Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(19):4124. doi:10.7150/jca.21725 This issue Cite
Research Paper
T-cell Acute Lymphoblastic Leukemia Cells Display Activation of Different Survival Pathways
Sausan A. Moharram1,2, Kinjal Shah1,2, Julhash U. Kazi1,2 ![]()
1. Division of Translational Cancer Research, Department of Laboratory Medicine, Lund University, Lund, Sweden;
2. Lund Stem Cell Center, Department of Laboratory Medicine, Lund University, Lund, Sweden.
Received 2017-7-2; Accepted 2017-9-9; Published 2017-11-6
Abstract
T-cell acute lymphoblastic leukemia (T-ALL) is a disease of the blood affecting T-lymphocytes. Although notable improvements have been achieved in T-ALL treatment, half of the adult T-ALL patients still experience treatment failure. In order to develop a targeted therapy, we need a better understanding of T-ALL pathogenesis. In this study, we used patient-derived cell lines which display resistance to glucocorticoids. We observed that different cell lines are dependent on different survival signaling pathways. Aberrant activation of AKT, p38, S6K or ERK signaling was not found to the same degree in all cell lines studied. To understand the molecular differences in T-ALL cells, we compared gene expression and somatic mutations. Gene set enrichment analysis showed enrichment of the mTORC1, MAPK or TGF-beta signaling pathways. Loss-of-function mutations in the TP53 and FBXW7 genes were identified in all cell lines investigated. Thus, we suggest that T-ALL cells from different patients are addicted to different mutations and thereby to different signaling pathways. Therefore, understanding the enrichment of molecular pathways for each individual patient will provide us with a more precise and specific treatment plan.
Keywords: T-ALL, Lymphoblast, MOLT-3, Jurkat, CCRF-CEM.
Introduction
Acute lymphoblastic leukemia (ALL) is a group of diseases affecting the lymphoblastic lineage of blood cells. Depending on the lymphoblastic lineage affected, ALL can be subdivided into T-cell ALL (T-ALL) and B-cell ALL (B-ALL). In the past years, a significant progress has been made in the treatment of T-ALL which has contributed to improved T-ALL patient survival. However, a portion of pediatric T-ALL patients still experiences treatment failure and in an even high proportion of adult patients [1]. Therefore, we need improved therapies for patients that are unresponsive to the conventional chemotherapy. Furthermore, patients, particularly pediatric patients, suffer from long term side effects after chemotherapy.
With genomic profiling and next generation sequencing, we know the majority of genetic alterations in T-ALL [2]. Mutations in the transmembrane receptor NOTCH1 and the E3 ubiquitin ligase FBXW7 are the most common alterations in T-ALL. A constitutively activating mutation in NOTCH1 can be found in as high as 50% of T-ALL patients [3], while FBXW7 mutations occur in around 9-12% of patients [4, 5]. Mutations in FBXW7 result in stabilization of NOTCH signaling components and thereby activate NOTCH signaling by releasing the ubiquitination-mediated balance of cell signaling [4]. Mutations in NOTCH1 induce ligand-independent proteolytic cleavage of its intracellular domain [6]. The intracellular NOTCH1 domain acts as a transcription factor that induces expression of genes regulating cell proliferation and differentiation [6, 7].
The prognostic significance of NOTCH1 and/or FBXW7 mutations is not conclusive. While some studies showed that patients carrying NOTCH1 or FBXW7 mutations display favorable prognosis, other studies claim no correlation or negative correlation with survival [8-12]. Therefore, it is likely that additional oncogenic mutations carried by the patients hold an important role in the disease progression, in addition to the NOTCH1/FBXW7 mutations. Besides NOTCH1 and FBXW7 mutations, a mutation in the glucocorticoid receptor (GR) or loss of GR expression results in resistance to the most commonly used clinical drugs dexamethasone and prednisolone [13]. Glucocorticoids bind to the GR and induce apoptosis of lymphoid cells. However, the exact mechanism by which glucocorticoids induce apoptosis, or the mechanism of resistance to glucocorticoids in ALL patients remains debated. Using a panel of glucocorticoids resistant T-ALL cell lines, we here show that different cells harbor different mutations and activate different survival signaling pathways.
