Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(12):2175-2182. doi:10.7150/jca.25428 This issue Cite
Research Paper
AMPH-1 is critical for breast cancer progression
Yajun Chen1*, Jian Liu2*, Lei Li3, Hefeng Xia4‡ ![]() , Zaijun Lin5‡
, Zaijun Lin5‡ ![]() , Tianying Zhong1‡
, Tianying Zhong1‡ ![]()
1. Department of Clinical Laboratory, Obstetrics and Gynecology Hospital Affiliated to Nanjing Medical University, Nanjing Maternity and Child Health Care Hospital, Nanjing 210004, China.
2. Reproductive & Developmental Biology Laboratory, National Institute of Environmental Health Sciences (NIEHS), Research Triangle Prk, NC 27709, USA.
3. East China Normal University, 500 Dongchuan Road, Shanghai 200241, China.
4. Dongtai People,s Hospital, The Affiliated Hospital of Nantong University, 2 Kangfu Road, Yancheng 224000, China.
5. Shanghai Municipal Hospital of Tranditional Chinese Medicine, 274 Zhijingzhong Road, Shanghai 200071, China.
*These authors contributed equally to this work.
‡Authors with equal contribution
Received 2018-2-7; Accepted 2018-5-11; Published 2018-5-25
Abstract
Amphiphysin 1 (AMPH-1) is a nerve terminals-enriched protein involved in endocytosis, and we observe that its expression is increased in breast cancer tumor in compared with normal breast. However, its function in breast cancer is unknown. Here we aim to explore the role of AMPH-1 in breast cancer cells. Knockdown of AMPH-1 in breast cancer cells promotes cell proliferation, cell cycle progression and cell migration, and attenuates cell apoptosis. Of note, knockdown of AMPH-1 promotes breast cancer progression in xenograft mouse model. These oncogenic phenotypes may be partially due to the activated EMT and ERK pathways after inhibition of AMPH-1. Oncomine analyses of multiple breast cancer patient datasets show that reduced AMPH-1 mRNA level is significantly associated with breast cancer patients having metastatic events, advanced stage, poor clinical outcomes, and Paclitaxel+FEC treatment resistance. In summary, our results identified the anti-oncogenic function of AMPH-1 in breast cancer in vitro and in vivo. Activation of AMPH-1 may be a promising approach to treat breast cancer patients.
Keywords: Amphiphysin 1, AMPH-1, breast cancer, apoptosis, migration, p-Erk1/2
Introduction
Breast cancer is one of the most challenging diseases, which has emerged as a major public health concern among women. Each year, about 1.7 million women worldwide are diagnosed with breast cancer; over half of all cases (53%) occur in less-developed regions 1. A number of exogenous and endogenous factors can stimulate the pathology of breast cancer and some side effects of conventional treatment such as chemotherapy and radiotherapy are increasing the burden more and making it more challenging to treat the patients of breast cancer 2-5.
Amphiphysin 1 (AMPH-1) is a nerve terminals-enriched protein 6. It is a 128 kD protein with three identified functional domains, including an N-terminal Bin/Amphiphysin/Rvs (BAR) domain, a middle Clathrin and adaptor binding domain (CLAP), and a C-terminal SH3 domain 7-9. Previous studies of AMPH-1 were mainly focused on its role in neuronal function 10, 11, which involves the recruitment of dynamin to sites of clathrin-mediated endocytosis 12, 13. Knockdown of AMPH-1 in the neurons decreases filopodia formation and leads to the collapse of the growth cones 14, 15. Reduced synaptic vesicle recycling efficiency and cognitive deficits were observed in Amph-1 knockout mice 16. Cdk5 and Calpain are identified as regulators of AMPH-1's function in neurodevelopmental disorder 11, 12.
Up to now, although no function study of AMPH-1 in cancer has been carried out, a few studies found that AMPH-1 was a dominant autoantigen in a small minority of stiff-person syndrome (SPS) patients associated with breast cancer 17-19, lung cancer, and melanoma 6. Stiff-person syndrome (SPS), formerly known as stiff-man syndrome, is a rare neuroimmunologic disease, the understanding and diagnosis of which greatly advanced with the identification of disease-associated autoantibodies 20. Amphiphysin Ab are frequently coexpressed with other paraneoplastic antibodies and amphiphysin Ab-associated SPS is a recognized clinical entity21. Some studies demonstrate that amphiphysin Ab-associated SPS is a cancer-associated syndrome 22. Other studies also showed that the yeast homologs of amphiphysin, Rvs161 and Rvs167 23, 24 were involved in the transition from exponential cell growth to stationary phase upon exposure to nutrient starvation, implicating a possible role in the biology of cancer.
