Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(12):2203-2210. doi:10.7150/jca.24024 This issue Cite
Research Paper
Predicting hepatocellular carcinoma development for cirrhosis patients via methylation detection of heparocarcinogenesis-related genes.
Yuan Huang1,#, Ling Wei1,#, Rong-Ce Zhao2, Wei-Bo Liang3, Jing Zhang4, Xue-Qin Ding4, Zhi-Long Li3, Cheng-Jun Sun4, Bo Li2, Qiu-Ying Liu1, Jing-Yang He1, Xiao-Qin Yu1, Bo Gao5, Ming-Mei Chen1, Ai-Min Sun5, ![]() , Yang Qin1,
, Yang Qin1, ![]()
1. Department of Biochemistry and Molecular Biology, West China School of Basic Medical Sciences & Forensic Medicine, Sichuan University, Chengdu 610041, China
2. Division of Liver Transplantation, Department of Liver Surgery, West China Hospital, Sichuan University, Chengdu, Sichuan Province,China
3. Department of Forensic Genetics, West China School of Basic Science and Forensic Medicine, Sichuan University, Chengdu 610041, China
4. West China School of Public Health, Sichuan University, Chengdu 610041, China
5. Analytical & testing center, Sichuan University, Chengdu, Sichuan Province, China
#These authors contributed equally to this work
Received 2017-11-26; Accepted 2018-4-4; Published 2018-6-4
Abstract
Background: Most hepatocellular carcinoma (HCC) patients have undergone a progression from chronic hepatitis, then liver cirrhosis (LC), and finally to carcinoma. The objective of this study was to elucidate risk factors to predict HCC development for cirrhosis patients.
Methods: Multiple methylated specific PCR (MSP) was applied to determine methylation status of heparocarcinogenesis-related genes in 396 tissue and plasma specimens and multivariate cox model was used to analyze the relationship between risk variables and HCC development among cirrhosis patients, followed up in a median period of 30 months.
Results: Among 105 LC cases, HCC incidence rate at 30 months was 30.48% (32/105), which were statistically associated with patients' age and aberrant methylation of p16, SFRP, and LINE1 (p<0.05). Receiver operating characteristic (ROC) curve showed the overall predictive accuracy reached the highest (90.7%) if the four risk variables were concurrent to predict HCC development. Moreover, along with the growth of age from 0-40, 40-55, to 55-70 years or the increased number of aberrantly-methylated gene from 0-1 to 2-3, the HCC incidence rate of cirrhosis patients rised from 10.00%, 12.28% to 82.14% and 17.44% to 89.47%, separately. Thus, based on combined analysis with diverse age and number of aberrantly-methylated gene, 105 cases were divided into five groups and computed their respective HCC incidecne rate to categorize them into different risk groups. Of note, A significant lifting of HCC incidence rate in the high-risk group (40-55 years coupled with 2-3 aberrantly-methylated genes, 55-70 years coupled with 0-1 aberrantly-methylated gene, 55-70 years coupled with 2-3 aberrantly-methylated genes; n=33) was observed compared with the low-risk group (0-40 years coupled with 0-1 aberrantly-methylated gene, 40-55 years coupled with 0-1 aberrantly-methylated gene; (n=72) (p<0.01).
Conclusions: Ultimately, high-risk cirrhosis patients with 55-over years or 2-3 aberrantly-methylated genes should be paid more attention to be regularly screened with HCC development.
Keywords: Liver cirrhosis, Methylation, Hepatocellular carcinoma, Prediction, HCC incidence, Biomarkers.
Introduction
Hepatocellular carcinoma (HCC) was the most frequent liver cancer affecting around 700,000 patients every year[1]. To date, their poor prognosis remained a problem due to intrahepatic spread and extrahepatic metastasis[2]. Furthermore, curative treatments, like surgical resection, radiofrequency ablation, liver transplantation, were only confined to early-stage cancer[3]. As we know, HCC always occurred in patients with underlying chronic liver disease, such as cirrhosis patients. Therefore, it is of clinical importance to identify non-invasive risk variables for monitoring and screening high-risk patients with cirrhosis.
To date, we still remain unclear about the molecular pathogenesis of heparocarcinogenesis. However, it has been revealed that epigenetic aberrance, especially global DNA hypomethylation concomitant with locus-specific DNA hypermethylation in gene promoters, plays vital roles in carcinoma progression[4-7]. Additionally, alterations in DNA methylation patterns contributes to early-stage hepatocarcinogenesis[8] and as compared to cirrhosis, aberrantly-methylated genes tested in HCC were enriched[9]. Moreover, DNA methylation markers could be utilized to detect human cancers in blood, plasma, secretion, or exfoliated cytology specimens and predict the risk of cancer development[10]. Thus, cell free DNA (cfDNA) circulating in plasma of cirrhosis patients may represent a promising non-invasive alternative for HCC screening and monitoring.
