Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(12):2072-2081. doi:10.7150/jca.23427 This issue Cite
Review
Histone methylation in DNA repair and clinical practice: new findings during the past 5-years
Shuhua Wei1, Chunxiao Li1, Zhongnan Yin2, Jie Wen1, Hui Meng1, Lixiang Xue1,2, ![]() , Junjie Wang1,
, Junjie Wang1, ![]()
1. Department of Radiation Oncology, Peking University Third Hospital, Beijing 100191, China
2. Medical Research Center, Peking University Third Hospital, Beijing 100191, China
Received 2017-10-21; Accepted 2018-3-31; Published 2018-5-24
Abstract
DNA double-strand breaks (DSBs) are highly toxic lesions that can impair cellular homeostasis and genome stability to result in tumorigenesis for inappropriate repair. Although DSBs are repaired by homologous recombination (HR) or non-homologous end-joining (NHEJ), the related mechanisms are still incompletely unclear. Indeed, more and more evidences indicate that the methylation of histone lysine has an important role in choosing the pathways of DNA repair. For example, tri-methylated H3K36 is required for HR repair, while di-methylated H4K20 can recruit 53BP1 for NHEJ repair. Here, we reviewed the recent progress in the molecular mechanisms by which histone methylation functions in DNA double-strand breaks repair (DSBR). The insight into the mechanisms of histone methylation repairing DNA damage will supply important cues for clinical cancer treatment.
Keywords: DNA repair, Double-Strand Breaks, Histone methylation.
Introduction
DNA is the most important bio-macromolecule throughout the life cycle of cells, which can be affected by exogenous insults factor and endogenous DNA damage stimulations during DNA metabolism, such as ionizing radiation(IR), ultraviolet radiation (UV), chemical agents, DNA depurination and oxidation [1]. As the results of the damage, DSBs (DNA double-strand breaks, DSBs) have major effects on eukaryotes, which can lead to autophagy, apoptosis, aging and even cancer without being well-repaired. Therefore, upon damages, it is immediately necessary for cells to identify DSBs and trigger a variety of processes to repair them. Eukaryotes have two main approaches to repair DSBs: HR (homologous recombination, HR) and NHEJ (non-homologous end-joining, NHEJ) [1]. However, it still needs further investigation to elucidate which pathway the cell would go. Emerging evidence indicates that the methylation of histone lysine can play more and more important roles in determining the repair pathway upon DSBs [2].
In eukaryotic cells, DNA is condensed into chromatin. The core unit of chromatin is nucleosome which consisted of approximately 146 bp of DNA wrapped around a nucleosome, an octamer composed of pairs of each of the four core histones (H2A, H2B, H3, and H4). Histones can be methylated on lysine and arginine residues only, but methylation is most commonly observed on lysine residues of histone tails H3 and H4. The tail end furthest from the nucleosome core is the N-terminal. Common sites of methylation associated with gene activation include H3K4, H3K36, and H3K79. Common sites for gene inactivation include H3K9 and H3K27 [3, 4]. The most extensively studied histone lysine methylation sites are listed in Table 1 [5-7]. Methylated lysine sites might serve as the docking sites for HR or NHEJ-related proteins. Besides, histone methylation could recruit chromatin remodeling complex and transcriptional repression complex to affect the expression of repair proteins and their loading onto DSB sites.
The methyltransferase and demethylase for histone 3 and 4. The specific human enzymes are distinguished by a prefix h. The yeast enzymes are distinguished by a prefix: Sc (Saccharomyces cerevisiae) or Sp (Saccharomyces pombe).
| Histone lysine locus | Methyltransferase | Demethylase |
|---|---|---|
| H3K4 | hMLL1 (KMT2A) hMLL2 (KMT2B) hMLL3 (KMT2C) hMLL4 (KMT2D) hMLL5 (KMT2E) hET1A (KMT2F) hSET1B (KMT2G) ASH1 (KMT2H) SMYD1 (KMT3D) SMYD2 (KMT3C) SMYD3 (KMT3E) Sc/Sp SET1 SET7/9 (KMT7) | LSD1(KDM1A) LSD2 (KDM1B) JHDM1A (KDM2A) JHDM1B (KDM2B) JARID1A (KDM5A) JARID1B (KDM5B) JARID1C (KDM5C) JARID1D (KDM5D) NO66 |
| H3K9 | SUV39H1 (KMT1A) SUV39H2 (KMT1B) G9a (KMT1C) GLP (KMT1D) SETDB1 (KMT1E) SETDB2 (KMT1F) RIZ1 (KMT8) CLL8 SpClr4 | LSD1 (KDM1A) JHDM2A (KDM3A) JHDM2B (KDM3B) JHDM3A (KDM4A) JMJD2B (KDM4B) JMJD2C (KDM4C) JMJD2D (KDM4D) JHDM1D (KDM7) PHF8 |
| H3K27 | EZH2 (KMT6) | UTX (KDM6A) JMJD3 (KDM6B) |
| H3K36 | Sc/Sp SET2 (KMT3A) SET3 NSD1 (KMT3B) NSD2 NSD3 SMYD2 (KMT3C) METNASE(SETMAR) | JHDM1A (KDM2A) JHDM1B (KDM2B) FBXL10 JMJD2A (KDM4A) JMJD2B (KDM4B) JMJD2C (KDM4C) JMJD5(KDM8) |
| H3K79 | Sc/Sp DOT1 h DOT1L (KMT4) | Not described |
| H4K20 | h MMSET Pr-SET7/8 (KMT5A) SpSet 9 SUV420H1(KMT5B) SUV420H2 (KMT5C) NSD1(KMT3B) | PHF8(KDM7B) |
Here, we reviewed the relationship between DNA damage repair and histone methylation. Especially, we focus on a series of discoveries that have shed light on how histone methylation determine the repair approach of DSBs and the potential applications of histone methylation in clinical trials for cancer.
Two major pathways involved in the DSBR
Eukaryotic cells have two main repair pathways to deal with DSBs: HR which requires extensive end resection at the DSB site to generate a novel single-strand DNA to invade an identical DNA sequence in the genome which serves as a template for DNA repair, and NHEJ which rejoins the broken ends of a severed DNA molecule without regard to homology [8, 9]. Besides, the recruitment of NHEJ-related proteins like Ku70, Ku80 and DNA-PKcs and HR-related proteins like BRCA1, Rad51 and RPA is associated with histone methylation.
