Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(8):1349-1356. doi:10.7150/jca.22390 This issue Cite
Research Paper
TRIM32 promotes proliferation and confers chemoresistance to breast cancer cells through activation of the NF-κB pathway
Ting-Ting Zhao1, Feng Jin1, Ji-Guang Li1, Ying-Ying Xu1, Hui-Ting Dong1, Qun Liu1, Peng Xing1, Guo-Lian Zhu2, Hao Xu3, Song-Cheng Yin4, Zhi-Feng Miao4 ![]()
1. Department of Breast Surgery, First Hospital of China Medical University, Shenyang, Liaoning Province, China.
2. Department of Breast Surgery, Fifth People's Hospital of Shenyang, Shenyang, Liaoning Province, China
3. Department of Medical Oncology, Shengjing Hospital of China Medical University, Shenyang, Liaoning Province, China
4. Department of Surgical Oncology, First Hospital of China Medical University, Shenyang, Liaoning Province, China.
Received 2017-8-15; Accepted 2018-1-30; Published 2018-4-5
Abstract
Dysregulation of TRIM32 has been implicated in several human cancers, however, its clinical significance and biological function in breast cancer have not been investigated. Using immunohistochemistry, we found that TRIM32 expression is upregulated in breast cancer tissues and that it correlates with advanced stage and poor prognosis. TRIM32 is also overexpressed in 4/7 breast cancer cell lines. CCK8 and colony formation assays showed that TRIM32 depletion inhibited proliferation and colony formation in the T47D cell line, while TRIM32 overexpression promoted MCF-7 cell growth and colony formation. Cell viability and Annexin V/PI staining demonstrated that TRIM32 maintained breast cancer cell survival and reduced apoptosis rate when cells were treated with cisplatin. Western blot analysis demonstrated that TRIM32 overexpression resulted in an upregulation of p-IκB, p-p65, cIAP1, and cIAP2 and a downregulation of p21 and p27 in MCF-7 cells. TRIM32 depletion in T47D cells demonstrated the opposite results, suggesting that TRIM32 may activate the NF-κB pathway. The NF-κB inhibitor BAY 11-7082 blocked the effects of TRIM32 on cisplatin resistance and cIAP1/2 protein regulation.
Taken together, the present study demonstrates that TRIM32 downregulates p21/p27 and upregulates IAP family proteins to facilitate breast cancer cell growth and inhibit drug-induced apoptosis, possibly through the NF-κB signaling pathway.
Keywords: breast cancer, TRIM32, NF-κB, apoptosis
Introduction
Breast cancer is one of the leading causes of death in women worldwide (1, 2). In the last decades, the survival rate for breast cancer patients has improved significantly due to the development of conventional therapies. However, breast cancer still ranks second among cancer-related deaths for women (3). Thus, finding new biomarkers to predict tumor malignancy and resistance to conventional therapies is of great importance (4-7).
TRIM32 is a member of the tripartite motif (TRIM) family proteins (8, 9). TRIM32 regulates skeletal muscle stem cell differentiation and is necessary for normal adult muscle regeneration (10). It was recently reported that TRIM32 is involved in carcinogenesis, with TRIM32 mRNA found to be highly expressed in skin cancer and head and neck cancer (11, 12). The function and mechanisms of TRIM32 are not well understood. TRIM32 was reported to have E3 Ubiquitin ligase activity, which may contribute to cell differentiation and carcinogenesis. TRIM32 could ubiquitinate and downregulate tumor suppressor Abi2 to exert its function as an oncogene (12).
To date, the clinical significance and biological functions of TRIM32 in breast cancer have not been well explored. Thus, in this study, we aim to examine TRIM32 expression in breast cancer tissues using immunohistochemistry. We further investigated the function and molecular mechanism of TRIM32 in cancer cell lines.
Materials and Methods
Patients and tissue samples
This study was conducted with the approval of the Ethics Committee at the Institutional Review Board of the First Affiliated Hospital of China Medical University. All participants provided their written informed consent and the investigation was conducted according to the principles expressed in the Declaration of Helsinki.