RAS association domain family 1A (RASSF1A), p16, Secreted frizzled-related protein 1 (SFRP1), Embryonic liver fodrin (ELF), Suppressor of cytokine signaling 3 (SOCS3), p53, Glutathione S-transferaseP1 (GSTP1), Hepatocellular carcinoma suppressor 1 (HCCS1), Doublecortin domain-containing 2 (DCDC2), Histidine triad nucleotide-binding protein 1 (Hint1) hypermethylation and Long interspersed nuclear elements (LINE1) hypomethylation have been demonstrated to be associated with hepatocarcinogenesis[11-19]. Multiplex methylated sepcific PCR (multiplex MSP) was implemented to detect methylation status of candidate genes elected from database. Afterwards, we took the initiative to follow up a cohort of patients with cirrhosis to elucidate contributing risk variables, predicting HCC development, to guide monitoring and surveillance for these high-risk individuals.
Materials and Methods
Clinical specimens
Plasma and tissues samples were obtained from the West China Hospital in Sichuan University with informed consent, comprising of 119 HCC, 105 liver cirrhosis, 52 benign lesion patients (liver angioma, etal) and 50 healthy people. We collected 326 plasma specimens from above these people and 70 tissue specimens, with 40 HCC and 30 paired non-HCC tissue included, from 119 HCC patients. Among the 105 LC patients, 77 were male and 28 were female, aged from 18 to 70 years with average age being 45 years and there were 78 HBV-positive cases and 6 serum AFP > 400ng/ml cases.
DNA extraction and Bisulfite modification
According to the manufacturer's protocol, genomic DNA was extracted from plasma samples with a commercial DNA-extraction kit (AxyPrep Body Fluid Viral DNA/RNA Miniprep Kit; AxyPrep, China). Likewise, DNA was isolated from tissue samples by TIANamp Genomic DNA Kit (TIANGEN, China). 200-500ng plasma or tissue DNA was subjected to sodium bisulfite modification conducted by the EZ DNA Methylation kit (Zymo Research).
Multiplex methylated sepcific PCR
To investigate the methylation status of CpG islands of RASSF1A, p16, SFRP1, SOCS3 and LINE1, multiplex MSP was performed in a 25µL-volume reaction system, consisted of 50ng sodium-bisulfite treated DNA, isometric mixture of gene primers 3µL, 2×Master Mix 12.5µL (Qiagen, Germany) and ddH2O. The multiplex MSP primer sequences for RASSF1A, p16, SFRP1, SOCS3 and LINE1 were described in Table S1[11,14,20-22]. The reaction conditions were listed as follows: denaturation at 95℃ for 15 min, 30 cycles of 94℃ for 30s, annealing for 90s and 72℃ for 90s, with an ultimate extension of 10 min at 72℃. The multiplex MSP products were analyzed by capillary electrophoresis (CE).
Follow-up
The LC patients were followed up through telephone calls and the average follow-up period was 30 months. They were inspected routinely with serum AFP level and abdominal ultrasonography in hospital. Also, computed Tomography (CT) was implemented together with chest radiographic examination to get aware of the disease progression.
Statistical data analysis
SPSS19.0 statistical software was applied for data analysis, including the Pearson's χ2 test or Fisher's exact test. Univariate and multivariate cox regression analysis were explored to estimate risk variables for patients. The overall predictive accuracy for HCC incidence was summarized by Receiver operating characteristic (ROC) curve. All P values were two-tailed and were considered significant when less than 0.05.
