Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(19):4048-4056. doi:10.7150/jca.21338 This issue Cite
Research Paper
The PARP-1 inhibitor Olaparib suppresses BRCA1 protein levels, increases apoptosis and causes radiation hypersensitivity in BRCA1+/- lymphoblastoid cells
Emma C. Bourton1*, Pia-Amata Ahorner1*, Piers N. Plowman2, Sheba Adam Zahir1, Hussein Al-Ali1, Christopher N. Parris3 ![]()
1. Division of Biosciences, Department of Life Sciences, College of Health and Life Sciences, Brunel University, Uxbridge Middlesex, UB8 3PH, UK.
2. Department of Radiotherapy, St. Bartholomew's Hospital, West Smithfield, London, EC1A 7BE, UK.
3. Department of Biomedical and Forensic Science, Faculty of Science and Technology, Anglia Ruskin University, East Road Cambridge, Cambridgeshire, CB1 1PT, UK.
*ECB and PA-A contributed equally to this work.
Received 2017-6-5; Accepted 2017-7-21; Published 2017-10-23
Abstract
The use of polyADPribose polymerase inhibitors in cancer treatment provides a unique opportunity to target DNA repair processes in cancer cells while leaving normal tissue intact. The PARP-1 enzyme repairs DNA single strand breaks (SSB). Therefore PARP-1 inhibition in BRCA1 negative cancers results in the formation of cytotoxic DNA double strand breaks (DSB) causing synthetic lethality. The use of PARP1 inhibitors is gaining momentum in the treatment of a variety of tumours with BRCA1 involvement including breast, ovarian, pancreatic and prostate cancer.
Our previous work showed that the PARP-1 inhibitor Olaparib causes both hypersensitivity of BRCA1+/- cells following exposure to gamma radiation due to the persistence of DNA strand breaks in cells, measured by the DNA damage biomarker γ-H2AX. Therefore dual treatment of cancers with radiotherapy and PARP1 inhibition may lead to cases of increased normal tissue toxicity in cancer patients.
In this study we exposed two normal lymphoblastoid cell lines and three heterozygous BRCA1 lymphoblastoid cell lines to the PARP-1 inhibitor Olaparib and gamma radiation and after measured BRCA1 protein expression and apoptosis levels following treatment. BRCA1 protein foci analysis was performed on cells exposed to 2 Gy radiation in the presence or absence of 5 μM Olaparib. Using immunofluorescence and imaging flow cytometry, foci were measured in untreated cells and at 0.5, 3, 5 and 24 hours post-irradiation. Exposing normal and BRCA1+/- cells to Olaparib followed by gamma radiation results in a dramatic change in BRCA1 protein foci expression, with a significant reduction in BRCA1 protein expression observed in the heterozygote cells, together with an increase in apoptosis levels in these cells.
In conclusion, combining PARP1 inhibitors with radiotherapy in treating of BRCA1-related cancers has clinical relevance, however this study and our previous publications serve to highlight the potential problems of increased side effects in these scenarios.
Introduction
The poly(ADP-ribose) polymerase (PARP) family of enzymes are involved in a number of cellular processes, including DNA transcription, genome maintenance (including DNA damage response and repair), cell cycle regulation and cell death. The PARP-1 enzyme, the most abundant and ubiquitous of these enzymes [1], identifies and repairs DNA single strand breaks (SSB) primarily via the base excision repair (BER) mechanism. Inhibition of PARP-1 results in DNA double strand breaks (DSB) forming from SSB during DNA replication [2].
The first molecular inhibitors of PARP, synthesised over 40 years ago, were nicotinamide analogues such as the benzamides e.g. 3-aminobenzamide. Further research and development yielded more potent PARP inhibitors that have recently entered the clinical setting e.g. Olaparib [3]. Facilitating the accumulation of DNA DSB in tumour cells by chemically inhibiting the PARP-1 enzyme has an important role in cancer treatment, particularly in treating cancers with mutations in BReast CAncer susceptibility genes 1 and 2 (BRCA1 and BRCA2, respectively) [4].
BRCA1 and BRCA2 are tumour suppressor genes involved in many cellular processes, especially DNA repair and transcriptional regulation in response to DNA damage [5]. Such DNA damage, particularly from the ionising radiation used in clinical radiotherapy, is repaired using one of two separate pathways, non-homologous end joining (NHEJ) and homologous recombination (HR). Whilst NHEJ, although error prone, repairs DNA DSB in all stages of the cell cycle, HR repair is the dominant repair mechanism in cycling (G2 and S stage) cells [6]. Investigations revealed that both BRCA1 and BRCA2 proteins are active in HR; BRCA1 is a signal mediator, and BRCA2 initiates repair by recruiting Rad51 to DSB [7].