NHEJ pathway. NHEJ, a pathway that repairs double-strand breaks in DNA, is referred to as "non-homologous" because the break ends are directly ligated without the need for a homologous template, in contrast to homology directed repair, which requires a homologous sequence to guide repair. The term "non-homologous end joining" was coined in 1996 by Moore and Haber. When DSBs occur on the G1 phase, the cells prefer NHEJ pathway against the damage [10]. Ku70-Ku80 heterodimer rapidly binds to the DSBs, and then ATM-related DNA-dependent protein kinase catalytic subunit (DNA-PKcs) is recruited to the sites and activated. DNA-PKcs is responsible for maintaining the broken DNA ends in close proximity, which is beneficial for rejoining and recruiting a variety of end-processing factors. Subsequently, DNA ligase complex promotes the relegation of the broken DNA ends [11].
HR pathway. Homologous recombination is a type of genetic recombination in which nucleotide sequences are exchanged between two similar or identical molecules of DNA. It is most widely used by cells to accurately repair harmful breaks that occur on both strands of DNA, known as double-strand breaks. When DSBs mainly occur on S/G2 phase, HR pathway is activated in response to the damage [10]. Following the replication of genome, sister chromatid is available in S/G2 phase and serves as a template chain for DNA repair [12]. MRE-11/RAD50/NBS1 (MRN) complex binds to the broken DNA sites, followed by the recruitment of C-terminal binding protein interacting protein (CtIP) and several nucleases, to promote a complex and highly regulated process of DNA end-resection [10]. Then, replication protein A (RPA) coats a long stretch of 3' single-stranded DNA (ssDNA). Subsequently, the key facilitator of HR, RAD51, displaces RPA to form a RAD51-ssDNA nucleofilament which assembles with the help of a few of proteins including BRCA1 (Breast Cancer 1), BRCA2 and several paralogues of RAD51. Finally, the RAD51-nucleofilament searches for the complementary DNA template in the genome to synthesize and synapse, and then to accomplish HR repair [13].
A tale of DNA repair pathway choice directed by histone methylation
The processes of DNA damage repair are very complicated. Once suffering the damage stimulations, cells open the “sluice” and initiate a rapid DNA damage response signaling “cataract” to sense and repair the DNA lesion. However, epigenetic modification exerts a pivotal role in these processes, especially for histone methylation. The different combinational methylation pattern of histone serves as a docking site for various effector proteins involved in either HR or NHEJ pathway, which might determine the final choice.
H3K4 methylation in DSBR
In budding yeast, the methylation of lysine 4 on histone H3 (H3K4me3) is involved in maintaining genome stability. In addition to its presence at promoters of highly transcribed genes, H3K4me3 is recruited to sites of newly created double-stranded breaks. Cells lacking this histone modification display the defects such as a significant decrease in the repair of DNA breaks by the NHEJ pathway and a difficulty to survive in presence of replication stresses [4]. Set mutants in yeast show sensitivity to several DNA damage agents, while set1△cells in budding yeast show deficiency in NHEJ and in response to republication stress. Experiments on Caenorhabditis elegans showed that SET1 orthologue SET-2 might maintain transgenerational genome stability by timely repair of induced DSBs downstream of DNA damage repair response [14]. Similarity in vertebrate and mammalian cells, once DSBs occurs, H3K4 is di- or tri-methylated by Set1 in the presence of E3 ubiquitin protein ligase RNF20 (ring finger protein 20, RNF20) as well [12-14]. Inconsistently, studies indicate that H3K4 methylation could also be down-regulated at newly generated DSB sites. For instance, the reduction of H3K4me2 occurs in late S/G2 cells when the level of lysine specific demethylase 1 (LSD1) increases. Meanwhile, the recruitment of 53BP1, which promotes NHEJ repair pathway, is also dependent on LSD1. However, knockdown of LSD1 causes an increase in HR and the status of H3K4 methylation may regulate whether HR is deployed for DNA repair [13, 15]. However, histone methyltransferase SMYD3 promotes HR via the H3K4 methylation at the HR-related gene promoters [16]. DNA damage-induced lysine-specific histone demethylase 5A (KDM5A) recruitment to DSB sites demethylates H3K4me3, which is required for the interactions between damaged chromatin and ZMYND8-NuRD, favors the repair of DSBs by HR. KDM5B, another member of KDM family, is required for efficient recruitment of Ku70 and BRCA1, which are essential components for NHEJ and HR [17, 18]. Therefore, H3K4 methylation associated HR or NHEJ is partially determined by different type of methyltransferase or demethylase and their co-factors. But more delicate observations are needed to deeply elucidate how methylated H3K4 influences DNA repair pathway choice.
H3K36 methylation in DSBR
H3K36 methylation plays dual roles in the DNA repair pathway choice, which depends on the total number of the added methyl group and the phase of cell cycle. On one hand, SETD2-dependent H3K36me3 is required for HR repair and for protecting genome stability [19, 20]. Interestingly, H3K36me3 is not induced by DNA damages, but rather, pre-existing [21]. Without DNA damage factors (Fig. 1), lens epithelium derived growth factor (LEDGF) masks H3K36me3 mark via PWWP domain [22]. After IR, LEDGF promotes CtIP to gather at DSBs to drive the end processing for Rad51 loading, which is necessary for HR repair [22]. Consistently, depletion of SETD2 which is the only reported methyltransferase for H3K36me3, can severely block HR repair [23]. Recent study indicates that DSBs located in transcriptional activation genes and H3K36me3-enriched loci prefer HR, while inactive genes and H3K36-depleted regions target to NHEJ repair [24]. Controversially, H3K36me3 is enriched in heterochromatin which is repaired mainly by Artemis-dependent HR pathway [25].
Different forms of methylated H3K36 prefer different DNA repair pathway. In the absence of DNA damage, tri-methylated H3K36 by SETD2 is masked by LEDGF. Once upon damage, LEDGF promotes the recruitment of CtIP at DSBs to drive the end processing for Rad51 loading, which is necessary for HR repair. Meanwhile, the phosphorylated METNASE on Ser495 by Chk1 catalyzes the di-methylation of H3K36. ATM mediates the phosphorylation of KDM2A on Thr632 to promote the dissociation of KDM2A from H3K36me2. Then, H3K36me2 is exposed to several proteins, such as Ku70-Ku80 heterodimer, PHRF1, NBS1 for NHEJ and MRN complex for HR.