Cell culture and transfection
MCF-7 and T47D cell lines were obtained from American Type Culture Collection (Manassas, USA). Cells were cultured in DMEM (Gibco, USA) with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA). The pCMV6-TRIM32 plasmid and empty vector were purchased from Origene (Origene, USA). TRIM siRNA and negative control siRNA were purchased from Dharmacon (GE healthcare, USA). siRNA1 sequence is GAUCAGGGGUGGUCAAAUA. siRNA2 sequence is GCAUAGCCCUAACUCCUAA. Transfection of miRNA and siRNA were performed with DharmaFECT transfection reagent (GE, USA). The pCMV6-TRIM32 plasmid was obtained from Origene (Origene, USA)
Real-Time PCR
RNA extraction was performed using RNAiso (TAKARA, China). Real-Time PCR was performed using SYBR Green Master Mix from ABI (Applied Biosystem, USA). Real-Time PCR was carried out using an ABI 7500 Real-Time PCR system (Applied Biosystems, USA). Relative quantification of target genes was calculated using the 2-ΔΔCt method.
Western blot analysis
Protein was extracted using cell lysis buffer (Thermo Fisher, USA). Protein quantification was performed using the Bradford method. 30 μg protein was transferred to PVDF membranes after separation via SDS-PAGE. The PVDF membrane was incubated at 4°Covernight with the following antibodies: p21, p27, p-p65,p-κB, pp65 or GAPDH (1:1000, Cell Signaling Technology, Boston, MA, USA). The TRIM32 antibody was purchased from Santa Cruz (USA). Membranes were then incubated with HRP-coupled anti-mouse/rabbit IgG (1:2000, Cell Signaling Technology, USA) at 37°C for 2 hours and the protein bands were visualized using an ECL kit (Thermo Fisher, IL, USA) with the DNR BioImaging System (DNR, Israel).
Colony formation and CCK8 assay
For colony formation assays, cells were seeded in culture dishes and cultured for 2 weeks. The plates were then stained using Giemsa.
We perform CCK8assays using the Cell Counting Kit-8 kit (Dojindo) with a plate reader. 96 well cell plates were examined at 490 nm.
Apoptosis detection
Cell apoptosis was detected using the Annexin V/PI double staining Kit (BD bioscience, USA). After treatment, cells were harvested with 0.25% trypsin, washed with PBS and resuspended in binding buffer. Staining solution containing Annexin V/FITC and propidium iodide (PI) was added to the cell suspension. After incubation in the dark for 30 minutes, the rate of apoptosis was analyzed via ACEA flow cytometer.
Statistical analysis
SPSS version 17 for Windows was used for all statistical analyses. The Chi-Square test was used to evaluate possible correlations between TRIM32 overexpression and clinicopathologic factors. A Student's t-test was used to compare data between control and transfected cells. All p values were based on the two-sided statistical analysis and a difference with p<0.05 was considered to be statistically significant.
Results
TRIM32 is overexpressed in breast cancers
We examined the protein expression of TRIM32 in normal breast tissues, intraductalpapillomas of the breast, ductal carcinoma in situ, and 96 cases of invasive ductal carcinoma (IDC). TRIM32 protein was mainly localized in the cytoplasmic compartment of cancer cells. Negative TRIM32 expression was detected in normal breast tissues (Figure 1A), and weak expression of TRIM32 was found in 11 cases of intraductalpapillomas (Figure 1B) and 8 cases of ductal carcinoma in situ (DCIS) (Figure 1C). We found high expression/overexpression of TRIM32 in 49 of 96 (51%) invasive ductal carcinoma specimens (Figure 1D).
Expression pattern of TRIM32 in breast cancer tissue samples. A. Negative TRIM32 staining is seen in the majority of normal breast tissue. B. Negative TRIM32 staining in a case of intraductal papilloma of the breast. C. Weak TRIM32 staining in a case of ductal carcinoma in situ. D. Strong nuclear staining of TRIM32 in invasive ductal carcinoma. (Magnification, 400X). E. Kaplan-Meier analysis showing that patients with high TRIM32 have worse overall survival than patients with low TRIM32 protein (Log-Rank test, p=0.0055).