PARP1 knockout mice (PARP-/-) are both viable and fertile, despite exhibiting defective DNA SSB repair, possibly because they also exhibit elevated levels of error-free HR [8]. In the majority of cell lines, treatment with a PARP inhibitor at concentrations that inhibit PARP activity does not automatically initiate cell death. However, breast cancer cells with homozygous mutations in either BRCA1 or BRCA2 exhibited extreme sensitivity to PARP inhibitors [8-9]. This is an example of synthetic lethality, where the simultaneous loss of two genes or proteins (in this case PARP-1 and BRCA) causes cell death that would not occur if singular gene or protein function was lost [10]. BRCA1 interacts with a wide range of DNA repair proteins, for example by directing the MRE11/RAD50/NBS1 complex to DSB sites, where it plays a role in DNA strand break resection. Therefore cells with homozygous mutations in BRCA1 are likely to have greatly decreased DNA DSB repair capacity [11]. This, together with increased DSB formation from PARP-1 inhibition, produces an accumulation of DSB that is lethal to the cell.
Fractionated radiotherapy is a standard treatment method for many locally advanced cancers, including breast cancer. Side effects, termed normal tissue toxicity (NTT), are reported in 15-20% patients undergoing such treatment, and in severe cases can limit the treatment effectiveness [12]. NTT has been standardised into scales for all tissue types [13-14]. It has also been observed that a small minority of patients exhibit side effects that are so severe as to fall outside of the established NTT scales - these individuals are referred to as 'over-reactors'. Frequently over-reactors possess one or more inherited genetic defects in the repair of DNA double strand breaks (DSB), for example Ataxia Telangiectasia (A-T) [15], and Severe Combined Immunodeficiency with Sensitivity to Ionising Radiation (RS-SCID) [16]. We have previously reported a clinical case of patient death resulting from radiotherapy-induced neurological trauma. The patient was found to be carrying an inactivating mutation in the DNA-PKcs gene (which is central to DSB repair), resulting in extreme cellular and clinical radiosensitivity [17].
The fact that the BRCA1 and BRCA2 proteins are central to proficient DNA DSB repair by either NHEJ or HR produces the possibility that individuals carrying mutations in either of these genes might represent a cohort of patients in which radiotherapy-induced NTT might be over and above the normal range expected. Indeed cellular clonogenic assays in cells derived from BRCA1 and BRCA2 heterozygotes reveal elevated radiosensitivity and chromosome aberrations when compared to cells derived from normal individuals, which is suggestive of a haplo-insufficient phenotype [18]. However, clinical evidence suggests that heterozygotes for both BRCA1 and BRCA2 do not experience abnormally severe NTT [19].
Our previous studies determined that lymphoblastoid cells heterozygous for BRCA1 exhibited hypersensitivity (in the form of decreased cell survival) when exposed to a combination of the PARP inhibitor Olaparib and gamma radiation [20]. This correlated with prolonged persistence of γ-H2AX foci (indicative of DNA DSB) within the same cells [21]. Whilst this therapeutic combination is undoubtedly promising effectiveness when treating tumours homozygous for BRCA1 mutations in pre-clinical trials [22], we cautioned that these patients may also experience increased NTT levels, caused by DNA damage and subsequent apoptosis of cells in normal tissue that are heterozygous for BRCA1 mutations.
We now report from an in-depth investigation using imaging flow cytometry that this hypersensitivity is due to reduced levels of BRCA1 protein in the cell, specifically the nucleus, and enhanced apoptosis of the cells.
Materials and methods
Cell lines
Human B lymphocytes which had been immortalised using the Epstein-Barr virus were purchased from Coriell Cell Repositories (Camden, New Jersey, USA). Details of the cell lines are shown in Table 1. Cell lines "BRCA1" were heterozygotes for mutations in the BRCA1 gene. The nature of these mutations is also detailed in Table 1.