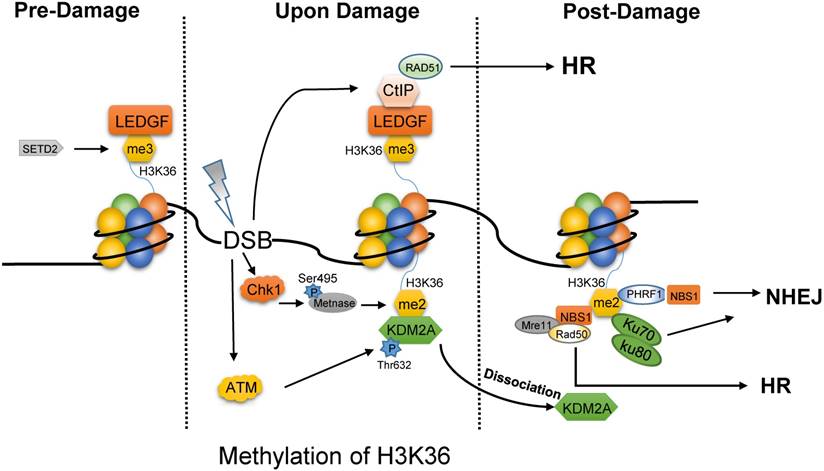
In contrast to tri-methylated H3K36, di-methylated H3K36 functions in the post-damage of DNA (Fig. 1) [26]. First, the activated Chk1 catalyzes the phosphorylation of METNASE on Ser495, and promotes its recruitment to DSBs [27]. Then, METNASE takes the responsibility for H3K36me2 which is rapidly induced after IR [26, 28]. However, JHDM1A/KDM2A, the specific demethylase of H3K36me2, masks this site to disturb the performance of H3K36me2. Interestingly, upon damages, the activated ATM would result in the disaggregation of KDM2A from H3K36me2 by mediating the phosphorylation of KDM2A on Thr632 [28]. Hence, H3K36me2 is exposed to other effector proteins, such as Ku70, PHRF1 (PHD and ring finger domain 1), and NBS1. As a result, PHRF1 may combine with H3K36 methylation and NBS1 to promote NHEJ and stabilize genomic integrity upon DNA damage insults [29]. It is more interesting that NBS1 directly bind to H3K36me2 through its BRCT2 domain, which is beneficial for the association of MRN complex with chromatin to complete HR repair [28].
Briefly, H3K36me2 promotes NHEJ [26], but H3K36me3 is required for HR pathway. Currently, it is still unknown whether H3K36me2 can also promote HR-based repair. It is worth mentioning that, in C. elegans, the demethylation of H3K36me2 by JMJD-5 can promote the resolution of recombination intermediates and the release of RAD-51, which convoys the late steps of HR [30]. Therefore, H3K36me2 might mediate both NHEJ and HR based on the context. Recently, another finding demonstrates that the modification of H3K36 could switch DSB repair pathway choice. H3K36me3 by SETD2 reduces chromatin accessibility to block end processing and promote NHEJ in G1 phase, while H3K36ac by acetyltransferase Gcn5 increases chromatin accessibility to facilitate end resection and HR in S/G2 phase [31].
H3K79 methylation in DSBR
All three forms of H3K79 methylation are catalyzed by the sole enzyme disruptor of telomeric silencing-1 (Dot1, known as DOT1L in human). Of note, Dot1/DOT1L is the only known non-SET domain containing histone lysine demethylase and methylated lysine 79 locates within the globular domain of histone H3. Moreover, H3K79 methylation is enriched in actively transcribed genes and involved in RNA polymerase II-associated transcriptional elongation and DNA repair [32]. The interaction between H3K79 methylation and 53BP1 represents a functional and physical barrier to end processing of DSBs and uncapped telomeres and controls ssDNA at DSB sites [33]. As a central component of chromatin-based DSB signaling, 53BP1 is implicated in the DNA repair by NHEJ during G1. So far, it has been found that the methylated H3K79, as a docking site, can recruit 53BP1 to drive NHEJ repair in G1/G2 phase in human cells when H4K20me2 levels are low [2, 34]. Consistently, the mutation of H3K79 or depletion of DOT1L can block the recruitment of 53BP1 to DSBs while H3K79 methylation is inhibited in both cases [35]. Intriguingly, a novel strategy Epi-ID, through directly assessing chromatin status, identified several HR-related genes as negative regulators of H3K79 methylation [36]. Recently, a study performed in Saccharomyces cerevisiae showed that di-methylated H3K79 rather than H3K79me3 plays a regulatory role in UV repair, specifically in error-free translesion synthesis but not in DSB repair [37]. It might contribute to DOT1L-mediated H3K79 methylation in binding and recruiting XPC to DNA damage sites for nucleotide excision repair [38].
H3K9 methylation in DSBR
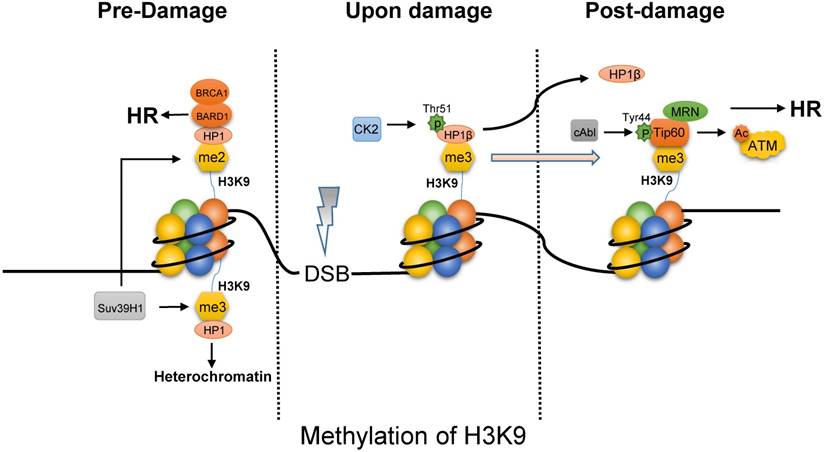
The methylation of H3K9 functions on choosing HR pathway via opening chromatin and recruiting repair proteins. First, the tri-methylated H3K9 by Suv39H1 maintains the stability of heterochromatin by binding HP1 (Fig. 2) [39, 40], the domain formation of which is mediated by phase separation [41]. Upon IR, CK2 (casein kinase 2) phosphorylates HP1β (subunit of HP1) on Thr51 to promote its dissociation from the H3K9me3 [42, 43]. In addition, the displacement of HP1β not only promotes chromatin relaxation but also facilitates the ability of Tip60 to access H3K9me3 via Tip60's chromo-domain. Besides, oncogene cAbl induced by IR also promotes Tip60 to incorporate onto H3K9me3 [44]. This interaction between Tip60 and H3K9me3 at DSBs activates the acetyltransferase activity of Tip60. And MRN complex that is HR-associated protein, targets Tip60 to H3K9me3 and is required to activate Tip60's acetyltransferase activity [45]. It is reported that inactivation of Tip60 leads to defective DNA repair [46]. However, the activated-Tip60 acetylates H4K16 to hinder the binding of 53BP1 to the neighboring H4K20me2, which is required for NHEJ repair [47]. In contrast to tri-methylated H3K9, the BARD1 (BRCA1-associated RING domain protein 1) was found to bind to the di-methylated H3K9 through the interaction of BARD1 and HP1, which is required for BRCA1 retention at DSBs [48]. Finally, as the characteristics of histone code, rapid decrease of H3K9 methylation can be observed after exposure to IR [49], which is probably resulted of expression of KDM4B and suppression of Suv39H1 mediated by p53. Chromatin relaxation due to the displacement of HP1 and the decrease of H3K9 methylation increases chromatin accessibility to facilitate end resection for HR. In addition, the binding of Tip60 to H3K9me3 with MRN complex and BRCA1 to H3K9me2 is required for HR.