Breast cancer is the most common malignancy among women and is the third highest leading cause of cancer mortality worldwide 25. The involvement of AMPH-1 in breast cancer is unknown. In this study, we explored the function of AMPH-1 in breast cancer cells. We knocked down AMPH-1 in two breast cancer cell lines and found that AMPH-1 silencing attenuated cell apoptosis, promoted cell cycle progression and cell migration. We further evaluated the in vivo growth of the AMPH-1-knockdown cells in a mouse xenograft model and addressed its expression and the involved signaling pathway, demonstrating a novel function for AMPH-1 in the suppression of breast cancer.
Materials and methods
Cell culture, RNA interference, antibody
The MCF-7 and MDA-MB-231 cells were purchased from ATCC. All the cells were maintained for no more than 50 passages before we thaw up a new stock. The stable cell lines were generated by integration of lentiviral shRNA vectors specific for AMPH-1 or a control gene. The shRNA against AMPH-1 (1#) shown as follows: 5'- GCGAGAACUCCGAGGAUAUTT-3'. The shRNA against AMPH-1 (2#) was 5'- GCAGGAAGCUAGUGGACUATT-3'. Antibodies were purchased from Sigma (β-actin), Abcom (AMPH-1), Cell Signaling (E-cadherin. N-cadherin, P-ERK1/2 and Cleaved Caspase-3).
Transwell migration assay, MTT assay, colony formation
To evaluate breast cancer cell migration, 8 nm pore-size culture inserts (Transwell, Costar) were placed into the wells of a 24-well culture plate, separating the upper and the lower chambers. 1x 10^4 control or AMPH-1 knockdown cells were seeded to the upper chamber in 200ul of serum-free medium. After 12 h of incubation at 37 °C in 5% CO2, the number of cells that migrated through the pores was counted in 3 independent visual fields. The cell morphology was observed by hematoxylin staining. The assay was performed in triplicate.
For MTT assay, 5×103 early log phase of control or AMPH-1 knockdown cells were seeded in 96-well plates for the MTT assay. Cell density was measured by Cell Viability Kit (MTT, Roche, Indianapolis, IN, USA) following the manufacturer's instructions. The absorbance value (OD) was read in a microtiter plate reader at wavelength of 570 nm. All experiments were repeated at least three times.
For colony formation assay, cells were seeded into 96 well dishes with density of 5×104 cells per well. Individual colonies were fixed and stained with a solution containing 0.2% crystal violet in 10% ethanol for 30 minutes. The amount of dye taken up by the monolayer is quantitated in a spectrophotometer or plate reader at 570 nm.
Apoptosis assay and Cell Cycle Analysis
Double-staining of Annexin V-FITC-Propidium iodide (PI) was used for analyzing apoptosis. Briefly, cells were resuspended by cold PBS at a density of 1×106 cells/ml. PI and Annexin V-Alexa Fluor488 conjugate were mixed and added into the cell suspensions, followed by incubation at room temperature for 15 min. Flow cytometry was used to analyze apoptosis.
Cells were washed twice with cold PBS, resuspended in cold 70% ethanol and stored at 4°C overnight. Then, the cells were washed three times in cold 1x PBS, and stained with a PI solution (50 μg/ml) for 30 mins. Each assay was repeated for three times. Fold change values were represented as mean of the three experiments.
Western blot
For Western blot, proteins were separated on 10% SDS-polyacrylamide gels and transferred to NC membranes. Membranes were blocked in 5% non-fat milk in Tris-buffered saline (plus 0.1% Tween-20). AMPH-1, E-cadherin, Caspase3- cleaved, P-ERK, Ki67and β-actin antibodies were purchased from Proteintech and Cell Signaling. Blots were detected by chemiluminescence and exposed on X-ray films.
Immunohistochemistry (IHC)
IHC was performed for continuous sections from paraffin-embedded blocks. Antigen retrieval was performed by microwaving for 3 min in citrate-buffered solution (pH 6.0). Blocking was done by incubation with 10% goat serum at room temperature for 30 min. Sections were incubated with AMPH-1 (1:500), p-ERK (1:300), Ki67(1:200) and E-cadherin (1:500) antibodies that are indicated in this study for overnight at 4°C. Staining with secondary antibody (conjugated with horseradish peroxidase) was performed for 1 hour at room temperature. Sections were finally stained with 3, 3- diaminobenzidine tetrahydrochloride (DAB) and counterstained with hematoxylin.
Mouse xenograft tumor model
The five female BALB/c nude mice at the age of 5 weeks were anesthetized. ShAMPH-1 and control cells were suspended in cold PBS (2×106 cells/100 µL) and injected into the mice subcutaneously on flanks. Mice were continuously monitored for 6 weeks. Mice bearing tumors were sacrificed for the assessment of final tumor size. Mice were bred in the Animal Core Facility by following procedures approved by the East China Normal University of Institutional Animal Care and Use Committee.