Description of B-Lymphoblastoid cell lines used in study
| Cell Line | Type | Details of BRCA Gene Mutation |
|---|---|---|
| GM00893 | Normal | None |
| GM05423 | Normal | None |
| GM13705 | BRCA1 | 4-base pair deletion in exon 11 = truncated protein. |
| GM14090 | BRCA1 | 2-base pair deletion in exon 3 = truncated protein. |
| GM16105 | BRCA1 | Base substitution in intron 8. |
Cell culture
The cell lines were initially established in T25 cell culture flasks (Sarstedt, Leicester, UK) in RPMI 1640 culture medium (Labtech International Ltd, East Sussex, UK) supplemented with 20% foetal bovine serum (Scientific Laboratory Supplies, Yorkshire, UK), 2.0 mM L-Glutamine (Labtech International Ltd.), 100 Uml-1 Penicillin and 100 µgml-1 Streptomycin (Labtech International Ltd). After two days cell lines were transferred to T75 flasks for ongoing culture. Cells were routinely incubated at 37°C in a humidified atmosphere of 5% CO2 in air.
Cell concentration values (number of cells per ml cell suspension) and viability values (% live cells/dead cells) were determined as required using a "Countess™" automated cell counter based upon the method of trypan blue exclusion (Invitrogen, Renfrewshire, UK). Cell cultures were maintained at approximately 2 × 105 viable cells/ml; for experiments this was increased to approximately 1 × 106 viable cells/ml. Cell lines were used over a restricted range of ten passages, during which cell viability was not less than 80%.
Exposure to radiation and the PARP inhibitor Olaparib
Prior to irradiation, the cell suspension of each cell line was divided in half; one suspension was incubated for 1 hour in 5.0 µM Olaparib (LC Laboratories Inc., Woburn, Massachusetts, USA) whilst the other remained in RPMI-1640 medium, supplemented as above. The Olaparib concentration used (5.0 µM) was found from previous work to be the maximum concentration that did not significantly decrease cell survival [20]. One aliquot of 10 ml from each cell suspension was retained as an un-irradiated control; the remaining cell suspensions were irradiated with 2 Gy gamma radiation from a 60Cobalt source (Puridec Technologies, Oxfordshire, UK) sited at a distance of 25 cm with a dose rate of 0.7 Gy per minute.
Cell fixation
Irradiated cells were fixed at 30 minutes, 3 hours, 5 hours and 24 hours post-irradiation, with two samples reserved as compensation samples fixed at the 30 minute time point where the level of DNA damage and therefore fluorescence was expected to be at the maximal level. The un-irradiated control cells were fixed using the same method. Cells were pelleted by centrifugation at 1500 rpm for 5 minutes at room temperature and the complete medium discarded, washed with phosphate buffered saline (PBS) (Severn Biotech Ltd, Worcestershire, UK) and fixed in ice-cold 50:50 (V:V) methanol:acetone. Samples were stored at -20°C until submission to the immunocytochemistry stage [21].
Immunocytochemistry
Cells were re-hydrated by washing in PBS (Severn Biotech Ltd) at 4°C then incubated with gentle agitation for 5 minutes at room temperature in permeabilisation buffer consisting of 0.5% Triton™ X-100 (Sigma-Aldrich, Dorset, UK) in PBS. Cells were subsequently incubated with gentle agitation for 1 hour at room temperature in blocking buffer consisting of 5.0% rabbit serum (Labtech International Ltd), with 0.1% Triton™ X-100 in PBS. After removal of the blocking buffer the cells were incubated with gentle agitation overnight at 4°C in primary antibody solution. The primary antibody solution consisted of an anti-BRCA1 mouse monoclonal antibody, clone number MS110 (Abcam, Cambridge, UK) diluted 1 in 2000 in blocking buffer. Wash buffer, consisting of 0.1% Triton™ X-100 in PBS, was used in two sequential steps to remove excess primary antibody. A secondary antibody solution, consisting of an Alexa Fluor®488 rabbit anti-mouse IgG antibody (Invitrogen) diluted 1 in 1000 in blocking buffer, was added to each sample, except the DRAQ5™ compensation samples. The cells were incubated with gentle agitation for 2 hours at room temperature, and excess secondary antibody was removed by washing twice with wash buffer. The cells were re-suspended in 100 µl Accumax™ solution (Labtech International Ltd), and left overnight at 4°C, but with no agitation. 1.0 µl of 5 mM DRAQ5™ solution (Biostatus Limited, Leicestershire, UK) was added to each sample, except the AlexaFluor®488 compensation samples, and the samples submitted for analysis by imaging flow cytometry.