The methylated H3K9 mainly promotes HR pathway upon DNA damage. In the absence of DNA damage, tri-methylated H3K9 by Suv39H1 can bind HP1 to maintain heterochromatin, while HP1 bind on di-methylated H3K9 site is combined with BARD1/BRCA1 to initiate HR pathway. Upon damage, phosphorylated HP1β on Thr51 by CK2 will dissociate from the H3K9me3 mark, which facilitates Tip60 to access H3K9me3. Besides, cAbl also promotes Tip60 to incorporate onto H3K9me3. And MRN complex can target Tip60 to H3K9me3 and is required to activate Tip60's acetyltransferase activity which activates ATM by acetylation.
H3K27 methylation in DSBR
The di- and tri-methylation of H3K27 are mediated by two catalytic subunits of Polycomb repressive complex 2 (PRC2), EZH1 and EZH2 [50]. Besides, the role that H3K27 methylation plays in DNA damage repair is related to PRC1-mediated silencing [51, 52]. Upon DNA damage, the PARP-dependent recruitment of EZH2 at DSBs facilitates the tri-methylation of H3K27, and then PRC1 can be recruited to trimethylated H3K27 to perform its repressive effect [53]. One subunit of PRC1 functions via the ubiquitination of H2AK119 and its depletion would lead to the impaired repair of DSBs by HR and cell arrest in G2/M [54]. In addition, H3K27me3 inhibits the expression of FBOX32 that could promote p21 degradation, while p21 is involved in p53-dependent DNA damage checkpoint. Therefore, H3K27me3 is closely linked to the regulation of cell cycle and DNA damage repair by regulating the expression of p21 [55]. Moreover, mutation of EZH2 in C.elegans and deletion of PcG protein in mammals show a DNA damage-sensitivity phenotype [56]. Similarly, it was reported that EZH2 binds to the ATM promoter and inhibits its expression through covalent modification of H3K27me3 in hepatocellular carcinoma (HCC) cell lines. Upon DNA damage mediated by chemotherapeutic drug, shRNA targeting EZH2 or its inhibitor GSK126 significantly promoted genotoxicity and increased the cell chemosensitivity [57]. The fact that EZH2 and MTA2 which is a subunit of chromatin remodeling complex with deacetylase activity are co-existed in the promoter of DSBR gene suggests that EZH2 and MTA2 may be associated with DNA damage repair [2]. Furthermore, upon IR, UTX and JMJD3 which are both the demethylases of H3K27 methylation can also gather at DSB sites to facilitate the expression of specific genes, such as Ku80 which is a key mediator protein in NHEJ pathway [58, 59]. Trimethylated H3K27 were enriched on the promoters of DNA-repaired-related gene RAD51 and BRAC1, which were deregulated in human uterine fibroids and restored by treatment with EZH2 inhibitor [60]. In a word, the methylation of H3K27 might suppress the expression of repair-related proteins by recruiting PRC1 to influence DNA damage repair. However, beyond transcription silencing, EZH2 also localizes at DNA damage sites and recruits MUS81 nuclease via trimethylation of H3K27 to degradate stalled replication forks and then, favor HR-mediated repair [61, 62]. These findings have provided the possibility for targeting PcG to sensitize the cancer cells upon DNA damage based on IR treatment and chemotherapy.
H4K20 methylation in DSBR
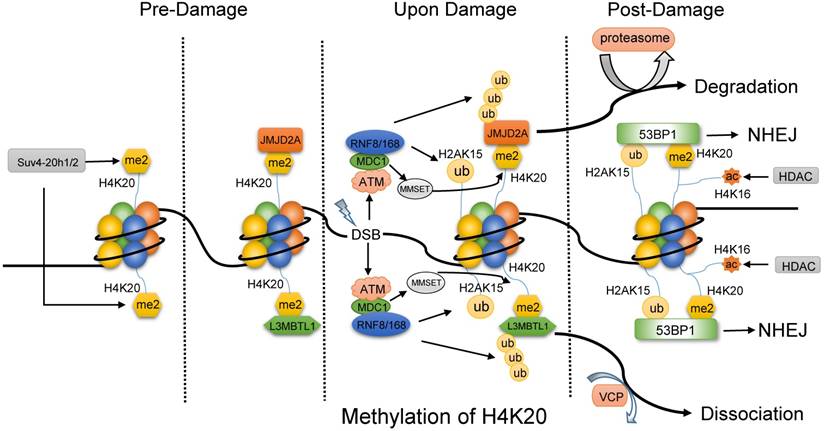
H4K20 methylation works its way into DNA repair mainly by 53BP1. Further, the generalist 53BP1 plays as a scaffold for DSB-responsive factors, promotes checkpoint signaling [63] and switches the pathway choice to NHEJ [64, 65]. 53BP1 binds to H4K20me2 and H2AK15ub simultaneously via Tandem Tudor domain to exert its function. H4K20me2 modified by Suv4-20h1/2 plays as a docking site in undamaged cells and is exposed to 53BP1 upon DNA damage, such as IR. In the absence of DNA damage (Fig. 3), a group of proteins mask H4K20me2 mark, including L3MBTL1 (lens malignant (3) brain tumor-like protein 1) [66] and histone demethylase JMJD2A [67]. Upon IR, ATM-dependent recruitment and Ser139 phosphorylation of mediator of DNA damage checkpoint 1 (MDC1) promotes RNF8 and RNF168 to accumulate at DSBs, which modify L3MBTL1 and JMJD2A with multi-ubiquitination. Thus, the ubiquitylated L3MBTL1 is evicted by ATPase valosin-containing protein (VCP) and nuclear protein localization cofactor protein4 (NPL4), and the ubiquitylated JMJD2A is degraded by the proteasomes. At the same time, RNF8/168 also mediates the ubiquitination of H2AK15. Even so, a stable subunit of Tip60 complex MBTD1 assists DNA repair by HR via binding to H4K20me2 and blocking H2AK15ub [68]. In addition, the activated ATM promotes the phosphorylation of MMSET (multiple myeloma SET protein) on Ser102. Subsequently, the phosphorylated MMSET is recruited to DSBs to mediate de novo di-methylation of H4K20 by binding to MDC1. Thus, the exposed H4K20me2 and prepared H2AK15ub provide 53BP1 with opportunity to incorporate and initiate NHEJ pathway [66, 67]. 53BP1 recruits its downstream effector RIF1 and MAD2L2 to counteract BRAC1-mediated DSB relocation and promotes NHEJ in G1-phase cells [69-71]. The recruitment of the complex protein 53BP1-RIF1-MAD2L2 is linked to the DNA replication-associated dilution of H4K20me2, which appropriately directs 53BP1 to pre-replicative chromatin [72, 73]. Most recently, a novel substrate-competitive inhibitor of SUV4-20 enzymes, A-196, inhibited 53BP1 formation at IR-induced DSB sites and impaired the DSB repair by NHEJ instead of the effect on HR repair. Moreover, depletion of PR-Set7 and deficiency of H4K20me1 leads to the accumulation of DNA damage and an ATR-dependent cell cycle arrest [74]. Therefore, the modification on H4K20 methylation has the potential to be the target for the DNA repair-based drugs [75].