Statistical measurements
Data are presented as mean ± SEM of three independent experiments. Significance of means between two groups is determined by student's t-test. A P value < 0.05 was considered significant.
Human Breast Tumor Expression Data Sets
Data sets in Oncomine (https://www.oncomine.org) were also examined followed previous methods 26.
Results
Knockdown of AMPH-1 promotes breast cancer cell growth
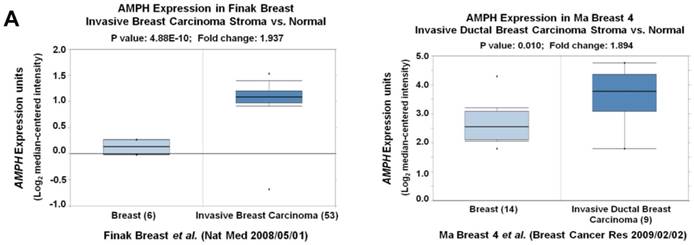
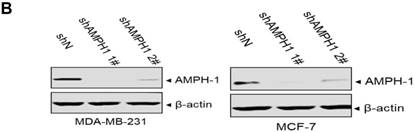
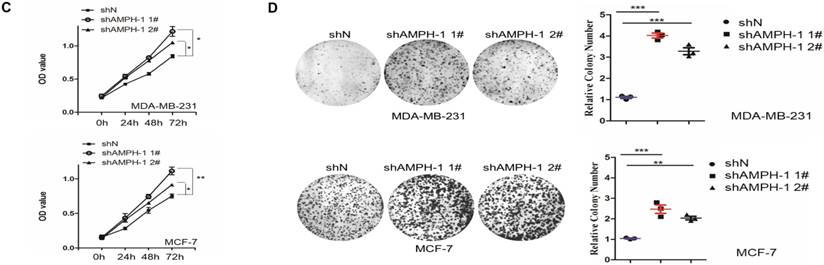
We noticed that the expression level of AMPH-1 was significantly increased in breast cancer compared to normal breast (Figure 1A). Therefore, we were curious about the role of AMPH-1 in breast cancer. First, we examined the expression of AMPH-1 in two breast cancer cell lines, MCF-7 and MDA-MB-231, and its role in regulation of breast cancer cell proliferation. Knock-down of AMPH-1 by two different shRNAs dramatically increased the cell growth in both cell lines (Figures 1A and 1B) using MTT assay. We further conducted soft agar colony formation assay to test the cell growth after AMPH-1 silencing in MDA-MB-231 and MCF-7 cells. Consistent with MTT results, knockdown of AMPH-1 significantly promoted cell colony formation compared with the controls (Figure 1C and 1D). Thus, knockdown of AMPH-1 promoted cell growth in human breast cancer cells, and these results implicated that AMPH-1 might be passively induced in breast cancer development.
Knockdown of AMPH-1 promotes breast cancer cell growth. A. Oncomine analysis of AMPH-1 mRNA expression in normal breast and breast cancer. B. shRNA knockdown of AMPH-1 in MDA-MB-231 cell and MCF-7 cell. C. MTT assay of cell growth. (**p <0.01, *p <0.05, p values were analyzed by two-tailed t test, Data are the mean ±SEM, representing three independent experiments). D. Colony formation assay was employed to measure the colony plate efficiency in control (shN) or AMPH-1 knockdown (shAMPH-1) cells. Quantitative assay was performed in control (shN) or AMPH-1 knockdown (shAMPH-1) cells. (**p <0.01, ***p <0.001, p values were analyzed by two-tailed t test, Data are the mean ±SEM, representing three independent experiments).
AMPH-1 silencing attenuates cell apoptosis and promotes cell cycle progression
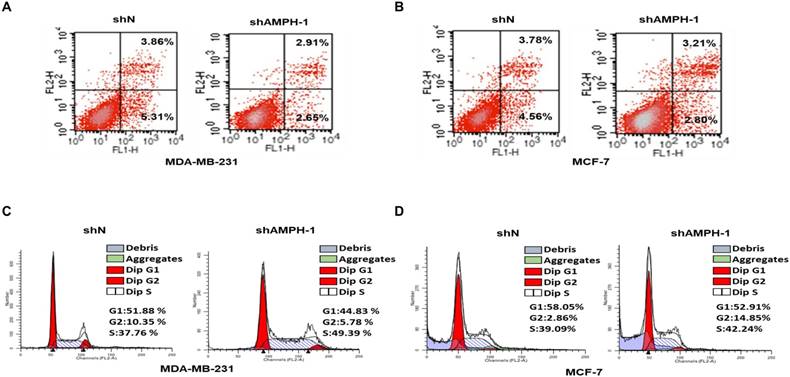
We next investigated the function of AMPH-1 in regulation of cell apoptosis, determined by Flow Cytometry analysis of MDA-MB-231 and MCF-7 cells stained with Annexin-V and propidium iodide (PI) (Figure 2A-B). The cell ratio undergoing late apoptosis was reduced from 3.86% to 2.91% after knockdown of AMPH-1 in MDA-MB-231 cells, and early apoptotic cell population was reduced from 5.31% to 2.65% (Figure 2A). Similar trend was observed in MCF-7 cells (3.78% to 3.21% (late apoptosis) and 4.56% to 2.8% (early apoptosis)) (Figure 2B). We then evaluated the effect of AMPH-1 on cell cycle distribution in the breast cancer cells. As demonstrated in Figure 2C, knockdown of AMPH-1 in cells caused a concomitant lower G1 population (44.83% and 52.91% for MDA-MB-231 and MCF-7, respectively) compared to the control group (51.88% and 58.05%, respectively). Meanwhile, the S phase cell population was elevated in both cell lines with AMPH-1 knockdown (49.39% vs 37.76% for MDA-MB-231 and 42.24% vs 39.09% for MCF-7) compared to control groups. Therefore, these data suggested that inhibition of AMPH-1 reduced breast cancer cell apoptosis and promoted cell cycle progression.