Imaging flow cytometry
Imaging flow cytometry was undertaken using the ImagestreamX Mark II system equipped with Inspire™ data acquisition software (Amnis Inc., Seattle, Washington, USA). This system captures images of each cell in flow using a maximum of six optical channels. Images of 10,000 cells were obtained on channel 1 for brightfield (BF); on channel 2 for Alexa Fluor®488 (AF) - this green stain indicating the presence of BRCA1 protein; and on channel 5 for DRAQ5™ (D5) - a red stain identifying the cell nucleus. Application of gating in real time permitted the capture of single cell images.
Image compensation
Image compensation is described in detail previously [21]. In brief, it was performed on the cell populations fixed 30 minutes post-irradiation where BRCA1 protein expression, and hence staining intensity, was highest. Images of cells stained with either antibody or DRAQ5™ were collected with the 488 nm laser only and were used to generate the compensation matrix. The software compensation wizard generated a table of coefficients whereby detected light displayed by each image was placed into the proper channel (channel 2 for antibody staining and channel 5 for DRAQ5) on a pixel-by-pixel basis. The coefficients were normalized to 1 and each coefficient represented the leakage of fluorescent signal into juxtaposed channels. Calculated compensation values were applied to all subsequent analyses as appropriate.
Analysis of cell images - calculation of foci number
Foci indicating the presence of the BRCA1 protein were quantified in approximately 10,000 images of cells per time point captured using the Inspire™ imaging flow cytometry software. Foci were quantified in a similar manner as described previously [23], with the spot counting wizard in the Ideas™ software permitting simplified foci quantitation. The wizard consists of sequential steps that identified and gated from all captured images those single cells that were in optical focus. Next, images of fifty cells from this population with varying numbers of clearly defined foci were selected to create a truth population. Images from the truth population were used to produce a mask that identified only those foci located within the cell nuclei of each image. The finalised truth population was then saved as a template and applied to quantitate foci present in all acquired cell images.
Analysis of apoptotic cell images
Significant changes in nuclear morphology are associated with apoptosis which include nuclear fragmentation and condensation of the DNA. Cellular morphological changes associated with apoptosis makes possible the automated identification of apoptotic cells. By measuring the area and the intensities of the brightest portions of the nuclear image, the bright, punctate nuclear imagery of apoptotic cells can be distinguished from the evenly stained nuclear imagery of a normal, healthy nucleus.
An analysis wizard which compares nuclear bright detail intensity and nuclear area effectively distinguishes apoptotic cells from healthy cells with a normal nuclear and cytoplasmic structure.
Statistical analysis
Data generated from the application of the Ideas™ software was further analysed using the data analysis programme in Microsoft Excel. A one way analysis of variance test was utilised to determine if there were significant differences in foci number within the cell populations at different time points after Olaparib and gamma radiation exposure. These populations consisted of cells treated or untreated with Olaparib prior to being irradiated and then fixed at specific time points post-irradiation.
Results
Quantitation of BRCA1 protein foci in normal and BRCA1+/- lymphoblastoid cells following gamma radiation exposure
Initial analysis was conducted to compare the number of BRCA1 protein foci in the nuclei of lymphoblastoid cells derived from two normal individuals (GM00893 and GM05423) with three lines derived from BRCA1+/- patients (GM13705, GM14090 and GM16105) over a period of 24 hours following the induction of DNA damage by exposure to 2 Gy gamma radiation. In Figure 1 the mean number of BRCA1 foci for the two normal cell lines and the three BRCA1+/- lines are plotted in a bar chart at each time point following exposure. Using a one way ANOVA test to determine differences in the mean number of foci at each time point we determined that there are significantly fewer foci in the BRCA1+/- cell lines when compared to normal cells (P = 0.0375).
shows the number of BRCA1 protein foci in the nuclei of lymphoblastoid cells derived from two normal individuals (GM00893 and GM05423) and from three BRCA1 heterozygote patients (GM14090, GM14090, GM16105). Bar charts are shown as normal or BRCA1 mutated (+/-) where the data form each cell line is combined. Error bars depict standard error of the mean of two independent experiments where images of at least 10,000 cells were captured and analysed.