Materials and Methods
Reagents and cell lines
The human MOLT3, RS4;11, CCRF-CEM and Jurkat cell lines were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany). Cells were cultured in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (Gibco, Thermo Scientific). Anti- AKT- phospho-Thr308, anti-AKT-phospho-Ser473 and anti-p70S6K-phospho-Thr389 were from Abcam. Anti-GR, anti-ERK1/2-phospho-Thr202/Tyr204 and anti-beta-Actin antibodies were from Santa Cruz Biotechnology. Anti-phospho-p38 was from BD Biosciences.
Microarray analysis
Cells were serum-starved overnight before collecting total RNA using RNeasy Mini Kit (Qiagen). RNA quality was measured by Bioanalyzer, and mRNA expression was measured by the Affymetrix GeneChip® Human Gene 2.0 ST Array. Data were normalized using Expression Console™ Software. Gene set enrichment analysis was done by using GSEA software developed by the Broad Institute.
Mutational data analysis
Mutational data for CCRF-CEM and Jurkat cell lines were obtained from the COSMIC database. Mutations described in the Cancer Gene Census were used for further analysis.
Cell viability assay
PrestoBlue (Thermo Scientific) cell viability assay was used to measure the cell viability as described previously [14, 15]. Cells were incubated in 96-well plates with or without different concentrations of inhibitors. After 46 hours of incubation, PrestoBlue was added (1:10). A multi-well plate reader was used to measure fluorescence.
Results
Glucocorticoid-resistant T-ALL cell lines display differential activation of PI3K/mTOR and MAPK signaling
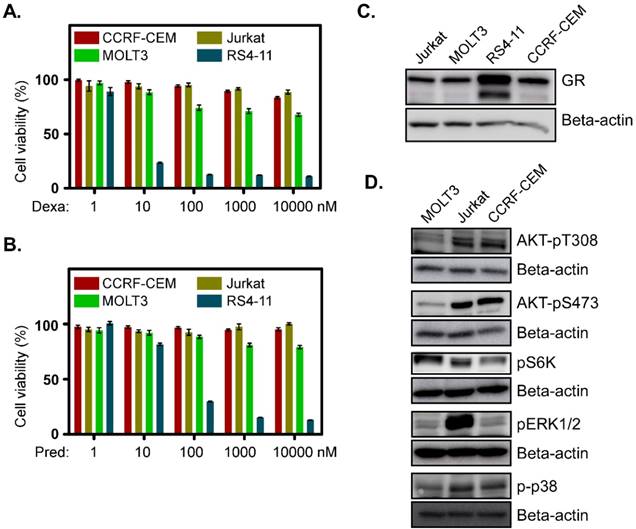
To understand the basic mechanism of the poor response to glucocorticoids, we used three T-ALL cell lines derived from relapsed T-ALL patients. MOLT3 cells were derived from a 19-year-old man with relapsed T-ALL. CCRF-CEM cells were obtained from a 3-year-old girl and Jurkat cells were from a 14-year-old boy who had experienced relapsed T-ALL. We observed that all the cell lines displayed resistance to the glucocorticoids dexamethasone (Fig. 1A) and prednisolone (Fig. 1B). These three cell lines also had a lower level of GR expression (Fig. 1C). However, activation of survival signaling pathways, such as the PI3K/mTOR and MAPK signaling pathways were different. MOLT3 cells displayed weaker phosphorylation of AKT-T308, AKT-S473, ERK1/2 and p38 (Fig. 1D, first panel), while CCRF-CEM cells achieved a decent level of AKT-T308, AKT-S473, and p38 phosphorylation but no ERK1/2 phosphorylation (Fig. 1D, 3rd panel). On the other hand, Jurkat cells showed a similar level of AKT-T308, AKT-S473 and p38 phosphorylation to that of CCRF-CEM but displayed strong ERK1/2 phosphorylation (Fig. 1D, 2nd panel). S6K phosphorylation was almost equal in all the three cell lines (Fig. 1D). Therefore, we suggest that MOLT3, Jurkat and CCRF-CEM cells have different driving forces for survival.