Results
Candidate genes screened from GEPIA database and pre-experiments
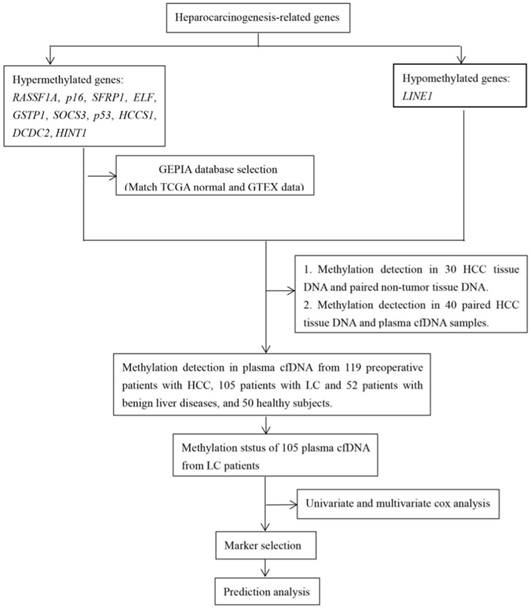
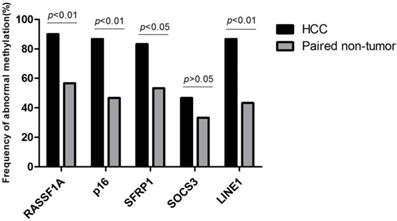
The workflow chart about candidate genes selection (RASSF1A, p16, SFRP1, ELF, SOCS3, GSTP1, HCCS1, DCDC2, Hint1 and LINE1) was shown in Fig. 1. Analyzed by The Gene Expression Profiling Interactive Analysis (GEPIA) database, RASSF1A, p16, SFRP1, SOCS3 was revealed as hypermethylated genes. Subsequently, we detected the methylation status of RASSF1A, p16, SFRP1, SOCS3, and LINE1 in 30 HCC and paired non-HCC tissue DNA. As shown in Fig. 2, abnormal methylation rate of RASSF1A, p16, SFRP1, SOCS3, and LINE1 were found in 27 of 30 (90%), 26 of 30 (86.67%), 25 of 30 (83.33%), 14 of 30 (46.67%), 26 of 30 HCCs (86.67%), and 17 of 30 (56.67%), 14 of 30 (46.67%), 16 of 30 (53.33%), 10 of 30 (33.33%), 13 of 30 paired non-HCCs (43.33%) (p<0.01; p<0.01; p<0.05; p>0.05; p<0.01), respectively. Thus, SOCS3 was removed for its insignificant difference between the two cohorts. Then, we compared the concordance of methylation status of RASSF1A, p16, SFRP1, and LINE1 in 40 HCC tissue DNA and paired plasma cfDNA. Accoding to the simple kappa coefficient test, RASSF1A was ruled out for its consistency lower than 0.4 (Table 1). All in all, p16, SFRP1, and LINE1 were enrolled in the prediction analysis.
Workflow chart of data generation and analysis.
Comparison of the concordance of methylation status for six genes in 40 HCC cases and tumor plasma (Simple kappa coefficient).
| Gene | Tumor tissue (n,%) | Corresponding tumor plasma (n,%) | P-value | Kappa |
|---|---|---|---|---|
| RASSF1A | 36 (90.0) | 29 (72.5) | 0.025 | 0.297 |
| p16 | 31 (77.5) | 27 (67.5) | 0.000 | 0.628 |
| SFRP1 | 29 (72.5) | 22 (55.0) | 0.000 | 0.634 |
| LINE1 | 30 (75.0) | 23 (57.5) | 0.000 | 0.514 |
Frequency of abnormal methylation in tissue DNA of RASSF1A, p16, SFRP1, SOCS3, and LINE1 among 30 HCC and paired non-HCC specimens. HCC, hepatocellular carcinoma; RASSF1A, RAS association domain family 1A; SFRP1, Secreted frizzled-related protein 1; SOCS3, Suppressor of cytokine signaling 3; LINE1, Long interspersed nuclear element.
Methylation frequency of p16, SFRP1, LINE1 in plasma samples
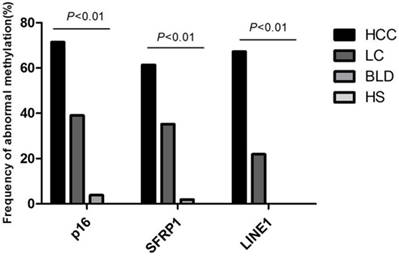
To determine whether methylation status in plasma could be employed for monitoring the multistep carcinogenesis, Multiplex MSP was applied to assay the methylation status for p16, SFRP1, and LINE1 in plasma specimens of 119 HCC patients, 105 LC patients, 52 patients with benign lesions and 50 healthy people (Figure S1). Aberrant methylation of p16, SFRP1, and LINE1 was measured in 85 of 119 (71.43%), 73 of 119 (61.34%), 80 of 119 (67.23%) HCCs, in 41 of 105 (39.05%), 37 of 105 (35.24%), 23 of 105 (21.90%) cirrhotic livers, in 2 of 52 (3.85%), 1 of 52 (1.92%), 0 of 52 (0%) patients with benign lesions, seperately, and no aberrant methylation was detected in normal people (p<0.01) (Figure 3). The average number of aberrantly-methylated gene in healthy subjects, benign lesions, LC, and HCC patients was enriched gradually from 0, 0.06, 0.96 to 2.33, certifying that hepatocarcinogenesis was associated with increasing DNA methylation.
Frequency of abnormal methylation in plasma DNA of p16, SFRP1 and LINE1 among 119 HCC, 105 LC, 52 benign liver disease patients and 50 healthy subjects. SFRP1, Secreted frizzled-related protein 1; LINE1, Long interspersed nuclear element; HCC, Hepatocellular carcinoma; LC, Liver cirrhosis; BLD, Benign liver diseases; HS, Healthy subjects.