AMPH-1 silencing attenuates cell apoptosis and promotes cell cycle progression. A-B. Cell apoptosis was evaluated by Flow cytometry. FL1-H is Annexin V and FL2-H is PI. C-D. Cell cycle distribution analysis for AMPH-1 knockdown (shAMPH-1) and control (shN) cells.
Inhibition of AMPH-1 promotes cell migration and activates EMT and ERK signal pathway
Metastasis accounts for the majority of cancer morbidity and mortality. Here we investigated the impact of AMPH-1 on the cell migration in MDA-MB-231 and MCF-7 cells. As shown in Figures 3A and 3B, an elevated invasive effect was observed in MDA-MB-231 cells as well as in MCF-7 cells after knockdown of AMPH-1.
Inhibition of AMPH-1 promotes cell migration and activates EMT and ERK signal pathway. A-B. Cell migration ability was determined using Transwell assays in A) MDA-MB-231 cells and B) MCF-7 cells. Quantify analysis of the migration cells in MDA-MB-231 and MCF-7 cells. (***p <0.001, p values were analyzed by two-tailed t test, Data are the mean ±SEM, representing three independent experiments). C. Reduced AMPH-1 mRNA expression level was observed in breast cancer patients with metastatic events compared no metastatic events (observed at 1-year, 3year and 5-year respectively). D. Advanced N1+ stage compared to N0 stage breast cancer patients. E. Western Blot of major components in EMT and ERK1/2 pathway. Immunoblot analysis of AMPH-1, E-cadherin, N-cadherin, P-ERK1/2 and Caspase3- cleaved expression in the AMPH-1 knockdown (shAMPH-1) and control (shN) cells. An anti-β-actin immunoblot was shown as the loading controls.
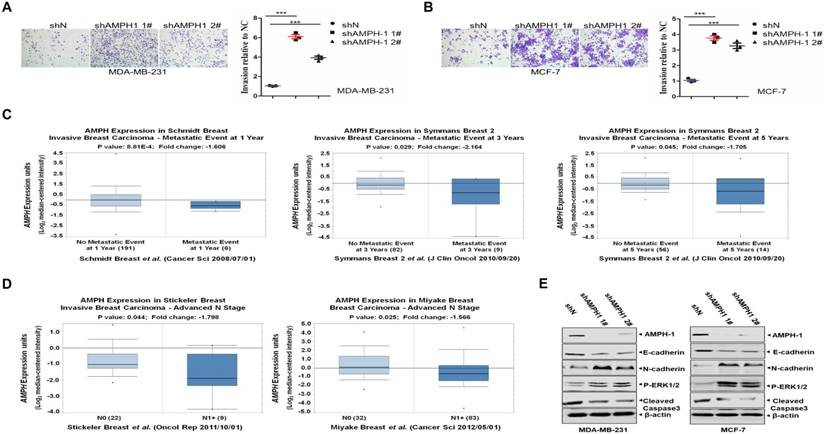
Next we explored the clinical significance of AMPH-1 expression in human invasive breast cancer patients. As indicated by Figure 3C 27, 28, the AMPH-1 expression level was got from the public cancer databases (Oncomine 3.0) and examined for patients with or without metastatic events at different time points. Corresponding to the metastatic events, reduced AMPH-1 mRNA expression level was observed at 1-year (-1.606 fold), 3-year (-2.164 fold) and 5-year (-1.705 fold) time points, respectively. We also checked the AMPH-1 level in advanced N1+ stage breast cancer patients 29, 30, and it had the lower expression compared to N0 stage (Figure 3D), indicating the involvement of AMPH-1 in the regulation of invasive/migration in advanced stage of human breast cancer.