Glucocorticoid-resistant T-ALL cell lines display upregulation of different survival signaling pathways: (A-B) CCRF-CEM, Jurkat and MOLT3 cells were treated with different concentrations of dexamethasone (A) or prednisolone (B). Two days after the treatment, cell viability was measured by PrestoBlue cell viability assay. RS4-11 cells were used as a control. (C) Total cell lysates from Jurkat, MOLT3, RS4-11 and CCRF-CEM cell lines were used for western blot analysis to detect GR. (D) Cell lysates from different cell lines were used for western blot analysis using different phospho-specific antibodies.
CCRF-CEM and Jurkat cells display differences in pathway enrichment
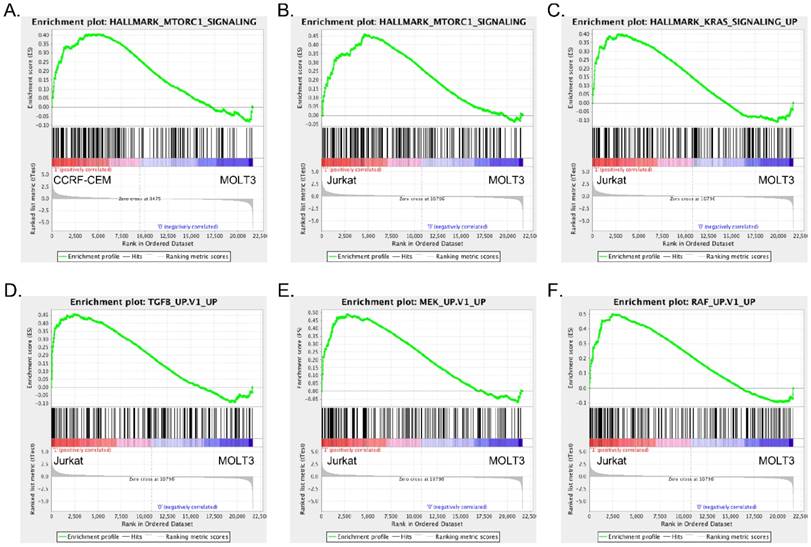
Since we observed differences in signaling events in the different cell lines, we wanted to understand the underlying mechanisms. We analyzed mRNA expression of MOLT3, CCRF-CEM and Jurkat cells using microarrays. Since MOLT3 cells displayed basal levels of AKT and ERK phosphorylation, we used gene expression of MOLT3 cells as a control to compare upregulated gene signature of CCRF-CEM and Jurkat cells. Gene set enrichment analysis identified mTORC1 pathway enrichment in CCRF-CEM cells (Fig. 2A, S1A and table S1-S2), while, Jurkat cells displayed enrichment of the TGF-beta, MEK, RAF and KRAS pathways in addition to the mTORC1 signaling pathway (Fig. 2B-F, S1B-D and table S3-S4).
CCRF-CEM and Jurkat cells differ in their enrichment of oncogenic signaling: Gene set enrichment analysis (GSEA) showed enrichment of mTORC1 (A) signaling in CCRF-CEM cells, mTORC1 (B), KRAS (C) TGF beta (D), MEK (E) and RAF (F) signaling in Jurkat cells.