The concurrent analysis of age and plasma p16, SFRP1, LINE1 methylation could promote the overall accuracy for HCC incidence prediction among LC patients
After the methylation status of p16, SFRP1, and LINE1 in plasma cfDNA was investigated, 105 cirrhosis patients were followed up in a median period of 30 months. 32 out of them developed HCC and the HCC incidence rate was 30.48%. Then, we summarized the risk factors (Age, gender, HBsAg, Anti-HCV, and AFP level) and abnormally-methylated genes (p16, SFRP1, and LINE1) to participate in the univariate and multivariate cox analysis (Table 2). As data demonstrated, patients' age and aberrant methylation of p16, SFRP1, and LINE1 were statistically related with HCC development among the 105 high-risk individuals. Aimed to explore whether the concurrent four risk variables were superior for HCC development, we analyzed the sensitivity, specificity and predictive accuracy with the use of singe or multi-risk panels based on Receiver operating characteristic (ROC) curve. Table 3 presented that along with the increased number of risk variables enrolled in HCC incidence prediction, the overall accuracy kept an elevating trend. Most importantly, the sensitivity, specificity, and overall predictive accuracy reached the highest with the prediction panel containing the four variables together (93.8%, 63.0%, 90.7%, respectively).
Univariate anaysis and multivariate analysis of 105 patients with cirrhosis liver in relation to HCC development.
| Variable (n=61) | No.of patients | No. of patients with HCC incidence | Univariate anaysis | multivariate analysis | |||
|---|---|---|---|---|---|---|---|
| Hazard ratio (95% CI) | p | Hazard ratio (95% CI) | p | ||||
| Age | >50 years | 44 | 25 | ||||
| ≤50 years | 61 | 7 | 6.203 (2.672-14.382) | 0.000 | 0.355 (0.144-0.874) | 0.024 | |
| Gender | Male | 77 | 23 | ||||
| Female | 28 | 9 | 0.937 (0.434-2.026) | 0.869 | 0.996 (0.437-2.270) | 0.992 | |
| AFP (μg/L) | ≥400 | 6 | 4 | ||||
| <400 | 99 | 28 | 5.081 (1.770-14.583) | 0.003 | 1.609 (0.405-6.399) | 0.500 | |
| HBsAg | + | 78 | 29 | ||||
| - | 27 | 3 | 3.768 (1.148-12.375) | 0.029 | 0.614 (0.165-2.286) | 0.467 | |
| Anti-HCV | + | 2 | 0 | ||||
| - | 103 | 32 | 0.048 (0.000-1744.723) | 0.570 | 14020.160 (0.000-) | 0.982 | |
| p16 | M | 41 | 19 | ||||
| U | 64 | 13 | 2.837 (1.398-5.754) | 0.004 | 0.327 (0.154-0.696) | 0.004 | |
| SFRP1 | M | 37 | 18 | - - | |||
| U | 68 | 14 | 2.975 (1.476-5.293) | 0.002 | 0.111 (0.037-0.327) | 0.000 | |
| LINE1 | M | 23 | 16 | ||||
| U | 82 | 16 | 0.186 (0.093-0.373) | 0.000 | 0.068 (0.022-0.209) | 0.000 | |
Comparison of the predictive accuracy of age and plasma p16, SFRP1, LINE1 methylation, when used alone or combined, in liver cirrhosis patients.
| Sensitivity | Specificity | AUC (Area under curve) | Youden index | |
|---|---|---|---|---|
| Age | 78.10% | 28.8% | 74.7% | 0.50 |
| SFRP1 | 56.3% | 26.0% | 65.1% | 0.50 |
| LINE1 | 50.0% | 8.2% | 70.9% | 0.50 |
| p16 | 59.4% | 31.5% | 63.9% | 0.50 |
| Age+SFRP1 | 87.5% | 34.2% | 82.4% | 0.23 |
| Age+LINE1 | 96.9% | 31.5% | 86.5% | 0.23 |
| Age+p16 | 78.1% | 39.7% | 77.2% | 0.22 |
| SFRP1+LINE1 | 87.5% | 34.2% | 82.4% | 0.23 |
| SFRP1+p16 | 75.0% | 54.8% | 70.3% | 0.23 |