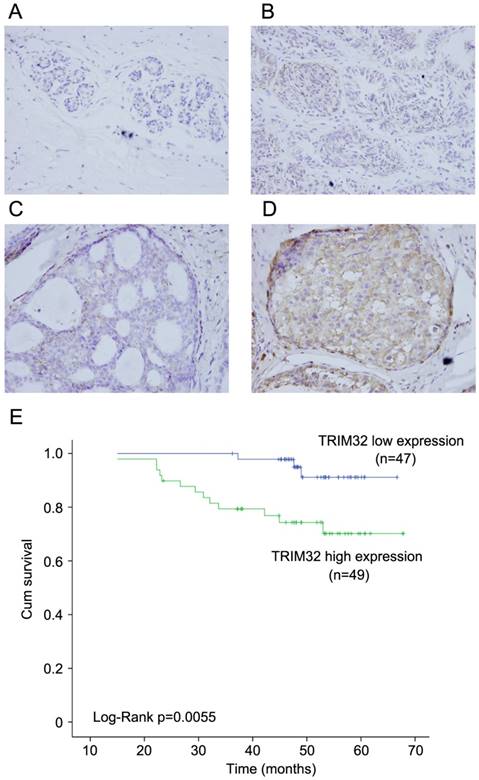
As shown in Table 1, no difference was observed between TRIM32 expression level and patient age (p=0.2887), tumor size (p=0.0605), estrogen receptor (ER) status (p=0.7728), progesterone receptor (PR) status (p=0.6918), or HER2 status (p=0.3137). High TRIM32 expression positively associated with advanced TNM stage (p=0.0129) and positive lymph node metastasis (p=0.0416). A Kaplan-Meier analysis showed that patients with high TRIM32 expression had shorter overall survival than those with low TRIM32 expression (Figure 1E) (Log-Rank test, p=0.0055).
Distribution of TRIM32 status in breast cancer according to clinicopathological characteristics.
| Characteristics | Number of patients | TRIM32 low expression | TRIM32 overexpression | P |
|---|---|---|---|---|
| Age | ||||
| <60 | 72 | 33 | 39 | 0.2887 |
| ≥60 | 24 | 14 | 10 | |
| TNM stage | ||||
| Ⅰ | 35 | 23 | 12 | 0.0129 |
| Ⅱ-Ⅳ | 61 | 24 | 37 | |
| Tumor size | ||||
| <2cm | 32 | 20 | 12 | 0.0605 |
| ≥2cm | 64 | 27 | 37 | |
| Lymph node metastasis | ||||
| Absent | 47 | 28 | 19 | 0.0416 |
| Present | 49 | 19 | 30 | |
| Estrogen receptor | ||||
| Absent | 32 | 15 | 17 | 0.7728 |
| Present | 64 | 32 | 32 | |
| Progesterone receptor | ||||
| Absent | 45 | 23 | 22 | 0.6918 |
| Present | 51 | 24 | 27 | |
| HER-2 | ||||
| Absent | 69 | 36 | 33 | 0.3137 |
| Present | 27 | 11 | 16 | |
| Triple-negative | ||||
| Absent | 78 | 37 | 41 | 0.5345 |
| Present | 18 | 10 | 8 |
TRIM32 expression is up-regulated in breast cancer cells
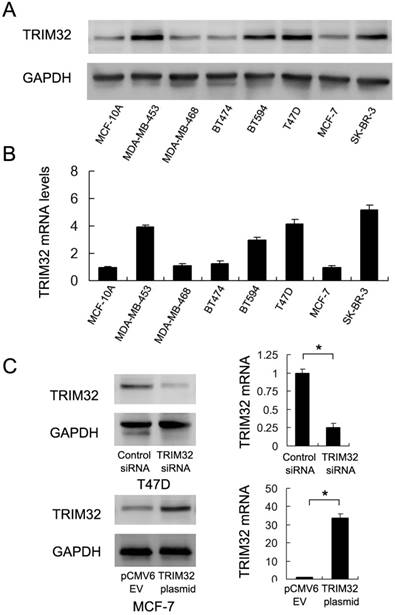
We used western blotting and Real-Time PCR to qualify TRIM32 protein and mRNA in the non-tumorigenic breast epithelial cell line MCF-10A and 7 breast cancer cell lines (Figure 2A&B). The results showed that TRIM32 was significantly elevated in 4 of the 7 cell lines (MDA-MB-453, BT594, T47D and SK-BR-3). Due to their relatively low and high levels of TRIM32 mRNA expression, we selected MCF-7 for TRIM32 overexpression via plasmid transfection and T47D for TRIM32 siRNA transfection. Transfection efficiency was examined and confirmed via PCR and western blot (Figure 2C). In addition, we used a second siRNA sequence to avoid off-target effects. The knockdown efficiency of siRNA2 was shown in Supplementary Figure 1A.