To dissect the signaling pathway regulated by AMPH-1 in breast cancer cells, western blot against major components of Epithelial-Mesenchymal Transition (EMT) and ERK pathway was carried out. As shown in Figure 3E, knockdown of AMPH-1 caused the decrease of E-cadherin and cleaved caspase-3 level, and the increase of p-ERK1/2 and N-cadherin in MDA-MB-23 and MCF-7 cells. Given that phosphorylated ERK1/2 augmented cell proliferation and reduced level of E-cadherin and abnormal activation of N-cadherin were essential in EMT-mediated cancer initiation, migration and progression 31, these oncogenic phenotypes (Figures 1-3) after knockdown of AMPH-1 in breast cancer cells were due to the activated EMT/ERK pathway. Of note, our results (Figures 1-3) suggested that AMPH-1 might be passively induced at the early stage of breast cancer, while AMPH-1 was inhibited during breast cancer development.
Knockdown of AMPH-1 promotes breast cancer growth in xenograft mouse model
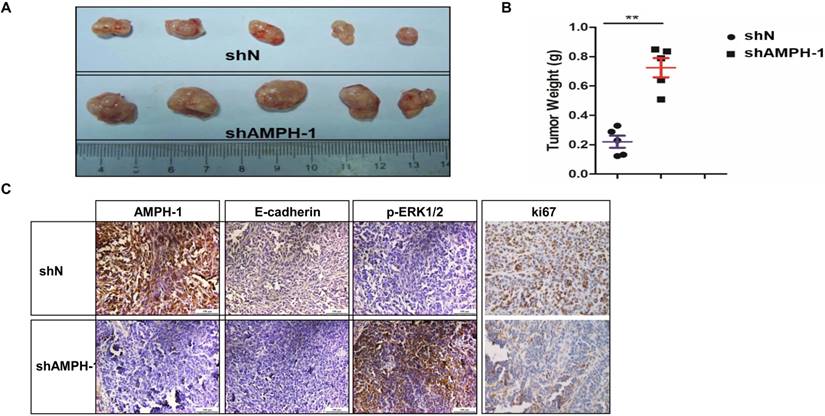
MDA-MB-231 (shN and shAMPH-1) cells were inoculated in nude mice, and the volume and weight of MDA-MB-231 tumors were measured at the final stage. The tumor volume and tumor weights in AMPH-1 silencing group were significantly increased compared with these of control group (Figures 4A-B). Immunohistochemical staining for Ki67 (a proliferation marker), E-cadherin and p-ERK1/2 in xenograft tumors was performed. The staining of Ki67 and E-cadherin was remarkably reduced and p-ERK1/2 was significantly increased in knockdown group compared to controls (Figure 4C). Thus, these in vivo results showed knockdown of AMPH-1 promoted breast cancer growth, probably dependent on the activation of ERK1/2 and EMT pathway in xenograft breast tumors after the inhibition of AMPH-1.
Knockdown of AMPH-1 promotes breast cancer progression in xenograft mouse model. A-B. The AMPH-1 silencing significantly increased A) the final tumor volume and B) tumor weights. C. Immunohistochemical staining for AMPH-1, E-cadherin, p-ERK1/2 and Ki67.
AMPH-1 is decreased in breast cancer patients with worse clinical outcome
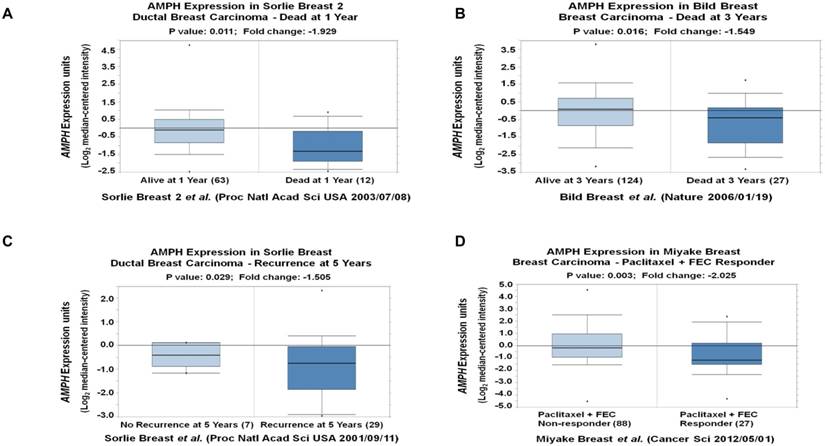
To explore the outcome of breast cancer patients with different AMPH-1 expression levels, we analyzed multiple breast cancer patient datasets using Oncomine 26, 32. The reduced mRNA expression of AMPH-1 (-1.929 fold, p=0.011) was observed in ductal breast carcinoma dead at 1 year compared to those still alive at 1 year (Figure 5A) 33. Similar reduction of AMPH-1 mRNA was observed in breast cancer patients dead at 3 years compared to those still alive at 3 years (Figure 5B) 34. The expression level of AMPH-1 was also negatively related to recurrence and certain drug response. As shown in Figure 5C 34, the recurrence at 5-year group had a significantly reduced AMPH-1 level compared to no recurrence group. Next, we evaluated patients' AMPH-1 expression level and chemotherapeutic efficacy of Paclitaxel followed by concurrent 5-fluorouracil, epirubicin, and cyclophosphamide (FEC) 29. A 2.025-fold (p=0.003) reduction was detected between Paclitaxel+FEC no responder vs Paclitaxel+FEC responder (Figure 5D). Taken together, the lower expression of AMPH-1 was associated with worse clinical outcome.