CCRF-CEM and Jurkat cells display distinct mutational patterns
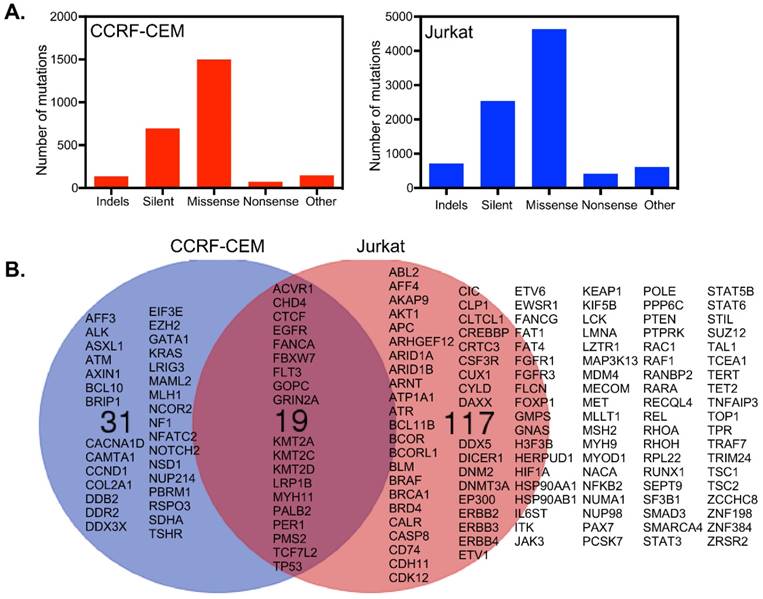
CCRF-CEM cells harbor a total of 2555 mutations in 1664 genes, while Jurkat cells have 8937 mutations in 4678 genes (Fig. 3A). Around 699 and 2540 mutations were found to be synonymous in CCRF-CEM and Jurkat cell lines respectively. We looked at genes described in the Cancer Gene Census of the COSMIC database and found 136 mutated genes in Jurkat cells and 50 mutated genes in CCRF-CEM. Nineteen mutated genes were common for both the cell lines which included mutations in receptors and genes encoding downstream signaling components (Fig. 3B). However, the role of all those mutations in cancer has not been defined. For example, the EGFR mutations observed in CCFR-CEM (A1118T) and Jurkat (R451C) cells are not known to be oncogenic. The role of FLT3 V643I (Jurkat) and A627T (CCRF-CEM) mutations is also unknown. Therefore, activation of the PI3K/mTOR and/or MAPK pathways that we observed are probably cell line specific and mediated through specific mutations in genes encoding other proteins. Although CCRF-CEM cells carry a KRAS-G12D mutation, it displays lower levels of ERK signaling. It has been shown that a single copy of KRAS mutation poorly activates ERK signaling [16]. The I323V mutation in ACVR1/ALK2 found in Jurkat cells (CCRF-CEM has a frame-shift mutation in the same gene) has not been previously studied. Since the I323V mutation occurs in the kinase domain and similar mutation, I328V, has a role in another disease, it can be speculated that ALK2 may contribute to ERK activation, that we also see in GSEA (Fig. 2D). Cell line specific mutations might play important roles in ERK activation. The A728V mutation in the BRAF gene strongly activates ERK1/2 [17] and is only observed in Jurkat cells. Jurkat cells also display KRAS (Fig. 2C), MEK (Fig. 2E) and RAF (Fig. 2F) pathway enrichment. The involvement of many other mutations in upstream kinases has not been studied. However, they might contribute to ERK1/2 phosphorylation. Another interesting mutated gene is FBXW7/CDC4 which encodes a ubiquitin ligase. CCRF-CEM cells carry an R465C mutation while Jurkat cells harbor the R505C mutation. The R465C mutation in FBXW7 plays an important role in ALL progression when combined with NOTCH1 mutation [18]. The R505C mutation in FBXW7 impairs NOTCH1 binding [4, 19]. The TP53 mutations, R248Q and R175H found in CCRF-CEM cells, are loss-of-function mutations [20]. The TP53-R248Q displays increased AKT signaling in a mouse model [21]. Jurkat cells carry the R196* mutation in TP53 that might also be a loss-of-function mutation but has not previously been studied.
