Sternin H. et al. Enhanced oxidative stress and altered antioxidants in brains of Bcl-2-deficient mice. J Neurochem. 1998;71:741-8
34. Fu ZQ, Yang Y, Song J. et al. LiCl attenuates thapsigargin-induced tau hyperphosphorylation by inhibiting GSK-3β in vivo and in vitro. J Alzheimers Dis. 2010;21:1107-17
35. 35.Arbab IA, Looi CY, Abdul AB. et al. Dentatin Induces Apoptosis in Prostate Cancer Cells via Bcl-2, Bcl-xL, Survivin Downregulation, Caspase-9, -3/7 Activation, and NF-κB Inhibition. Evid Based Complement Alternat Med. 2012;2012:856029
Author contact
![]() Corresponding author: Changhui Wang. Shanghai Tenth People's Hospital, Tongji University, Shanghai 200072, China. Email: wang-chang-huicom; Fax number: 86-021-66301685, Telephone: 86-021-66301685
Corresponding author: Changhui Wang. Shanghai Tenth People's Hospital, Tongji University, Shanghai 200072, China. Email: wang-chang-huicom; Fax number: 86-021-66301685, Telephone: 86-021-66301685