AMPH-1 is decreased in breast cancer patients with worse clinical outcome. A-B. The reduced mRNA expression of AMPH-1 was observed in ductal breast carcinoma A) dead at 1 year, and B) dead at 3 years C) with recurrence at 5 years compared to the control. D. The expression level of AMPH-1 was reduced in breast cancer patients with no chemotherapeutic efficacy.
Discussion
Human amphiphysin 1 is a protein highly concentrated in nerve terminals 13. Previous study showed that AMPH-1 was also detected in breast cancer cell line Hs578T, in samples from a patient with breast cancer and paraneoplastic sensory neuropathy, as well as in normal human mammary tissue 35. Here we demonstrated that AMPH-1 was detected in both breast cancer cell lines, MDA-MB-231 and MCF-7 (Figure 1A). Our results, together with previous observations, showed that AMPH-1 was expressed in breast cell lines. These results suggested that the presence of AMPH-1 in human mammary tissue was not attributed to the peripheral nerves, implicating it had multiple functions other than in neuron cell synaptic vesicle recycling. From an evolutionary perspective, Drosophila amphiphysin is broadly expressed during development and it is required for normal locomotion but not endocytosis 36. Yeast amphiphysins, Rvs161 and Rvs167, are involved in exponential to stationary growth phage transition 23, 24.
AMPH-1 plays an important role in tumor cell growth suppression, which is supported by the elevated cell proliferation and enhanced survival after knockdown of AMPH-1 (Figures 1B-D-). This might attribute to the attenuated spontaneous apoptosis rate and aberrant cell cycle progression (Figure 2). MDA-MB-231 was a highly metastatic cell line as indicated in our transwell assay (Figure 3A). AMPH-1 silencing further promoted its migration capacity (Figure 3A). Of note, the knockdown also boosted the migration of the non-metastatic cells (MCF-7, Figure 3B) to the extent similar to MDA-MD-231, suggesting its potent anti-migration function in breast cancer cells.
To further validate the cellular function of AMPH-1 in vivo, xenograft mouse model was employed to study the role of AMPH-1 in breast cancer growth. There was good agreement between in vivo and in vitro data: silencing of AMPH-1 significantly increased the final tumor volume and weight (Figure 4A-B) and cell growth (Figure 1), highlighting the constitutive protection function of AMPH-1 in breast cancer.
EMT plays an important role in tumor migration and metastasis 37, 38. ERK signaling regulates various cellular processes including cell motility, survival, and migration in cancer. While the EMT pathway and ERK pathway were aberrantly activated in AMPH-1 knockdown group revealed by western blot (Figure 3E) and immunohistochemistry (Figure 4C), the underlying mechanism still awaits further study. Studies showed that the AMPH-1 and AMPH-2 form heterodimers that bind to Clathrin associated GTPase Dynamin via their SH3 domains 8. It is interested to check whether AMPH-2 forms dimer involved in breast cancer suppression.
Patients' poor response to chemotherapy often correlates to high risk of recurrence and mortality 39. Our data indicated that reduced AMPH-1 mRNA level was strongly associated with metastatic events, advanced stage, poor clinical outcomes, and resistant to Paclitaxel+FEC treatment. Combined with these clinical data, the in vivo anti-oncogenic function of AMPH-1 in breast cancer was also detected. Furthermore, our results suggested that AMPH-1 might be passively induced in early stage of breast cancer, while loss of AMPH-1 might serve as a biomarker of advanced breast cancer development. Some published studies 17-19 showed that AMPH-1 was an autoantigen in a few breast cancer and SPS patients, suggesting that the altered or modified AMPH-1 was probably not only responsible for the development of breast cancer but also triggered the neurological autoimmune paraneoplastic syndrome.
Collectively, our study demonstrates a novel function of AMPH-1 in suppression of breast cancer cells, indicating a tumor suppressor role in breast cancer development. While the signaling pathway involved in this regulation needs further study, our preliminary data showed that EMT pathway and ERK1/2 pathway were activated in AMPH-1 knockdown cells, and both pathways might be involved in the regulation. Therefore, therapeutic approaches aiming at activating AMPH-1 might be highly useful in the treatment of human breast cancer.