The methylation of H4K20 might primarily favor NHEJ pathway. Before DNA damage, the di-methylated H4K20 by Suv4-20h1/2 is combined with L3MBTL1 and JMJD2A. Upon damage, ATM facilitates the recruitment of MDC1, and then MDC1 promotes RNF8 and RNF168 to accumulate at DSBs, which modify L3MBTL1, JMJD2A and H2AK15. Then, ubiquitylated L3MBTL1 is evicted by VCP and ubiquitylated JMJD2A is degraded by the proteasomes. In addition, phosphorylated MMSET by ATM is recruited to DSBs to mediate de novo dimethylation of H4K20 via binding to MDC1. As a result, 53BP1 is accessible to both the di-methylated H4K20 and ubiquitylated H2AK15 and initiate NHEJ pathway, while the deacetylation of H4K16 by HDAC might promote the binding of 53BP1.
A symphony of histone methylation in DSBR
Methylations among different histones and their performances are not individual, but connected. They have been indicated to act in concert with each other in a context-dependent manner in DSBR.
Firstly, the methylation of different histone lysine sites collaboratively regulates DSBR in an intricate manner and could serve as the dorking sites for the same effector protein. For instance, both methylated H3K79 and di-methylated H4K20 can bind to 53BP1 to channel specific DNA repair pathway, but H4K20 has higher affinity to 53BP1 [2]. In Saccharomyces cerevisiae, the H3K4 and H3K36 methylation independently recruit histone acetyltransferase NuA3 to the actively transcribed genes, which also provide an accessible chromatin state for DNA repair [76].
Secondly, many other types of histone modifications such as acetylation, deacetylation and ubiquitination can regulate DSBR in cooperation with histone methylation. Moreover, the proteins which interact with methylated histones may also mediate the sequential histone methylation as well as other modifications. For instance, the focus formation of 53BP1 on DSBs requires the recognition of both H4K20me2 and H2AK15ub [77], while the DNA damage-dependent H4K16ac by lysine acetyltransferase KAT5 hinders the association of 53BP1 Tudor domain with H4K20me2 [13, 78]. Histone deacetylase 1(HDAC1) and HDAC2 could reverse this process following DSBs induction and promote the binding of 53BP1 to H4K20me2 [79]. Strikingly, the acetylation of H4K16 blocks Sir3 (a deacetylase) binding and stimulates nearby H3K79 methylation [80]. For H4K16ac that is aforementioned repeatedly, there is a crosstalk between H3K36me3 and H4K16ac in HR of DSB repair. DNA damage-induced tri-methylation of H3K36 stimulates H4K16 acetylation through the interaction of H3K36me3 reader LEDGF with KAT5 [81]. Furthermore, the monoubiquitination of H2B-K123 by Rad6 ubiquitin-conjugating E2 enzyme and RNF20/Bre1 ubiquitin E3 ligase is regarded as a prerequisite for both H3K4 and H3K79 methylation. In line with these findings, the deletion of Rad6 has been identified to protect H3K4 and H3K79 from methylation [82, 83]. The ubiquitylation of H2B at lysine 120 induces H3K79 methylation by corralling the methyltransferase hDot1L on the nucleosome surface [84]. Therefore, the orchestrated histone modification centered on methylation plays harmoniously upon DSBR.
Thirdly, besides the cooperation within histone modification on different sites mentioned above, histone methylation, may be in either independent way or interplay with DNA methylation within a specific genomic region and even some small RNAs [40, 85].
Other histone methylation in DSBR
Moreover, a number of other histone lysine methylations identified by mass spectrometry were reported to participate in DSBR. The dimethylation of H2AX on lysine 134 by SUV39H2 is critical for γ-H2AX production, which was a marker of DNA damage and triggered DSBR [86]. H3K125me was identified as a novel histone PTM around by chromatin affinity purification with mass spectrometry (ChAP-MS) and probably participated in HR repair through blocking K125 ubiquitination [87]. Trimethylation of H3K23 by Ezl3p coordinates the localization of meiosis-induced DSBs to protect heterochromatin from damages [88]. The roles of these newly featured histone lysine methylation on DSBR need further investigation.
Application perspectives of histone methylation in cancer
As the key reason of tumorigenesis, the mechanism of DNA damage repair from histone methylation point of view has been focused in the past decade. First, accumulated evidences show that ectopic expression of histone methyltransferases and demethylases are often found in a variety of cancer [6, 89-91]. Therefore, targeting on histone methylation might represent a promising strategy for anticancer treatment. For instance, histone methyltransferase MMSET is overexpressed in several types of tumor, all of which are associated with a poorer prognosis. Studies performed in U2OS cell showed that MMSET is required for efficient HR and NHEJ. Besides, the loss of MMSET resulted in loss of expression of several DNA repair proteins and decreased recruitment of these proteins to DSB sites. The fact that MMSET enhances DNA damage repair leading to an increased resistance to chemotherapeutic agents offers a potential target for therapy [92]. More interestingly, siRNA-mediated knockdown of MMSET in combination with metformin treatment led to further reduction in migration and invasion of prostate cancer cells [93].
Second, several histone methylation regulators are found significantly dysregulated in metastasis. Overexpression of EZH2 in prostate cancer inhibits the expression of TIMPs to shift the MMPs/TIMPs balance in favor of MMPs activity and promote ECM degradation and the invasion of cancer cells [94]. The expression of EZH2 is also identified as a prognostic biomarker of BRCA1-deficient tumor to chemotherapy [62]. SETDB1 might be a bona fide oncogene and its up-regulation in human hepatocellular carcinoma (HCC) promotes cancer metastasis [95]. And histone demethylase KDM2A promotes the tumorigenesis and metastasis of cancer cells in gastric cancer by down-regulating the expression of programmed cell death 4 (PDCD4) and in non-small cell lung cancer (NSCLC) by activating ERK1/2 signaling via epigenetic repression of DUSP3 expression [96]. Mutant IDH1 downregulated ATM, through increasing the H3K9 methylation on the promoter of ATM, to impair DNA repair and enhance the sensitivity to DNA damage, which implicated a strategy for IDH-mutant leukemia [97]. Of note, some of the histone methyltransferases mentioned above were found not only to methylate histone, but also to be non-histone proteins as the substrate, such as 53BP1, during carcinogenesis. Lysine methylation as a dynamic event that is involved in DNA damage repair is beyond the histone code [98].