Expression of TRIM32 in breast cancer cell lines. A. Western blot and B. PCR analysis of TRIM32 expression in normal cell line MCF-10A and 7 breast cancer cell lines (MDA-MB-453, MDA-MB-468, BT474, BT594, T47D, MCF-7 and SK-BR-3). TRIM32 protein and mRNA levels are significantly upregulated in MDA-MB-453, BT594, T47D and SK-BR-3 cell lines. C. Western blot and PCR analysis demonstrating that TRIM32 siRNA treatment markedly decreases its mRNA and protein levels in T47D cells. Transfection of the TRIM32 plasmid significantly upregulates its mRNA and protein expression in the MCF-7 cell line. * p<0.05.
TRIM32 promotes breast cancer cell proliferation and colony formation
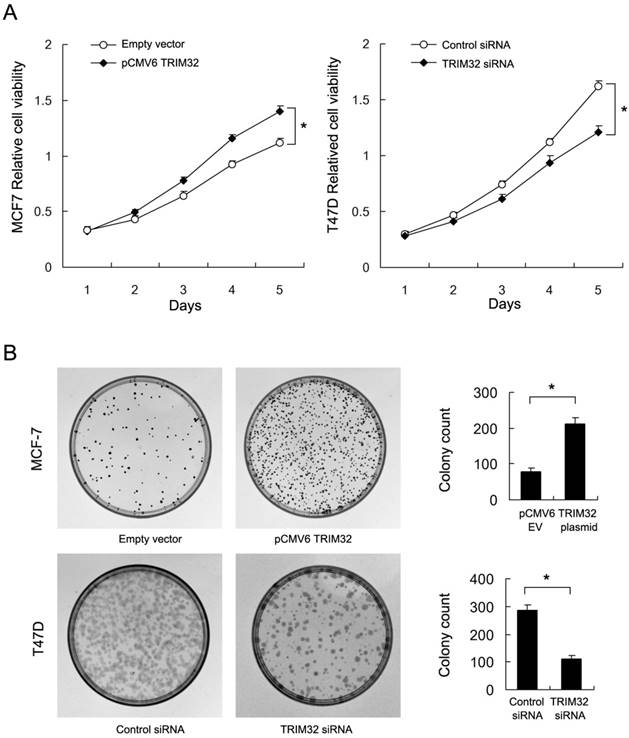
By use of CCK8 and colony formation assays, we found that TRIM32 overexpression significantly upregulated cancer cell growth rate and colony formation ability (empty vector vs. TRIM32: 78.3±6.5 vs. 211±16.7, respectively) in the MCF-7 cell line. Conversely, TRIM32 depletion in T47D cells blocked cell growth and downregulated colony formation ability (control siRNA vs. TRIM32 siRNA: 285±17.9 vs. 112±11.1) (Figure 3A&B). Use of siRNA2 also inhibited colony formation ability in T47D cell line (Supplementary Figure 1B).