Author Contributions
L.L., Y.j.C. and H.f.X. conducted the experiments and provided experimental methods and data. J.L. provided Oncomine analyses. T.y.Z, J.L., Z.j.L. and L.L. designed the study, analyzed the data and wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Altobelli E, Rapacchietta L, Angeletti PM, Barbante L, Profeta FV, Fagnano R. Breast Cancer Screening Programmes across the WHO European Region: Differences among Countries Based on National Income Level. Int J Environ Res Public Health. 2017:14
2. Li Y, Li S, Meng X, Gan RY, Zhang JJ, Li HB. Dietary Natural Products for Prevention and Treatment of Breast Cancer. Nutrients. 2017:9
3. Coates A, Abraham S, Kaye SB, Sowerbutts T, Frewin C, Fox RM, Tattersall MH. On the receiving end-patient perception of the side-effects of cancer chemotherapy. Eur J Cancer Clin Oncol. 1983;19:203-8
4. Clarke M, Collins R, Darby S, Davies C, Elphinstone P, Evans V, Godwin J, Gray R, Hicks C, James S, MacKinnon E, McGale P. et al. Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;366:2087-106
5. Mitra S, Dash R. Natural Products for the Management and Prevention of Breast Cancer. Evid Based Complement Alternat Med. 2018;2018:8324696
6. Pittock SJ, Lucchinetti CF, Parisi JE, Benarroch EE, Mokri B, Stephan CL, Kim KK, Killmann MW, Lennon VA. Amphiphysin autoimmunity: Paraneoplastic accompaniments. Annals of Neurology. 2005;58:96-107
7. David C, McPherson PS, Mundigl O, de Camilli P. A role of amphiphysin in synaptic vesicle endocytosis suggested by its binding to dynamin in nerve terminals. Proc Natl Acad Sci U S A. 1996;93:331-5
8. Wigge P, Kohler K, Vallis Y, Doyle CA, Owen D, Hunt SP, McMahon HT. Amphiphysin heterodimers: potential role in clathrin-mediated endocytosis. Mol Biol Cell. 1997;8:2003-15
9. Slepnev VI, Ochoa GC, Butler MH, De Camilli P. Tandem arrangement of the clathrin and AP-2 binding domains in amphiphysin 1 and disruption of clathrin coat function by amphiphysin fragments comprising these sites. J Biol Chem. 2000;275:17583-9
10. De Jesus-Cortes HJ, Nogueras-Ortiz CJ, Gearing M, Arnold SE, Vega IE. Amphiphysin-1 protein level changes associated with tau-mediated neurodegeneration. Neuroreport. 2012;23:942-6
11. Sekiguchi M, Katayama S, Hatano N, Shigeri Y, Sueyoshi N, Kameshita I. Identification of amphiphysin 1 as an endogenous substrate for CDKL5, a protein kinase associated with X-linked neurodevelopmental disorder. Archives of Biochemistry and Biophysics. 2013;535:257-67
12. Wu Y, Matsui H, Tomizawa K. Amphiphysin I and regulation of synaptic vesicle endocytosis. Acta Med Okayama. 2009;63:305-23
13. Takei K, Slepnev VI, Haucke V, De Camilli P. Functional partnership between amphiphysin and dynamin in clathrin-mediated endocytosis. Nature Cell Biology. 1999;1:33-9
14. Mundigl O, Ochoa GC, David C, Slepnev VI, Kabanov A, De Camilli P. Amphiphysin I antisense oligonucleotides inhibit neurite outgrowth in cultured hippocampal neurons. J Neurosci. 1998;18:93-103
15. Yoo J, Jeong MJ, Kwon BM, Hur MW, Park YM, Han MY. Activation of dynamin I gene expression by Sp1 and Sp3 is required for neuronal differentiation of N1E-115 cells. J Biol Chem. 2002;277:11904-9
16. Di Paolo G, Sankaranarayanan S, Wenk MR, Daniell L, Perucco E, Caldarone BJ, Flavell R, Picciotto MR, Ryan TA, Cremona O, De Camilli P. Decreased synaptic vesicle recycling efficiency and cognitive deficits in amphiphysin 1 knockout mice. Neuron. 2002;33:789-804
17. Folli F, Solimena M, Cofiell R, Austoni M, Tallini G, Fassetta G, Bates D, Cartlidge N, Bottazzo GF, Piccolo G, De Camilli P. et al. Autoantibodies to a 128-kd synaptic protein in three women with the stiff-man syndrome and breast cancer. N Engl J Med. 1993;328:546-51
18. De Camilli P, Thomas A, Cofiell R, Folli F, Lichte B, Piccolo G, Meinck HM, Austoni M, Fassetta G, Bottazzo G, Bates D, Cartlidge N. et al. The synaptic vesicle-associated protein amphiphysin is the 128-kD autoantigen of Stiff-Man syndrome with breast cancer. J Exp Med. 1993;178:2219-23