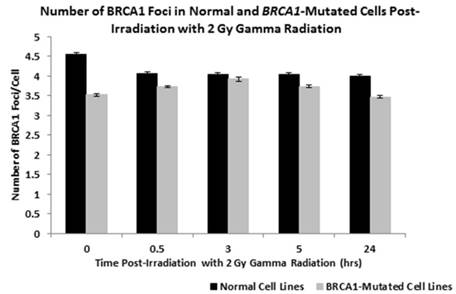
Figure 2 examines the number of BRCA1 foci in each of the three BRCA1+/- cell lines in comparison to the two normal cell lines. In each of the three BRCA1 heterozygotic cell lines we unsurprisingly observed fewer BRCA1 foci when compare to cells derived from normal individuals. This reduction of foci is most pronounced in the GM16105 BRCA1+/- line where it can be observed that at all time points here are significantly fewer foci compared to normal (ANOVA: P = 3.14 x 10-6). Additionally in the two other BRCA1+/- lines (GM13705 and GM14090) we observe a significant reduction in foci when compared to normal cells (ANOVA P = 0.000114 and P = 0.006 respectively).
shows the number of BRCA1 protein foci in two normal lymphoblastoid cell lines and three cell lines derived from BRCA1 heterozygotes as indicated in the figure. Error bars depict standard error of the mean of two independent experiments where 10,000 cell images where captured in each experiment.
BRCA1 protein expression in normal and BRCA1+/- cells following exposure to Olaparib and gamma radiation
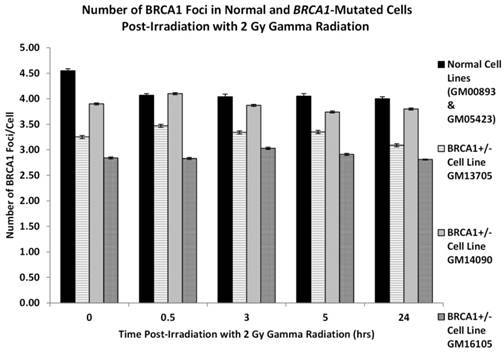
Having established the base level of BRCA1 protein/foci expression in the panel of cell lines following radiation exposure we wished to determine if the intracellular levels of BRCA1 protein were altered by the prior incubation with the PARP-1 inhibitor Olaparib. This experiment was driven by the fact that in previous observations [20] we observed hypersensitivity to ionising radiation in BRCA1 heterozygote cells after Olaparib treatment. Figure 3 depicts the mean levels of BRCA1 protein foci in two normal lymphoblastoid cell lines after 2 Gy gamma radiation in the absence or presence of Olaparib. In addition we show the mean number of BRCA1 foci in three BRCA1+/- cell lines after radiation exposure with and without Olaparib treatment. We observed that in all cell lines there is a reduction of BRCA1 protein foci when the cells are pre-treated with a non-cytotoxic dose of 5 μM Olaparib. In the normal cells there is minor but significant reduction in the number of BRCA1 protein foci induced by Olaparib incubation prior to gamma irradiation when compared to normal cells irradiated without PARP1 inhibition by Olaparib (ANOVA, P = 0.031). In addition in the BRCA1+/- cell lines the reduction on BRCA1 protein foci is more pronounced with on average two fewer BRCA1 protein foci per cell (ANOVA, P = 0.00051). The most dramatic effect of BRCA1 protein foci expression can be observed when normal cells are compared to BRCA1+/- cells after Olaparib exposure. A significant reduction in foci is seen in the heterozygote cells where on average there are 3 fewer foci per cell (ANOVA, P = 8.22 x 10-6).
shows the number of BRCA1 protein foci in two normal lymphoblastoid cell lines (GM00893 and GM05423) and three BRCA1 heterozygote cell lines (GM14090, GM14090, GM16105). Foci numbers are given for each cell type following radiation exposure with or without prior Olaparib treatment. Error bars depict standard error of the mean of two independent experiments where 10,000 cell images where captured in each experiment.
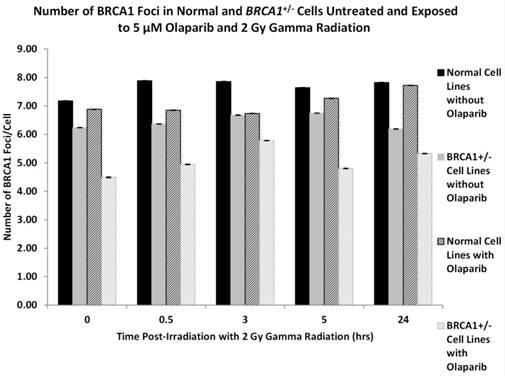
To demonstrate that appearance of BRCA1 protein foci as derived in imaging flow cytometry, representative examples of multispectral images of cells with increasing foci are shown in Figure 4.
show multispectral images of cells where channel 1 shows a brightfield image; channel 2, BRCA1 protein foci and channel 3 shows a nuclear image of each cell.