Finally, histone methylation is associated with de novo DNA methylation. By recruiting DNA methyltransferase 1 (DNMT-1) to the target chromatin complex to orchestrate the epigenetic effect is taking effect into surveillance of DNA damage and carcinogenesis. Therefore, the combination with both epigenetic strategy has become the trend [85, 99].
Clinical trial: encouraging plots indicate a promising future
DNA damage and inappropriate repair are the main causes for tumorigenesis. Histone methylation is regarded as a pivotal character in choosing DNA damage repair pathway, thus it might offer the huge room for the discovery and pharmacological intervention of cancer treatment.
Indeed, a number of small molecule inhibitors targeting histone methyltransferases and dimethylases have already reached the first stages of clinical trials in cancer treatment [6, 89]. Regarding the EZH2 inhibitor EPZ-6438 (tazemetostat), the first part of its clinical phase 1/2 trial in patients with advanced solid tumors or with relapsed or refractory B cell lymphomas, has already completed with a favorable safety and tolerability. In addition, nine of fifteen NHL patients in group achieve an objective response with the majority of adverse events evaluated at grade 1 or 2. Interestingly, a preclinical data indicated a synergism between EPZ-6438 with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone). Due to the data that the LSD1 inhibitor TCP can re-sensitize non-APL AML cells to ATRA (all-trans retinoic acid), three independent groups are testing the TCP/ATRA combination in AML patients. What's more, the synergistic lethal effect showed by the combination of SP2509 (LSD1 inhibitor) and panobinostat (pan-HDAC inhibitor). In contrast to each agent alone, the combination remarkably improves the survival of the mice engrafted with human AML cells without any detectable toxicity [100]. In addition, the combination by integrating DNA methylation inhibitors with inhibitors of histone modifiers may also provide the strategy of complementary therapies [85].
Although the aforementioned evidences shed light on the novel therapeutic targets, the field of epigenetic therapy still began to be addressed. Many issues remain to be solved. For example, subtype-selective inhibitors of lysine methyltransferase are difficult to identify [6]. In addition, the combining effect with other chemotherapeutic agents and even radiotherapy still need to be observed and evaluated to achieve more effective outcome clinically.
Acknowledgements
This work is supported by National Natural Science Foundation of China (No.81541142, No.81672091), the China Postdoctoral Science Foundation (2017M620545), and Beijing Natural Science Foundation (No.7172232).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ciccia A, Elledge SJ. The DNA Damage Response: Making it safe to play with knives. Molecular cell. 2010;40:179-204
2. Chen Y, Zhu WG. Biological function and regulation of histone and non-histone lysine methylation in response to DNA damage. Acta Biochimica et Biophysica Sinica. 2016;48:603-16
3. Ng SS, Yue WW, Oppermann U. et al. Dynamic protein methylation in chromatin biology. Cellular & Molecular Life Sciences Cmls. 2008;66:407-22
4. Freitag M. Histone Methylation by SET Domain Proteins in Fungi. Annual review of microbiology. 2017;71:413-439
5. Kinnaird A, Zhao S, Wellen KE. et al. Metabolic control of epigenetics in cancer. Nature Reviews Cancer. 2016;16:694-707
6. Morera L, Lübbert M, Jung M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clinical Epigenetics. 2016;8:1-16
7. Hyun K, Jeon J, Park K. et al. Writing, erasing and reading histone lysine methylations. Experimental & Molecular Medicine. 2017;49:e324
8. Chapman JR, Taylor MR, Boulton SJ. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Molecular cell. 2012;47:497-510
9. Clouaire T, Legube G. DNA double strand break repair pathway choice: a chromatin based decision? Nucleus. 2015;6:107-13
10. Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Genetics. 2011;45:247-71
11. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Biochemistry. 2010;79:181-211
12. Heyer WD, Ehmsen KT, Jie L. Regulation of homologous recombination in eukaryotes. Genetics. 2010;44:113-39
13. Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nature Reviews Molecular Cell Biology. 2014;15:7-18
14. Herbette M, Mercier MG, Michal F. et al. The C. elegans SET-2/SET1 histone H3 Lys4 (H3K4) methyltransferase preserves genome stability in the germline. DNA Repair. 2017;57:139-50
15. Mosammaparast N, Kim H, Laurent B. et al. The histone demethylase LSD1/KDM1A promotes the DNA damage response. Journal of Cell Biology. 2013;203:457-70
16. Chen YJ, Tsai CH, Wang PY. et al. SMYD3 Promotes Homologous Recombination via Regulation of H3K4-mediated Gene Expression. Scientific Reports. 2017;7:3842
17. Li X, Liu L, Yang S. et al. Histone demethylase KDM5B is a key regulator of genome stability. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:7096-101
18. Gong F, Clouaire T, Aguirrebengoa M. et al. Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. The Journal of cell biology. 2017;216:1959-74
19. Pfister S, Ahrabi S, Zalmas LP. et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Reports. 2014;7:2006-18
20. Hacker KE, Fahey CC, Shinsky SA. et al. Structure/Function Analysis of Recurrent Mutations in SETD2 Protein Reveals a Critical and Conserved Role for a SET Domain Residue in Maintaining Protein Stability and Histone H3 Lys-36 Trimethylation. Journal of Biological Chemistry. 2016;291:21283-21295
21. Aymard F, Bugler B, Schmidt CK. et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nature Structural & Molecular Biology. 2014;21:366-74
22. Daugaard M, Baude A, Fugger K. et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nature Structural & Molecular Biology. 2012;19:803-10
23. Ernst J, Kheradpour P, Mikkelsen TS. et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43-9
24. Aymard F, Bugler B, Schmidt CK. et al. Transcriptionally active chromatin recruits homologous recombination at DNA double strand breaks. Nature Structural & Molecular Biology. 2014;21:366-74
25. Beucher A, Birraux J, Tchouandong L. et al. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. Embo Journal. 2009;28:3413-27