| LINE1+p16 | 78.1% | 39.7% | 77.2% | 0.22 |
| Age+SFRP1+LINE1 | 96.9% | 31.5% | 88.8% | 0.17 |
| Age+SFRP1+p16 | 93.8% | 63.0% | 86.8% | 0.08 |
| Age+LINE1+p16 | 96.9% | 31.5% | 89.6% | 0.19 |
| SFRP1+LINE1+p16 | 93.8% | 63.0% | 86.8% | 0.19 |
| Age+SFRP1+p16+LINE1 | 93.8% | 63.0% | 90.7% | 0.19 |
HCC incidence rate elevated along with the growth of age or increased number of abnormally-methylated gene
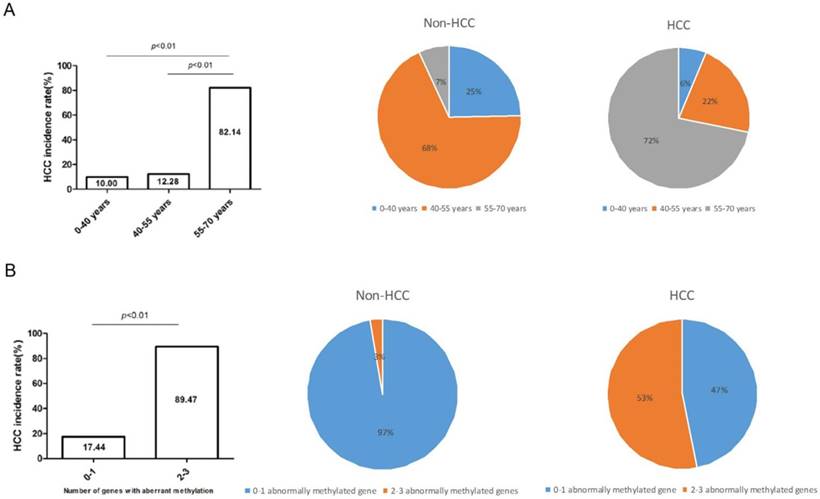
According to the diverse age, we divided high-risk populations into three groups, namely, 0-40 years group (n=20), 40-55 years group (n=57), 55-70 years group (n=28), and calculated their respective HCC incidence rate. As the histogram showed, HCC incidence rate gradually lifted from 10%, 12.28%, to 82.14% (P<0.01) along with the older age. Subsequently, HCC incidence rate of the three groups was computed in non-HCC and HCC cohorts and was dramatically raised in 55-70 years group (7%-72%) (Figure 4A). Analogously, HCC incidence rate lifted along with the increased number of aberrantly-methylated gene. Compared with patients with 0-1 aberrantly-methylated gene (17.44%), patients with 2-3 aberrantly-methylated genes were more likely to suffer from HCC development (89.47%). Moreover, in the fan chart of HCC group, patients with 2-3 aberrantly-methylated genes took up a thumping majority (53%), in comparison with non-HCC group (3%) (Figure 4B).
HCC incidence rate elevated along with the growth of age (A) or increased number of abnormally-methylated gene (B). The proportion of patients' age and the number of abnormally methylated genes were distinctly different between non-HCC and HCC group among 105 cases and the HCC incidence rate dramatically lifted among LC patients with ≥55 years or more than 2 abnormally-methylated genes.
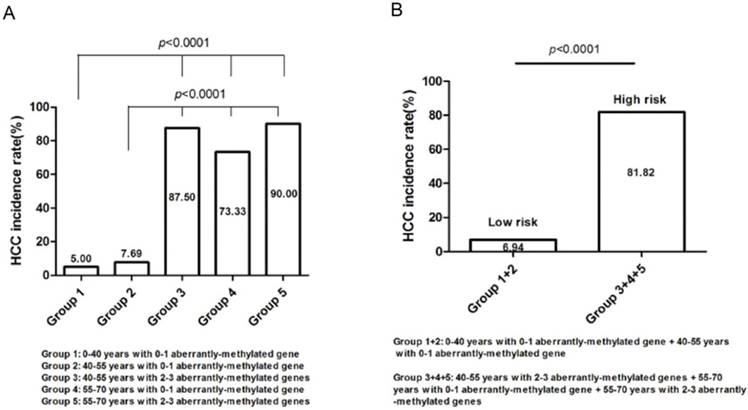
Risk group classification based on the concurrent analysis of age and number of abnormally-methylated gene
105 high-risk populations were separated into five groups (Table 4) and computed the respective HCC incidence rate (Figure 5), that is, group 1 (0-40 years with 0-1 aberrantly-methylated gene, n=20; 5.00%), group 2 (40-55 years with 0-1 aberrantly-methylated gene, n=52; 7.69%), group 3 (40-55 years with 2-3 aberrantly-methylated genes, n=8; 87.50%), group 4 (55-70 years with 0-1 aberrantly-methylated gene, n=15; 73.33%), and group 5 (55-70 years with 2-3 aberrantly-methylated genes, n=10; 90.00%). The incidence rate of group 3, 4, and 5 was statistically significant than group 1 and 2 (P<0.0001) (Figure 5A). Hence, we regarded patients with the age of 40-55 years coupled with 2-3 aberrantly-methylated genes, 55-70 years coupled with 0-1 aberrantly-methylated gene, and 55-70 years coupled with 2-3 aberrantly-methylated genes as the high-risk group (n=33), and patients with the age of 0-40 years coupled with 0-1 aberrantly-methylated gene and 40-55 years coupled with 0-1 aberrantly-methylated gene as the low-risk group (n=72). The incidence rate between the two risk groups was statistically significant (P<0.0001) (Figure 5B). Ultimately, high-risk cirrhosis patients with 55-over years or 2-3 aberrantly-methylated genes should be paid more attention with regular monitoring and screening of HCC development.