Increased apoptosis in BRCA1 heterozygote lymphoblastoid cells following Olaparib and gamma radiation exposure
In our previous investigations [20-21] we observed increased radiosensitivity in BRCA1 heterozygotic cells when compared to normal cells following Olaparib exposure and that this elevated sensitivity was due to a reduction in the repair of DNA DSB following radiation exposure. In order to determine the mechanism of cell death responsible for this enhanced sensitivity we examined the levels of apoptosis induction using the analysis wizard in the Ideas™ software package.
In Figure 5 it can be observed in both the normal and BRCA1+/- cells we observed an increase in apoptosis in those cultures that were treated with Olaparib and gamma radiation compared to those cultures that received no apriori Olaparib treatment. In the normal cells however, this increase in the overall levels of apoptosis was not significant (ANOVA, P = 0.731). This increase in apoptosis was most pronounced in the BRCA1+/- cultures where apoptosis increased to between 40-50% in all BRCA1+/- cell lines over a 24 hour time period after Olaparib exposure. In normal lymphoblastoid cell lines and non-Olaparib treated BRCA1+/- lymphoblastoid cell lines apoptosis ranged between 10-25% (ANOVA, P = 2.54 x 10-5).
shows the number of apoptotic cells in two normal and three BRCA1 heterozygotes lymphoblastoid cell lines. Error bars represent standard error of the mean of two independent experiments where 10,000 cell images were captured.
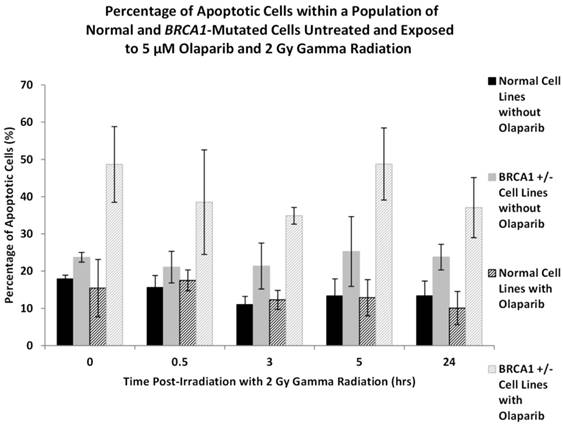
The levels of apoptosis in the normal and BRCA1+/- cells are shown in Figure 5 and are presented as percentages of apoptotic cells within the entire culture of cells.
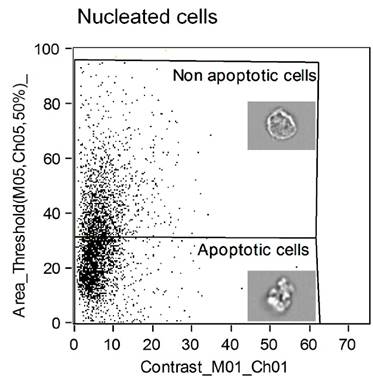
Figure 6 is a representative example of a dot plot from the Ideas™ software demonstrating the identification of apoptotic cells using the Ideas™ wizard tool. The region identified as non-apoptotic is shown together with a representative cell within the image and the apoptotic cell region is shown together with an image of a cell undergoing apoptosis with extensive membrane blebbing characteristic of apoptotic cells.
shows a prototypical imaging flow cytometric dot plot of apoptotic and non-apoptotic cells. Each dot within the graph represents an individual cell and apoptotic cells are distinguished from non-apoptotic cells by nuclear area and brightfield image contrast.
Conclusion
Our previous studies have demonstrated that lymphoblastoid cells derived from BRCA1 heterozygotes, when compared to normal or BRCA2 heterozygote cells, are hypersensitive to gamma radiation if pre-treated with the PARP1 inhibitor Olaparib [20-21]. In this study we extend these findings and show that there is reduced BRCA1 protein in heterozygote cells from BRCA1 carriers and that BRCA1 protein levels are further reduced by Olaparib exposure. Moreover, we show that the increased hypersensitivity in BRCA1 carrier cells is due to elevated apoptosis after Olaparib and radiation exposure.