26. Fnu S, Williamson EA, De Haro LP. et al. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:540-5
27. Hromas R, Williamson EA, Fnu S. et al. Chk1 phosphorylation of Metnase enhances DNA repair but inhibits replication fork restart. Oncogene. 2012;31:4245-54
28. Cao LL, Wei F, Du Y. et al. ATM-mediated KDM2A phosphorylation is required for the DNA damage repair. Oncogene. 2016;35:301-13
29. Chang CF, Chu PC, Wu PY. et al. PHRF1 promotes genome integrity by modulating non-homologous end-joining. Cell Death & Disease. 2015;6:e1716
30. Amendola PG, Zaghet N, Ramalho JJ. et al. JMJD-5/KDM8 regulates H3K36me2 and is required for late steps of homologous recombination and genome integrity. Plos Genetics. 2017;13:e1006632
31. Pai CC, Deegan RS, Subramanian L. et al. A histone H3K36 chromatin switch coordinates DNA double-strand break repair pathway choice. Nature Communications. 2014;5:4091
32. Krogan NJ, Dover J, Wood A. et al. The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Molecular cell. 2003;11:721-9
33. Lazzaro F, Sapountzi V, Granata M. et al. Histone methyltransferase Dot1 and Rad9 inhibit single-stranded DNA accumulation at DSBs and uncapped telomeres. Embo Journal. 2008;27:1502-12
34. Huyen Y, Zgheib O Jr DT. et al. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406-11
35. Farooq Z, Banday S, Pandita TK. et al. The many faces of histone H3K79 methylation. Mutation Research Reviews in Mutation Research. 2016;768:46-52
36. Vlaming H, Molenaar TM, Van TW. et al. Direct screening for chromatin status on DNA barcodes in yeast delineates the regulome of H3K79 methylation by Dot1. eLife. 2016;5:e18919
37. Boudoures AL, Pfeil JJ, Steenkiste EM. et al. A Novel Histone Crosstalk Pathway Important for Regulation of UV-Induced DNA Damage Repair in Saccharomyces cerevisiae. Genetics. 2017;206:1389-1402
38. Zhu B, Chen S, Wang H. et al. The protective role of DOT1L in UV-induced melanomagenesis. Nature communications. 2018;9:259
39. Stewart MD, Li J, Wong J. Relationship between Histone H3 Lysine 9 Methylation, Transcription Repression, and Heterochromatin Protein 1 Recruitment. Molecular & Cellular Biology. 2005;25:2525-38
40. Qian Z, Zhang J, Chen R. et al. Dissecting the precise role of H3K9 methylation in crosstalk with DNA maintenance methylation in mammals. Nature communications. 2016;7:12464
41. Strom AR, Emelyanov AV, Mir M. et al. Phase separation drives heterochromatin domain formation. Nature. 2017;547:241-245
42. Ayoub N, Jeyasekharan AD, Bernal JA. et al. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature. 2008;453:682-6
43. Mori E. A method for systematic mapping of protein lysine methylation identifies functions for HP1β in DNA damage response. Molecular cell. 2013;50:723-35
44. Kaidi A, Jackson SP. KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature. 2013;498:70-4
45. Sun Y, Jiang X, Xu Y. et al. Histone H3 methylation links DNA damage detection to activation of the Tip60 tumor suppressor. Nature Cell Biology. 2009;11:1376-82
46. Murr R, Loizou JI, Yang YG. et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nature Cell Biology. 2006;8:91-9
47. Hsiao KY, Mizzen CA. Histone H4 deacetylation facilitates 53BP1 DNA damage signaling and double-strand break repair. Journal of Molecular Cell Biology. 2013;5:157-65
48. Wu W, Nishikawa H, Fukuda T. et al. Interaction of BARD1 and HP1 Is Required for BRCA1 Retention at Sites of DNA Damage. Cancer Research. 2015;75:1311-21
49. Young LC, Mcdonald DW, Hendzel MJ. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following γ-irradiation. Journal of Biological Chemistry. 2013;288:21376-88
50. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343-9
51. Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nature Reviews Molecular Cell Biology. 2005;6:838-49
52. Sak A, Kübler D, Bannik K. et al. Epigenetic silencing and activation of transcription: influence on the radiation sensitivity of glioma cell lines. International Journal of Radiation Biology & Related Studies in Physics Chemistry & Medicine. 2017;93:494-506
53. Fischle W, Wang Y, Jacobs SA. et al. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes & Development. 2003;17:1870-81
54. Ginjala V, Nacerddine K, Kulkarni A. et al. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Molecular & Cellular Biology. 2011;31:1972-82
55. Wu Z, Lee ST, Qiao Y. et al. Polycomb protein EZH2 regulates cancer cell fate decision in response to DNA damage. Cell Death & Differentiation. 2011;18:1771-9
56. Chou DM, Adamson B, Dephoure NE. et al. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18475-80
57. Gao SB, Li KL, Qiu H. et al. Enhancing chemotherapy sensitivity by targeting PcG via the ATM/p53 pathway. American journal of cancer research. 2017;7:1874-1883
58. Williams K, Christensen J, Rappsilber J. et al. The Histone Lysine Demethylase JMJD3/KDM6B Is Recruited to p53 Bound Promoters and Enhancer Elements in a p53 Dependent Manner. Plos One. 2014;9:e96545
59. Zhang C, Hong Z, Ma W. et al. Drosophila UTX Coordinates with p53 to Regulate ku80 Expression in Response to DNA Damage. Plos One. 2013;8:e78652
60. Yang Q, Sangeeta N, Laknaur A. et al. The Polycomb Group Protein EZH2 Impairs DNA Damage Repair Gene Expression in Human Uterine Fibroids. Biology of reproduction. 2016;94:69 1-15
61. Campbell S, Ismail IH, Young LC. et al. Polycomb repressive complex 2 contributes to DNA double-strand break repair. Cell cycle (Georgetown, Tex). 2013;12:2675-83
62. Rondinelli B, Gogola E, Yücel H. et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nature cell biology. 2017;19:1371-1378
63. Wang B, Matsuoka S, Carpenter PB. et al. 53BP1, a Mediator of the DNA Damage Checkpoint. Science. 2002;298:1435-8
64. Bunting SF, Callén E, Wong N. et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243-54
65. Tuzon C, Spektor T, Kong X. et al. Concerted Activities of Distinct H4K20 Methyltransferases at DNA Double-Strand Breaks Regulate 53BP1 Nucleation and NHEJ-Directed Repair. Cell reports. 2014;8:430-8
66. Acs K, Luijsterburg MS, Ackermann L. et al. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nature Structural & Molecular Biology. 2011;18:1345-50