19. Schmierer K, Grosse P, De Camilli P, Solimena M, Floyd S, Zschenderlein R. Paraneoplastic stiff-person syndrome: no tumor progression over 5 years. Neurology. 2002;58:148
20. Levy LM, Dalakas MC, Floeter MK. The stiff-person syndrome: an autoimmune disorder affecting neurotransmission of gamma-aminobutyric acid. Ann Intern Med. 1999;131:522-30
21. Pittock SJ, Lucchinetti CF, Parisi JE, Benarroch EE, Mokri B, Stephan CL, Kim KK, Kilimann MW, Lennon VA. Amphiphysin autoimmunity: paraneoplastic accompaniments. Ann Neurol. 2005;58:96-107
22. Murinson BB, Guarnaccia JB. Stiff-person syndrome with amphiphysin antibodies: distinctive features of a rare disease. Neurology. 2008;71:1955-8
23. Crouzet M, Urdaci M, Dulau L, Aigle M. Yeast mutant affected for viability upon nutrient starvation: characterization and cloning of the RVS161 gene. Yeast. 1991;7:727-43
24. Bauer F, Urdaci M, Aigle M, Crouzet M. Alteration of a yeast SH3 protein leads to conditional viability with defects in cytoskeletal and budding patterns. Mol Cell Biol. 1993;13:5070-84
25. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-86
26. Liu J, Cho SN, Akkanti B, Jin N, Mao J, Long W, Chen T, Zhang Y, Tang X, Wistub II, Creighton CJ, Kheradmand F. et al. ErbB2 Pathway Activation upon Smad4 Loss Promotes Lung Tumor Growth and Metastasis. Cell Rep. 2015
27. Schmidt M, Bohm D, von Torne C, Steiner E, Puhl A, Pilch H, Lehr HA, Hengstler JG, Kolbl H, Gehrmann M. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008;68:5405-13
28. Symmans WF, Hatzis C, Sotiriou C, Andre F, Peintinger F, Regitnig P, Daxenbichler G, Desmedt C, Domont J, Marth C, Delaloge S, Bauernhofer T. et al. Genomic index of sensitivity to endocrine therapy for breast cancer. J Clin Oncol. 2010;28:4111-9
29. Miyake T, Nakayama T, Naoi Y, Yamamoto N, Otani Y, Kim SJ, Shimazu K, Shimomura A, Maruyama N, Tamaki Y, Noguchi S. GSTP1 expression predicts poor pathological complete response to neoadjuvant chemotherapy in ER-negative breast cancer. Cancer Sci. 2012;103:913-20
30. Stickeler E, Pils D, Klar M, Orlowsk-Volk M, Zur Hausen A, Jager M, Watermann D, Gitsch G, Zeillinger R, Tempfer CB. Basal-like molecular subtype and HER4 up-regulation and response to neoadjuvant chemotherapy in breast cancer. Oncol Rep. 2011;26:1037-45
31. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97-110
32. Huang Q. et al. Gene Expression Analysis Identifies Common and Distinct Signatures Underlying Ductal and Lobular Breast Cancers. Journal Of Reproductive Medicine. 2017:62
33. Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, Olson JA Jr, Marks JR. et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353-7
34. Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H. et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869-74
35. Floyd S, Butler MH, Cremona O, David C, Freyberg Z, Zhang X, Solimena M, Tokunaga A, Ishizu H, Tsutsui K, De Camilli P. Expression of amphiphysin I, an autoantigen of paraneoplastic neurological syndromes, in breast cancer. Mol Med. 1998;4:29-39
36. Leventis PA, Chow BM, Stewart BA, Iyengar B, Campos AR, Boulianne GL. Drosophila Amphiphysin is a post-synaptic protein required for normal locomotion but not endocytosis. Traffic. 2001;2:839-50
37. Barker N, Clevers H. Tumor environment: a potent driving force in colorectal cancer? Trends Mol Med. 2001;7:535-7
38. Rokavec M, Kaller M, Horst D, Hermeking H. Pan-cancer EMT-signature identifies RBM47 down-regulation during colorectal cancer progression. Sci Rep. 2017;7:4687
39. Jaiyesimi IA, Buzdar AU, Hortobagyi G. Inflammatory breast cancer: a review. J Clin Oncol. 1992;10:1014-24
Author contact
![]() Corresponding authors: Tianying Zhong, Email: 13851875320com, Zaijun Lin, Email: zjlin_doccom and Hefeng Xia, Email: hefeng_xia1226com
Corresponding authors: Tianying Zhong, Email: 13851875320com, Zaijun Lin, Email: zjlin_doccom and Hefeng Xia, Email: hefeng_xia1226com