Poly(ADP-ribose) polymerase 1 (PARP1) is an important enzyme involved in the repair of DNA single strand breaks. Therapeutic inhibition of PARP1 in a BRCA1-/- tumour leads to the formation of a potentially cytotoxic DNA double strand break at stalled replication forks resulting in synthetic lethality and tumour cell kill [10]. Therefore the clinical use of PARP1 inhibition in the treatment of BRCA1 negative breast, ovarian, pancreatic and prostate cancers is gaining momentum as a viable and effective treatment option [4].
While such cancers lend themselves to therapeutic inhibition of PARP1, more typical treatments utilise traditional cytotoxic radiotherapy and chemotherapy approaches [24]. Our research group has been interested in the factors controlling normal tissue toxicity (NTT) as a result of clinical radiotherapy. Some 50% of all cancer patients, irrespective of the histological cancer type, will receive radiotherapy (teletherapy or internal brachytherapy) as a part of their treatment [25] and evidence dictates that some 20% of all patients will experience side effects to a greater or lesser level in the form of NTT [26]. NTT has been classified according to tissue type and symptoms (acute or chronic) into standardised scales of NTT. These include the Radiation Therapy Oncology Group/European Organisation for Research and Treatment of Cancer scale (RTOG/EORTC) and the Late Effects Normal Tissue Task Force subjective, objective, management and analytic (LENT/SOMA) scale [27]. While the level of NTT in the majority of radiotherapy can be predicted, some individuals experience NTT which is so severe that it falls outside the normal standardised scale of responses and such patients are regarded as “over-reactors”. In such cases treatment with normal therapeutic doses of radiotherapy evoke dramatic NTT sequelae that can lead to death. Individuals with inherited defects in DNA repair genes such as the ataxia telangiectasia mutated gene (ATM) where a defect in cell cycle checkpoint control leads to extreme radiotherapy-induced NTT [15] would constitute an over-reactor. In addition, our group has demonstrated that a patient who over-reacted to radiotherapy, resulting in death, harboured a mutation in the DNA-PKcs gene, which is central to efficient non-homologous end joining (NHEJ) repair of DNA strand breaks [17]. Therefore while there are many factors which may dictate NTT in a radiotherapy patient, it is undisputed that occult defects and deficiencies in the repair of DNA strand break induced by radiotherapy are central to NTT [26-28].
Both the BRCA1 and BRCA2 genes are central to the repair of DNA DSB by both non homologous end joining (NHEJ) and homologous recombination (HR) and consequently the DNA repair function of these proteins may render patients susceptible to abnormal levels of NTT following radiotherapy if the patient is heterozygous for these genes in non-cancer normal tissue. Conversely it may be assumed that since it is only the tumour that is fully BRCA1-/- or BRCA2-/- while the patient is monoallelic for these genes then NTT should not be expected. However, the phenomenon of haploinsufficiency of DNA repair genes leading to increased NTT has been demonstrated in some scenarios. For example in Li-Fraumeni patients with an inherited mutation in the P53 gene, increased cancer incidence and sensitivity to DNA damaging agents at the cellular and clinical level can be observed [29]. Moreover we demonstrated in a patient that experienced extreme cellular radiation hypersensitivity and NTT resulting in death, a monoallelic mutation in the DNA-PKcs DNA repair gene was responsible. Correspondingly, we previously hypothesised that BRCA1+/- individuals receiving radiotherapy for BRCA1-/- cancer might experience abnormally high levels of NTT. Moreover, this might be exacerbated in combinations of treatment where radiotherapy and PARP1 inhibitors are used. While there is conflicting evidence that patients who are BRCA1+/- experience NTT, we have demonstrated that lymphoblastoid cells derived from BRCA1+/- heterozygotes are hypersensitive to radiotherapy if PARP1 has been inhibited by prior exposure to the inhibitor Olaparib [20] and that this sensitivity was due to a failure to efficiently repair DNA DSB evidenced by a retention of γ-H2AX foci in the nuclei of cells following 2 Gy gamma irradiation and Olaparib exposure [21].