Effects of TRIM32 on cell proliferation in breast cancer cells. A. CCK8 assay demonstrating that TRIM32 plasmid transfection facilitates increased cell growth in MCF-7 cells while TRIM32 siRNA decreases growth rate in T47D cells. B. TRIM32 plasmid transfection increases colony forming number in MCF-7 cells. TRIM32 siRNA decreases colony number in T47D cells. * p<0.05.
TRIM32 confers cisplatin resistance to breast cancer cells
To investigate the effect of TRIM32 on chemoresistance in breast cancer cells, we treated cancer cells with cisplatin and performed the CCK8 viability assay to examine survival rates (Figure 4A). TRIM32 overexpression increased cell viability in MCF-7 cells after 24 and 48 hours of treatment with cisplatin at 0.5μg/ml and 1μg/ml concentrations, while TRIM32 depletion reduced cisplatin resistance, resulting in decreased T47D cell viability.
TRIM32reduces chemosensitivity to cisplatin in breast cancer cells. A. CCK8 assay showing that TRIM32 significantly upregulates MCF-7 cell viability after 24 and 48 hours of cisplatin treatment (0.5μg/ml and 1μg/ml). TRIM32 siRNA shows the opposite effect by downregulating T47D cell viability B. Annexin V/PI staining shows that TRIM32 overexpression significantly downregulates apoptosis rate in MCF-7 cells while TRIM32 siRNA upregulates apoptosis in T47D cells. * p<0.05.
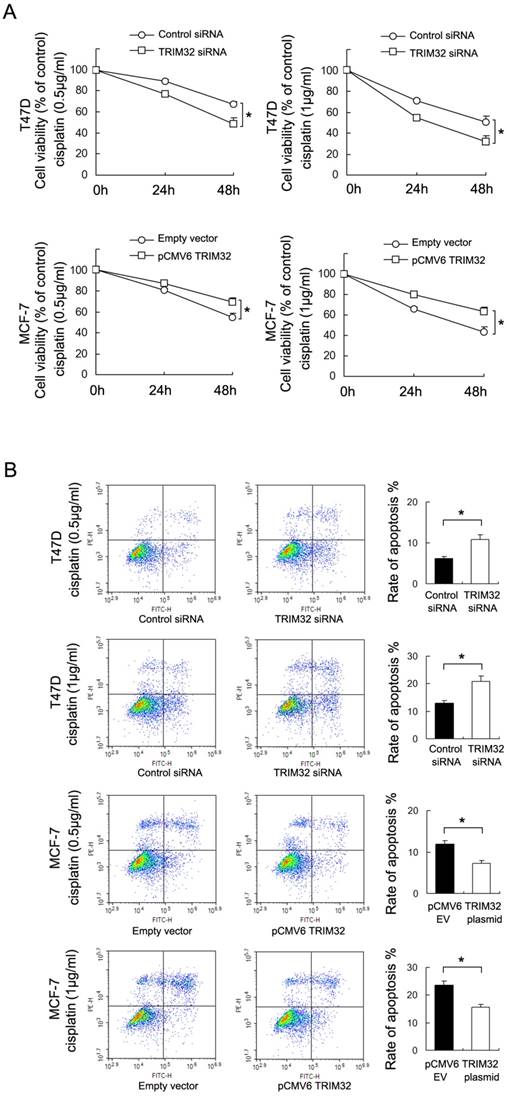
Annexin V/PI staining was employed to examine the rate of apoptosis. As shown in Figure 4B, TRIM32 overexpression significantly downregulated apoptosis rate in MCF-7 cells following 24 hours of cisplatin (0.5μg/ml and 1μg/ml) treatment, while TRIM32 siRNA upregulated apoptosis rate in T47D cells treated with 24 hours of cisplatin (0.5μg/ml and 1μg/ml). In addition, treatment with siRNA2 in T47D cells yields similar results (Supplementary Figure 1C). The combined results demonstrate that TRIM32 supports cisplatin resistance in breast cancer cells.