Groups classification based on concurrent analysis with diverse age and number of aberrantly-methylated gene.
| Group | 105 cases (n) | HCC incidence (n) | HCC incidence rate (%) |
|---|---|---|---|
| 0-40 years with 0-1 aberrantly-methylated gene | 20 | 1 | 5.00% |
| 0-40 years with 2-3 aberrantly-methylated genes | 0 | - | - |
| 40-55 years with 0-1 aberrantly-methylated gene | 52 | 4 | 7.69% |
| 40-55 years with 2-3 aberrantly-methylated genes | 8 | 7 | 87.50% |
| 55-70 years with 0-1 aberrantly-methylated gene | 15 | 11 | 73.33% |
| 55-70 years with 2-3 aberrantly-methylated genes | 10 | 9 | 90.00% |
Risk group classification based on the concurrent analysis of age and number of abnormally-methylated gene. (A). The relationship between HCC incidence rate and groups divided by diverse age and number of aberrantly-methylated gene. (B). High-risk and low-risk groups were defined according to the distinct difference of HCC incidence rate.
Discussion
In previous investigations, older age[23-24], male sex[25-26], severity of compensated cirrhosis at presentation, and sustained activity of liver disease[27] are important predictors of HCC incidence. Whereas, in this study, significant efforts had been put emphasis on the discovery and detection of other biomakers for early warning of high-risk individuals with cirrhosis for HCC incidence. It seemed that Promoter methylation appeared to be one of the earliest epigenetic abnormalities in human hepatocarcinogenesis[28-29]. And the molecular alteration could be already detectable in cirrhosis, representing a premalignant liver condition as overwhelming majority of HCC arised in the context of liver cirrhosis[30]. Above all, thus far, fewer than 20% of cirrhosis patients were efficaciously enrolled in surveillance programs[31]. These findings demonstrated the promise of gene promoter aberrant methylation in plasma as a molecular marker for identifying high-risk cirrhosis patients, more likely to suffer from cancer development[32].
Present studies have suggested that expression alterations of p16, SFRP1, and LINE1 were intimately implicated in hepatocarcinogenesis. Thus, the aberrant methylation of the three genes was included as biomarkers for screening and monitoring high-risk patients with cirrhosis. We observed that the average number of aberrantly methylated gene showed an increase from the progression of cirrhosis to HCC, consistent with the previous reports[11]. Whereafter, among 105 LC cases, who were investigated the three genes' methylation status, 32 patients developed HCC in a short follow-up period. Multivariate analysis suggested that HCC incidence was significantly depending on age and the aberrant methylation of p16, SFRP1, and LINE1, which could be considered as candidate contributing variables for HCC development. ROC analysis indicated that concurrent analysis of the four variables had a overall 90.7% predictive accuracy. Addtionally, HCC incidence rate was greatly in line with the increase of aberrantly-methylated gene' s number, which was in accordance with the result that genes epigenetically altered in HCC were significantly enriched along with HCC development[30]. Identification of risk variables for HCC incidence among high-risk patients with cirrhosis was extremely momentous, because they can be screened carefully in case of HCC incidence and given potentially curative treatments. According to the data suggested, patients with ≥55 years old or 2-3 abnormally-methylated genes may have the higher risk with HCC incidence, which may be valuable in assessing the risk of HCC incidence among high-risk individuals during a short period.
Conclusions
This is the first study to prospectively examine the relationship between epigenetic alterations in heparocarcinogenesis-related genes, clinical characteristics and HCC incidence among cirrhosis patients to guide monitoring and surveillance for these high-risk populations.
Abbreviations
HCC, hepatocellular carcinoma; LC, liver cirrhosis; Multiple MSP, multiple methylated specific PCR; RASSF1A, RAS association domain family 1A; SFRP1, Secreted frizzled-related protein 1; ELF, Embryonic liver fodrin; SOCS3, Suppressor of cytokine signaling 3; GSTP1, Glutathione S-transferaseP1; HCCS1, Hepatocellular carcinoma suppressor 1; DCDC2, doublecortin domain-containing 2; Hint1, Histidine triad nucleotide-binding protein 1; LINE1, Long interspersed nuclear element; AFP, alpha-foetoprotein; CT, Computed Tomography; ROC, Receiver operating characteristic; AUC, Area under the ROC curve.
Supplementary Material
Supplementary figure and table.
Acknowledgements
This study was supported by “Science & Technology Department of Sichuan Province (no. 2013SZ0048)
Ethics approval and consent to participate
Medical administration office of Sichuan University had approved the study and plasma specimens were obtained with patients' constent. All experiments were performed in accordance with the relevant guidelines and regulations.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Augusto Villanueva, Josep M. Llovet. Mutational landscape of HCC—the end of the Beginning. Nat Rev Clin Oncol. 11(2014)73-74. doi: 10.1038/nrclinonc. 2013 243
2. Lv X. et al. HOXD9 promotes epithelial-mesenchymal transition andcancer metastasis by ZEB1 regulation in hepatocellular carcinoma. Journal of Experimental & Clinical Cancer Research. 34.1(2015)1-12. doi: 10.1186/s13046-015-0245-3.