CCRF-CEM and Jurkat cells differ in their somatic mutations: (A) Jurkat cells acquired more mutations over CCRF-CEM cells. (B) Considering the Cancer Gene Census, CCRF-CEM and Jurkat cells have 19 common genes mutated. CCRF-CEM has 31 unique mutations while Jurkat has 117 unique mutations.
Discussion
Due to the genetic and epigenetic heterogeneity, a portion of T-ALL patients display resistance to the conventional treatment of cancer and therefore relapse. Mutation in GR or loss of GR expression is one major cause of resistance to glucocorticoids. However, other oncogenic mutations also play important roles in disease progression. The major T-ALL specific mutations include activating mutation in NOTCH1, loss of CDK inhibitory proteins and PTEN function, loss-of-function mutations in FBXW7, activating mutations in kinases and overexpression of cyclins [22]. Herein, using different patient-derived cell lines from relapsed T-ALL patients, we show that T-ALL cells differ in survival signaling due to the difference in mutations as well as gene expression.
T-ALL cell lines derived from relapsed patients display resistance to glucocorticoids probably due to the lower expression of GR. Jurkat cells also carry a heterozygous mutation in NR3C1 gene (R478H mutation) that encodes the GR protein [13]. Besides, the loss-of GR expression, these cells differentially activate survival pathways. CCRF-CEM and Jurkat cells aberrantly activate PI3K/AKT pathway most likely due to mutations in the different driver genes. For example, Jurkat cells have a frameshift mutation in PTEN, while CCRF-CEM cells display several mutations in genes that encode the upstream kinases of the PI3K/AKT pathway. Mutations in RAS family proteins potentiate MAPK signaling [23, 24]. However, although CCRF-CEM cells harbor a KRAS activating mutation, it has a lower level of ERK1/2 activation in comparison to Jurkat cells. This can be due to the fact that Jurkat cells have an extremely strong ERK1/2 signaling. Besides, ERK1/2 activation, KRAS can also activate the PI3K/mTOR signaling pathway [25]. Collectively, these findings suggest that CCRF-CEM and Jurkat cells differ in their way of molecular signaling although both cell lines carry mutations in many overlapping genes.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Professor Lars Rönnstrand for the critical comments on the manuscript. This research was funded by the Swedish Childhood Cancer Foundation (PR2014-0059), the Crafoord Foundation (20160643), Per-Eric och Ulla Schybergs Stiftelse (20160413), Kungliga Fysiografiska Sällskapet in Lund (20161117), Stiftelsen Clas Groschinskys Minnesfond (M16 29) and Ollie and Elof Ericssons Stiftelse (20160908). JUK is a recipient of an Assistant Professorship (forskarassistenttjänst) grant from the Swedish Childhood Cancer Foundation (TJ2014-0047).