TRIM32 regulates cIAP1, cIAP2, p21, and p27 protein expression and NF-κB signaling
Since TRIM32 inhibits apoptosis and facilitates cell growth, we monitored related proteins to identify affected pathways. We found that TRIM32 overexpression caused upregulated protein expression of cIAP1 and cIAP2, both of which are inhibitors of apoptosis. TRIM32 also downregulated cell cycle inhibitors p21 and p27 (Figure 5).
TRIM32 regulates protein expression of cIAP1, cIAP2, p21, p27, p-IκB, and p-p65. Western blotting demonstrating that protein expression of cIAP1, cIAP2, p-IκB and p-p65 are increased while p21 and p27 protein are decreased in MCF-7 cells after TRIM32 overexpression. TRIM32 siRNA displays the opposite effects on these proteins.
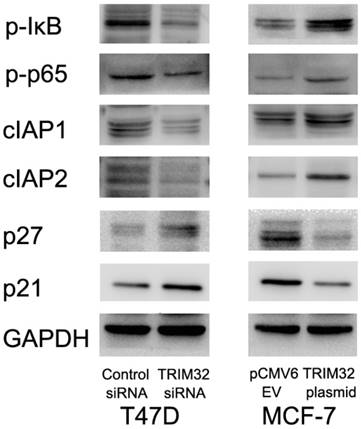
Involvement of TRIM32 in NF-κB signaling has been implicated in previous studies. We tested whether this was the case in breast cancer cells, and found that TRIM32 overexpression activated NF-κB signaling by upregulating p-κB and p-p65 levels, while its depletion downregulated NF- κB signaling via the same proteins. Treatment of T47D cells siRNA2 also downregulated p-κB, p-p65, cIAP1 and cIAP2 levels with upregulated p21 and p27 (Supplementary Figure 1D).
TRIM32 regulates cisplatin resistance through NF-κB signaling
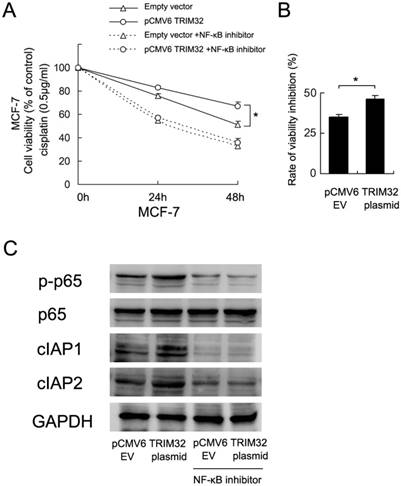
To validate the involvement of NF-κB in TRIM32-regulated cisplatin resistance, we employed BAY 11-7082 (5μM, 12 hours) to inhibit NF-κB signaling in MCF-7 cells overexpressingTRIM32. As shown in Figure 6A, blockage of NF-κB signaling downregulated cell viability and abolished the effect of TRIM32. The rate of inhibition (decreased cell viability/cell viability before treatment) by NF-κB inhibitor was higher in TRIM32 overexpressing cells than control cells (Figure 6B). NF-κB inhibition also blocked the expression of cIAP1 and cIAP2 induced by TRIM32 (Figure 6C).
TRIM32 regulates cisplatin resistance through NF-κB signaling. A. CCK8 assay demonstrates that NF-κB inhibitor treatment significantly downregulates cell viability. In cells with treated with the NF-κB inhibitor BAY 11-7082, TRIM32 fails to improve cell viability. B. The rate of cell viability inhibition by NF-κB inhibitor (decreased cell viability/cell viability before treatment) was higher in TRIM32 overexpressing cells than control cells. C. The NF-κB inhibitor abolishes the effect of TRIM32 on cIAP1 and cIAP2 upregulation.