3. Mazziotti G, Sorvillo F, Morisco F. et al. Serum insulin-like growth factor I evaluation as a useful tool for predicting the risk of developing hepatocellular carcinoma in patients with hepatitis C virus-related cirrhosis: a prospective study.[J]. Cancer. 2002;95(12):2539-2545 doi: 10.1002/cncr.11002
4. Chen K, Huang W, Huang B. et al. BORIS, Brother of the Regulator of Imprinted Sites, Is Aberrantly Expressed in Hepatocellular Carcinoma[J]. Genetic Testing & Molecular Biomarkers. 2013;17(2):160-5 doi: 10.1089/gtmb.2012.0242
5. Anwar S L, Lehmann U. DNA methylation, microRNAs, and their crosstalk as potential biomarkers in hepatocellular carcinoma[J]. World Journal of Gastroenterology. 2014;20(24):7894-7913 doi: 10.3748/wjg.v20.i24.7894
6. Dong Y, Wang A. Aberrant DNA methylation in hepatocellular carcinoma tumor suppression (Review)[J]. Oncology Letters. 2014;8(3):963-968 doi: 10.3892/ol.2014.2301
7. Zhu C, Utsunomiya T, Ikemoto T. et al. Hypomethylation of long interspersed nuclear element-1 (LINE-1) is associated with poor prognosis via activation of c-MET in hepatocellular carcinoma[J]. Annals of Surgical Oncology. 2014;21(4):729-735.doi 10.1245/s10434-014-3874-4
8. Sun L, Lee H J, Kim J H. et al. Aberrant CpG Island Hypermethylation Along Multistep Hepatocarcinogenesis[J]. American Journal of Pathology. 2003;163(4):1371-8 doi: 10.1016/S0002-9440(10)63495-5
9. Roncalli M, Bianchi P, Bruni B. et al. Methylation framework of cell cycle gene inhibitors in cirrhosis and associated hepatocellular carcinoma[J]. Hepatology. 2002;36(2):427-32 doi: 10.1053/jhep.2002.34852
10. Herbst A, Rahmig K, Stieber P. et al. Methylation of NEUROG1 in Serum Is a Sensitive Marker for the Detection of Early Colorectal Cancer[J]. American Journal of Gastroenterology. 2011;106(6):1110-1118 doi: 10.1038/ajg.2011.6
11. Huang W, Li T, Yang W. et al. Analysis of DNA methylation in plasma for monitoring hepatocarcinogenesis.[J]. Genetic Testing & Molecular Biomarkers. 2015;19(6):295-302 doi: 10.1089/gtmb.2014.0292
12. Zhao N, Bell D A, Maity A. et al. Global analysis of methylation profiles from high resolution CpG data.[J]. Genetic Epidemiology. 2015;39(2):53-64 doi: 10.1002/gepi.21874
13. Huang J, Zhang Y L, Teng X M. et al. Down-regulation of SFRP1 as a putative tumor suppressor gene can contribute to human hepatocellular carcinoma.[J]. BMC Cancer. 2007;7(1):1-15 doi: 10.1186/1471-2407-7-126
14. Wei L, Huang Y, Zhao R. et al. Detection of promoter methylation status of suppressor of cytokine signaling 3 (SOCS3) in tissue and plasma from Chinese patients with different hepatic diseases[J]. Clinical & Experimental Medicine. 2017;7:1-9 doi: 10.1007/s10238-017-0473-2
15. Yang B, Guo M. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma.[J]. American Journal of Pathology. 2003;163(3):1101-1107 doi: 10.1016/S0002-9440(10)63469-4
16. Wang J, Qin Y, Li B. et al. Detection of aberrant promoter methylation of GSTP1 in the tumor and serum of Chinese human primary hepatocellular carcinoma patients.[J]. Clinical Biochemistry. 2006;39(4):344. doi: 10.1016/j.clinbiochem.2006.01.008
17. Gan Y, Gu J, Cai X. et al. Adenovirus-mediated HCCS1 overexpression elicits a potent antitumor efficacy on human colorectal cancer and hepatoma cells both in vitro and in vivo.[J]. Cancer Gene Therapy. 2008;15(12):808. doi: 10.1038/cgt.2008.52
18. Inokawa Y, Nomoto S, Hishida M. et al. Detection of doublecortin domain-containing 2 ( DCDC2 ), a new candidate tumor suppressor gene of hepatocellular carcinoma, by triple combination array analysis[J]. Journal of Experimental & Clinical Cancer Research. 2013;32(1):65. doi: 10.1186/1756-9966-32-65
19. Zhang Y J, Wu H C, Shen J. et al. Silencing of Hint1, a novel tumor suppressor gene, by promoter hypermethylation in hepatocellular carcinoma[J]. Cancer Letters. 2009;275(2):277. doi: 10.1016/j.canlet.2008.10.042
20. Honda S. et al. The methylation status of RASSF1A promoter predicts responsiveness to chemotherapy and eventual cure in hepatoblastoma patients. Int J Cancer. 123(2008)1117-1125. doi: 10.1002/ijc.23613.