Author Contributions
SAM and KS performed experiments, analyzed data and prepared manuscript. JUK designed experiments, analyzed data and prepared manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gökbuget N, Stanze D, Beck J, Diedrich H, Horst HA, Hüttmann A. et al. Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood. 2012;120:2032-41
2. Mullighan CG. The genomic landscape of acute lymphoblastic leukemia in children and young adults. Hematology Am Soc Hematol Educ Program. 2014;2014:174-80
3. Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C. et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269-71
4. O'Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C. et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204:1813-24
5. Baldus CD, Thibaut J, Goekbuget N, Stroux A, Schlee C, Mossner M. et al. Prognostic implications of NOTCH1 and FBXW7 mutations in adult acute T-lymphoblastic leukemia. Haematologica. 2009;94:1383-90
6. Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2006;6:347-59
7. Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A. et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006;103:18261-6
8. Breit S, Stanulla M, Flohr T, Schrappe M, Ludwig WD, Tolle G. et al. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood. 2006;108:1151-7
9. Zhu YM, Zhao WL, Fu JF, Shi JY, Pan Q, Hu J. et al. NOTCH1 mutations in T-cell acute lymphoblastic leukemia: prognostic significance and implication in multifactorial leukemogenesis. Clin Cancer Res. 2006;12:3043-9
10. van Grotel M, Meijerink JP, van Wering ER, Langerak AW, Beverloo HB, Buijs-Gladdines JG. et al. Prognostic significance of molecular-cytogenetic abnormalities in pediatric T-ALL is not explained by immunophenotypic differences. Leukemia. 2008;22:124-31
11. Larson Gedman A, Chen Q, Kugel Desmoulin S, Ge Y, LaFiura K, Haska CL. et al. The impact of NOTCH1, FBW7 and PTEN mutations on prognosis and downstream signaling in pediatric T-cell acute lymphoblastic leukemia: a report from the Children's Oncology Group. Leukemia. 2009;23:1417-25
12. Zuurbier L, Homminga I, Calvert V, te Winkel ML, Buijs-Gladdines JG, Kooi C. et al. NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on DCOG or COALL protocols. Leukemia. 2010;24:2014-22
13. Schmidt S, Rainer J, Ploner C, Presul E, Riml S, Kofler R. Glucocorticoid-induced apoptosis and glucocorticoid resistance: molecular mechanisms and clinical relevance. Cell Death Differ. 2004;11(Suppl 1):S45-55
14. Moharram SA, Shah K, Khanum F, Marhäll A, Gazi M, Kazi JU. Efficacy of the CDK inhibitor dinaciclib in vitro and in vivo in T-cell acute lymphoblastic leukemia. Cancer Lett. 2017;405:73-8
15. Gazi M, Moharram SA, Marhäll A, Kazi JU. The dual specificity PI3K/mTOR inhibitor PKI-587 displays efficacy against T-cell acute lymphoblastic leukemia (T-ALL). Cancer Lett. 2017;392:9-16
16. Vartanian S, Bentley C, Brauer MJ, Li L, Shirasawa S, Sasazuki T. et al. Identification of mutant K-Ras-dependent phenotypes using a panel of isogenic cell lines. J Biol Chem. 2013;288:2403-13
17. Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM. et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855-67
18. King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P. et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell. 2013;153:1552-66
19. Yeh CH, Bellon M, Pancewicz-Wojtkiewicz J, Nicot C. Oncogenic mutations in the FBXW7 gene of adult T-cell leukemia patients. Proc Natl Acad Sci U S A. 2016;113:6731-6
20. Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268-86
21. Hanel W, Marchenko N, Xu S, Yu SX, Weng W, Moll U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013;20:898-909
22. Graux C, Cools J, Michaux L, Vandenberghe P, Hagemeijer A. Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: from thymocyte to lymphoblast. Leukemia. 2006;20:1496-510
23. Collins MA, Pasca di Magliano M. Kras as a key oncogene and therapeutic target in pancreatic cancer. Front Physiol. 2013;4:407
24. Knight T, Irving JA. Ras/Raf/MEK/ERK Pathway Activation in Childhood Acute Lymphoblastic Leukemia and Its Therapeutic Targeting. Front Oncol. 2014;4:160
25. Gilbert D, Rapley E, Shipley J. Testicular germ cell tumours: predisposition genes and the male germ cell niche. Nat Rev Cancer. 2011;11:278-88
Author contact
![]() Corresponding author: Julhash U. Kazi (kazi.uddinlu.se), Division of Translational Cancer Research, Department of Laboratory Medicine, Lund University, Medicon Village 404:C3, Scheelevägen 8, 22363 Lund, Sweden.
Corresponding author: Julhash U. Kazi (kazi.uddinlu.se), Division of Translational Cancer Research, Department of Laboratory Medicine, Lund University, Medicon Village 404:C3, Scheelevägen 8, 22363 Lund, Sweden.