Discussion
The E3-ubiquitin ligase TRIM32 has been reported to be overexpressed in head and neck squamous cell carcinomas (11). Overexpression of TRIM32 was shown to promote degradation of Abi2, which promoted cell growth and motility (12), and TRIM32 can also negatively regulate p53 and p53-mediated cellular stress responses (13). Overexpression of TRIM32 was further found in hepatocellular carcinoma and correlated with histological grade, tumor size, and poor patient survival (14). These data suggest that TRIM32 acts as a potential oncoprotein in human cancer development. Nevertheless, the expression pattern and biological roles of TRIM32 in human breast cancer remained unexplored. Our immunohistochemical data showed that TRIM32 is elevated in breast cancer tissues, compared to normal tissues, and that its expression significantly correlates with advanced TNM stage, nodal metastasis and poor patient survival, indicating that TRIM32 functions as a tumor oncogene in breast cancer development.
CCK8 and colony formation assays demonstrated that TRIM32 upregulates cell growth rate and colony formation ability in breast cancer cells. Accordingly, p21 and p27 protein were downregulated after overexpression of TRIM32. p21 and p27 are both cell cycle inhibitors which block cell cycle transitions (15, 16). p21 is also a downstream target of p53 signaling (17). Since TRIM32 interacts with p53, promoting its degradation through ubiquitination, it is possible that TRIM32 controls p21/p27 status indirectly through its regulation of the p53 pathway.
Next, we checked the role TRIM32 plays on cisplatin resistance and cisplatin-induced apoptosis. Our results indicate that TRIM32 induces cisplatin resistance and apoptosis inhibition. To find the underlying mechanism, we examined several related proteins and found that TRIM32 positively regulates cIAP1 and cIAP2 proteins. cIAP1/2 belongs to the inhibitor of apoptosis protein (IAP) family, which consists of an evolutionarily conserved group of apoptosis inhibitors. cIAP1/2 proteins directly interact with and inhibit the activity of caspases including caspase-3, 7, and 9 (18). In addition to the change in cIAP1/2 expression, we also found that TRIM32 could activate NF-κB signaling via upregulation of p-IκB and p-p65 expression. Activation of NF-κB signaling has been shown to lead to upregulation of IAP family proteins including cIAP1 and cIAP2 (19, 20).
To further confirm the association of TRIM32-induced cisplatin resistance and activation of NF-κB signaling, we used BAY 11-7082, an NF-κB inhibitor, to block NF-κB signaling in MCF-7 cancer cells. NF-kB inhibitor inhibited the cell viability of both control and TRIM32 cells to the same levels. However, the rate of inhibition (decreased cell viability/cell viability before treatment) was higher in TRIM32 overexpressing cells than control cells. Thus we assumed that the effect of NF-kB inhibitor was more potent in TRIM32-expressing cells, which indicates that biological effects of TRIM32 are mediated, at least partly, by NF-kB pathway. The results also showed that NF-κB blockage decreases cIAP1/2 protein expression and abolishes the effect of TRIM32 on cIAP1/2 upregulation. TRIM32 failed to upregulate cell viability in MCF-7 cells treated with the NF-κB inhibitor, suggesting that TRIM32 regulates cisplatin resistance possibly through NF-κB activation and cIAP1/2 upregulation.
In conclusion, this study demonstrates that TRIM32 is overexpressed in human breast cancers and mediates cisplatin resistance through the NF-κB signaling pathway. Our results provide insight into novel biomarkers which could predict breast cancer chemoresistance.