67. Mallette FA, Mattiroli F, Cui G. et al. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. Embo Journal. 2012;31:1865-78
68. Jacquet K, Fradet-Turcotte A, Avvakumov N. et al. The TIP60 Complex Regulates Bivalent Chromatin Recognition by 53BP1 through Direct H4K20me Binding and H2AK15 Acetylation. Molecular cell. 2016;62:409-21
69. Zhang H, Liu H, Chen Y. et al. A cell cycle-dependent BRCA1-UHRF1 cascade regulates DNA double-strand break repair pathway choice. Nature communications. 2016;7:10201
70. Boersma V, Moatti N, Segurabayona S. et al. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5' end resection. Nature. 2015;521:537-40
71. Michal Z, Francisca L, Buonomo SB. et al. 53BP1 regulates DSB repair using Rif1 to control 5' end resection. Science. 2013;339:700-4
72. Simonetta M, De IK, Serrat J. et al. H4K20me2 distinguishes pre-replicative from post-replicative chromatin to appropriately direct DNA repair pathway choice by 53BP1-RIF1-MAD2L2. Cell cycle (Georgetown, Tex). 2017:1-29
73. Pellegrino S, Michelena J, Teloni F. et al. Replication-Coupled Dilution of H4K20me2 Guides 53BP1 to Pre-replicative Chromatin. Cell reports. 2017;19:1819-1831
74. Li Y, Armstrong RL, Duronio RJ. et al. Methylation of histone H4 lysine 20 by PR-Set7 ensures the integrity of late replicating sequence domains in Drosophila. Nucleic acids research. 2016;44:7204-18
75. Bromberg KD, Mitchell TRH, Upadhyay AK. et al. The SUV4-20 inhibitor A-196 verifies a role for epigenetics in genomic integrity. Nature Chemical Biology. 2017;13:317-324
76. Martin BJ, Mcburney KL, Maltby VE. et al. Histone H3K4 and H3K36 Methylation Independently Recruit the NuA3 Histone Acetyltransferase in Saccharomyces cerevisiae. Genetics. 2017;205:1113-1123
77. Li J, Xu X. DNA double-strand break repair: a tale of pathway choices. Acta Biochimica Et Biophysica Sinica. 2016;48:641-646
78. Tang J, Cho NW, Cui G. et al. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nature Structural & Molecular Biology. 2013;20:317-25
79. Miller KM, Tjeertes JV, Coates J. et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nature Structural & Molecular Biology. 2010;17:1144-51
80. Altaf M, Utley RT, Lacoste N. et al. Interplay of Chromatin Modifiers on a Short Basic Patch of Histone H4 Tail Defines the Boundary of Telomeric Heterochromatin. Molecular cell. 2007;28:1002-14
81. Li L, Wang Y. Crosstalk between the H3K36me3 and H4K16ac histone epigenetic marks in DNA double-strand break repair. Journal of Biological Chemistry. 2017;292:11951-11959
82. Ng HH, Xu RM, Zhang Y. et al. Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J. Biol. Chem. 277, 34655-34657. Journal of Biological Chemistry. 2002;277:34655-7
83. Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002;418:104-8
84. Zhou L, Holt MT, Ohashi N. et al. Evidence that ubiquitylated H2B corrals hDot1L on the nucleosomal surface to induce H3K79 methylation. Nature communications. 2016;7:10589
85. Liang G, Weisenberger DJ. DNA methylation aberrancies as a guide for surveillance and treatment of human cancers. Epigenetics. 2017;12:416-432
86. Sone K, Piao L, Nakakido M. et al. Critical role of lysine 134 methylation on histone H2AX for γ-H2AX production and DNA repair. Nature communications. 2014;5:5691
87. Wang P, Byrum S, Fowler FC. et al. Proteomic identification of histone post-translational modifications and proteins enriched at a DNA double-strand break. Nucleic acids research. 2017;45:10923-10940
88. Papazyan R, Voronina E, Chapman JR. et al. Methylation of histone H3K23 blocks DNA damage in pericentric heterochromatin during meiosis. eLife. 2014;3:e02996
89. Song Y, Wu F, Wu J. Targeting histone methylation for cancer therapy: enzymes, inhibitors, biological activity and perspectives. Journal of Hematology & Oncology. 2016;9:1-21
90. Boscolorizzo P, Furlan C, Lupato V. et al. Novel insights into epigenetic drivers of oropharyngeal squamous cell carcinoma: role of HPV and lifestyle factors. Clinical epigenetics. 2017;9:124
91. Gusyatiner O, Hegi ME. Glioma epigenetics: From subclassification to novel treatment options. Seminars in cancer biology. 2017 pii: S1044-579X(17)30258-4
92. Shah MY, Martinezgarcia E, Phillip JM. et al. MMSET|[sol]|WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene. 2016;35:5905-5915
93. Habeeb WA, Garcia J, Fleshner N. et al. Metformin elicits antitumor effects and downregulates the histone methyltransferase multiple myeloma SET domain (MMSET) in prostate cancer cells. Prostate. 2016;76:1507-18
94. Shin YJ, Kim JH. The Role of EZH2 in the Regulation of the Activity of Matrix Metalloproteinases in Prostate Cancer Cells. Plos One. 2012;7:e30393
95. Wong CM, Lai W, Law CT. et al. Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology. 2016;63:474-87
96. KW W, H A, SS D. et al. KDM2A promotes lung tumorigenesis by epigenetically enhancing ERK1/2 signaling. Journal of Clinical Investigation. 2013;123:5231-46
97. Inoue S, Li WY, Tseng A. et al. Mutant IDH1 downregulates ATM and alters DNA repair and sensitivity to DNA damage independent of TET2. Cancer cell. 2016;30:337-48
98. Wu Z, Connolly J, Biggar KK. Beyond histones: the expanding roles of protein lysine methylation. Febs Journal. 2017:2732-44
99. Mateen BA, Hill CS, Biddie SC. et al. DNA methylation: Basic biology and application to traumatic brain injury. Journal of neurotrauma. 2017;34:2379-88
100. Fiskus W, Sharma S, Shah B. et al. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia. 2014;28:2155-64
Author contact
![]() Corresponding authors: Lixiang Xue, Ph.D., Medical Research Center, Peking University Third Hospital, Beijing 100191, China. Tel & Fax: (86)-010-82267715. E-mail: lixiangxuepku.edu.cn and Junjie Wang, M.D., Department of Radiation Oncology, Peking University Third Hospital, No.49 Huayuan North Road, Beijing, 100191, P.R. China, Tel & Fax.: (+86)-010-82265198. E-mail: junjiewang_educn
Corresponding authors: Lixiang Xue, Ph.D., Medical Research Center, Peking University Third Hospital, Beijing 100191, China. Tel & Fax: (86)-010-82267715. E-mail: lixiangxuepku.edu.cn and Junjie Wang, M.D., Department of Radiation Oncology, Peking University Third Hospital, No.49 Huayuan North Road, Beijing, 100191, P.R. China, Tel & Fax.: (+86)-010-82265198. E-mail: junjiewang_educn