21. Veeck J, Niederacher D, An H. et al. Aberrant methylation of the Wnt antagonist SFRP1 in breast cancer is associated with unfavourable prognosis.[J]. Oncogene. 2006;25(24):3479-3488 doi: 10.1038/sj.onc.1209386
22. Wang G. et al. Expression of a LINE-1 endonuclease variant in gastric cancer: its association with clinicopathological parameters. BMC Cancer. 13(2013)265. doi: 10.1186/1471-2407-13-265.
23. Asahina Y, Tsuchiya K, Tamaki N. et al. Effect of aging on risk for hepatocellular carcinoma in chronic hepatitis C virus infection.[J]. Hepatology. 2010;52(2):518-27 doi: 10.1002/hep.23691
24. Tokita H, Fukui H, Tanaka A. et al. Risk factors for the development of hepatocellular carcinoma among patients with chronic hepatitis C who achieved a sustained virological response to interferon therapy.[J]. Journal of gastroenterology and hepatology. 2005;20(5):752-8 doi: 10.1111/j.1440-1746.2005.03800.x
25. Sasaki R, Yamasaki K, Abiru S. et al. Serum Wisteria Floribunda Agglutinin-Positive Mac-2 Binding Protein Values Predict the Development of Hepatocellular Carcinoma among Patients with Chronic Hepatitis C after Sustained Virological Response[J]. Plos One. 2015;10(6):e0129053. doi: 10.1371/journal.pone.0129053
26. Velosa J, Serejo F, Marinho R. et al. Eradication of hepatitis C virus reduces the risk of hepatocellular carcinoma in patients with compensated cirrhosis[J]. Digestive Diseases & Sciences. 2011;56(6):1853. doi: 10.1007/s10620-011-1621-2
27. Perz J F, Armstrong G L, Farrington L A. et al. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide ☆[J]. Journal of Hepatology. 2006;45(4):529-538 doi: 10.1016/j.jhep.2006.05.013
28. Khan F S, Ali I, Afridi U K. et al. Epigenetic mechanisms regulating the development of hepatocellular carcinoma and their promise for therapeutics[J]. Hepatology International. 2017;11(1):1-9 doi: 10.1007/s12072-016-9743-4
29. Kondo Y, Kanai Y, Sakamoto M. et al. Genetic instability and aberrant DNA methylation in chronic hepatitis and cirrhosis—A comprehensive study of loss of heterozygosity and microsatellite instability at 39 loci and DNA hypermethylation on 8 CpG islands in microdissected specimens from patient[J]. Hepatology. 2000;32(5):970-9 doi: 10.1053/jhep.2000.19797
30. Ammerpohl O, Pratschke J, Schafmayer C. et al. Distinct DNA methylation patterns in cirrhotic liver and hepatocellular carcinoma.[J]. International Journal of Cancer. 2012;130(6):1319-28 doi: 10.1002/ijc.26136
31. Cadier B, Bulsei J, Nahon P. et al. Early detection and curative treatment of hepatocellular carcinoma: A cost-effectiveness analysis in France and in the United States[J]. Hepatology. 2017 doi: 10.1002/hep.28961
32. Zhang Y J, Wu H C, Shen J. et al. Predicting hepatocellular carcinoma by detection of aberrant promoter methylation in serum DNA.[J]. Clinical Cancer Research. 2007;13(8):2378-84 doi: 10.1158/1078-0432.CCR-06-1900
Author contact
![]() Corresponding authors: Department of Biochemistry and Molecular Biology, West China School of Basic Medical Sciences & Forensic Medicine, Sichuan University, Chengdu 610041, China. Tel: (028) 85501243; Fax: +86 028 85503204; E-mail address: qinyangedu.cn (Y. Qin). Analytical & testing center, Sichuan University, Chengdu, Sichuan Province, China. Tel: 028-028-85416260; Fax: +028-85412316; E-mail address: 2640842542com (Ai-Min Sun).
Corresponding authors: Department of Biochemistry and Molecular Biology, West China School of Basic Medical Sciences & Forensic Medicine, Sichuan University, Chengdu 610041, China. Tel: (028) 85501243; Fax: +86 028 85503204; E-mail address: qinyangedu.cn (Y. Qin). Analytical & testing center, Sichuan University, Chengdu, Sichuan Province, China. Tel: 028-028-85416260; Fax: +028-85412316; E-mail address: 2640842542com (Ai-Min Sun).