Supplementary Material
Supplementary figure S1.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant numbers 81272718, 81302125 and 81372550). The sponsors played no role in the study design, data collection, or analysis, or decision to submit the article for publication.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277-300
2. Permuth-Wey J, Sellers TA. Epidemiology of ovarian cancer. Methods Mol Biol. 2009;472:413-437
3. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA: a cancer journal for clinicians. 2012;62:10-29
4. Ahn S, Cho J, Sung J. et al. The prognostic significance of tumor-associated stroma in invasive breast carcinoma. Tumour Biol. 2012;33:1573-1580
5. Kabbage M, Trimeche M, Ben Nasr H. et al. Expression of the molecular chaperone alphaB-crystallin in infiltrating ductal breast carcinomas and the significance thereof: an immunohistochemical and proteomics-based strategy. Tumour Biol. 2012;33:2279-2288
6. Elfagieh M, Abdalla F, Gliwan A, Boder J, Nichols W, Buhmeida A. Serum tumour markers as a diagnostic and prognostic tool in Libyan breast cancer. Tumour Biol. 2012;33:2371-2377
7. Kurbel S. In search of triple-negative DCIS: tumor-type dependent model of breast cancer progression from DCIS to the invasive cancer. Tumour Biol. 2013;34:1-7
8. Malfavon-Borja R, Sawyer SL, Wu LI, Emerman M, Malik HS. An evolutionary screen highlights canonical and noncanonical candidate antiviral genes within the primate TRIM gene family. Genome biology and evolution. 2013;5:2141-2154
9. Meroni G. Genomics and evolution of the TRIM gene family. Advances in experimental medicine and biology. 2012;770:1-9
10. Nicklas S, Otto A, Wu X. et al. TRIM32 regulates skeletal muscle stem cell differentiation and is necessary for normal adult muscle regeneration. PloS one. 2012;7:e30445
11. Horn EJ, Albor A, Liu Y. et al. RING protein Trim32 associated with skin carcinogenesis has anti-apoptotic and E3-ubiquitin ligase properties. Carcinogenesis. 2004;25:157-167
12. Kano S, Miyajima N, Fukuda S, Hatakeyama S. Tripartite motif protein 32 facilitates cell growth and migration via degradation of Abl-interactor 2. Cancer research. 2008;68:5572-5580
13. Liu J, Zhang C, Wang XL. et al. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell death and differentiation. 2014;21:1792-1804
14. Cui X, Lin Z, Chen Y. et al. Upregulated TRIM32 correlates with enhanced cell proliferation and poor prognosis in hepatocellular carcinoma. Molecular and cellular biochemistry. 2016;421:127-137
15. Jain MV, Jangamreddy JR, Grabarek J. et al. Nuclear localized Akt enhances breast cancer stem-like cells through counter-regulation of p21(Waf1/Cip1) and p27(kip1). Cell cycle. 2015;14:2109-2120
16. Milde-Langosch K, Bamberger AM, Methner C, Rieck G, Loning T. Expression of cell cycle-regulatory proteins rb, p16/MTS1, p27/KIP1, p21/WAF1, cyclin D1 and cyclin E in breast cancer: correlations with expression of activating protein-1 family members. International journal of cancer. 2000;87:468-472
17. Fischer M, Quaas M, Steiner L, Engeland K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic acids research. 2016;44:164-174
18. Sanna MG, da Silva Correia J, Ducrey O. et al. IAP suppression of apoptosis involves distinct mechanisms: the TAK1/JNK1 signaling cascade and caspase inhibition. Molecular and cellular biology. 2002;22:1754-1766
19. Malinge S, Monni R, Bernard O, Penard-Lacronique V. Activation of the NF-kappaB pathway by the leukemogenic TEL-Jak2 and TEL-Abl fusion proteins leads to the accumulation of antiapoptotic IAP proteins and involves IKKalpha. Oncogene. 2006;25:3589-3597
20. Zou T, Rao JN, Guo X. et al. NF-kappaB-mediated IAP expression induces resistance of intestinal epithelial cells to apoptosis after polyamine depletion. American journal of physiology Cell physiology. 2004;286:C1009-1018
Author contact
![]() Corresponding author: Prof. Zhi-Feng Miao, Department of Surgical Oncology, First Hospital of China Medical University, Shenyang, Liaoning Province, China. E-mail: zfmiaoedu.cn; Phone: 8624-83283556; Fax: 8624-22703576, Zip Code: 110001
Corresponding author: Prof. Zhi-Feng Miao, Department of Surgical Oncology, First Hospital of China Medical University, Shenyang, Liaoning Province, China. E-mail: zfmiaoedu.cn; Phone: 8624-83283556; Fax: 8624-22703576, Zip Code: 110001