To understand the mechanism of sensitivity in the lymphoblastoid cells derived from BRCA1+/- patients, we used imaging flow cytometry to investigate the effects of Olaparib exposure on BRCA1 protein levels and evaluated the mechanism of cell kill underlying the observed hypersensitivity. Imaging flow cytometry is a novel technique combining microscopic image acquisition of cells in flow across multiple channels including bright, darkfield and fluorescence image capture. We have extensive experience in this technique and have developed protocols for the determination of DNA damage [26-30], apoptosis in human cells [31] and identification of circulating tumour cells (Al-Ali et al., 2017 paper submitted).
In this study we were able to demonstrate that in lymphoblastic cells derived from BRCA1 heterozygotes there were fewer BRCA1 protein foci in the nuclei of cells when compared to cells derived from normal individuals. Prima facie, such an observation would be expected and support the notion of haploinsufficiency observed with many DNA repair genes. However, previous investigations have demonstrated that reduced BRCA1 protein in heterozygotic cells is very much mutation specific, [e.g. 32] which demonstrated that carriers with mutations in exon 11 had reduced BRCA1 mRNA transcripts compared to other carrier mutations. In the three BRCA1+/- cell lines used here, GM13705 and GM14090 both have base pair deletions (exon 11 and 3 respectively) leading to a truncated protein while GM16105 harbours a missense mutation in exon 5. We observed the most pronounced reduction in protein levels in the GM16105 cell line compared to those harbouring base pair deletions.
A second fundamental finding from this investigation was the significant reduction in BRCA1 protein foci levels when cells were treated with Olaparib prior to 2 Gy gamma radiation exposure. A slight but significant reduction was seen in cells derived from two normal individuals when Olaparib treatment occurred prior to radiation exposure but this was more pronounced in cells carrying a BRCA1 mutation. While this observation would provide a mechanistic corroboration of why BRCA1+/- cells exhibit hypersensitivity to Olaparib and radiation compared to normal cells and those cells derived from BRCA2 carriers, the reasons for this reduction in BRCA1 protein levels is not known. However, off-target effects have been observed with PARP1 inhibitors leading to the suppression of a number of kinases including Chk1a and ATR leading to replication stress, collapsed replication forks and a potential failure to recruit BRCA1 protein to sites of DNA damage during HR [33].
Finally, using imaging flow cytometry we have been able to demonstrate that the hypersensitivity of the BRCA1+/- carrier cells to radiation and Olaparib is due to increased levels of apoptosis. This finding supports the observations of other investigators who similarly show elevated rates of apoptosis following Olaparib exposure in BRCA1-/- negative breast cancer cells [34].
In conclusion, the use of PARP1 inhibitors in the treatment of BRCA1-related cancers will no doubt gain momentum and moreover combinatorial treatments being evaluated and applied in clinical practice will expand. This investigation and our previous publications serve to highlight the potential problems of increased NTT in these scenarios.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Michels J, Vitale I, Saparbaev M. et al. Predictive biomarkers for cancer therapy with PARP inhibitors. Oncogene. 2014;33(30):3894-07
2. McGlynn P, Lloyd B. Recombinational repair and restart of damaged replication forks. Nat Rev Mol Cell Biol. 2002;3(11):859-70
3. Rouleau M, Patel A, Hendzel MJ. et al. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293-301
4. Benafif S, Hall M. An update on PARP inhibitors for the treatment of cancer. Onco Targets Ther. 2015;8:519-28
5. Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004;95:866-71
6. Mao Z, Bozzella M, Seluanov A. et al. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 2008;7(18):2902-06
7. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;12:68-78
8. Bryant HE, Schultz N, Thomas HD. et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913-17
9. Farmer H, McCabe N, Lord CJ. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917-21
10. Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5:387-93
11. Liu Y, West SC. Distinct functions of BRCA1 and BRCA2 in double-strand break repair. Breast Cancer Res. 2002;4(1):9-13
12. Lawrence TS, Ten Haken RK, Giaccia A. Principles of radiation oncology. In: (ed.) DeVita VT Jr, Lawrence TS, Rosenberg SA. Cancer: principles and practice of oncology. Philadelphia: Lippincott Williams and Wilkins. 2008:307-36
13. Cox JD, Stetz J, Pajak TF. Toxicity criteria of the Radiation Therapy Oncology Group (RTOG) and the European Organization for Research and Treatment of Cancer (EORTC). Int J Radiat Oncol Biol Phys. 1995;31(5):1341-46
14. Trotti A, Colevas AD, Setser A. et al. CTCAE v3.0: Development of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol. 2003;13(3):176-81
15. Lavin MF, Shiloh Y. The genetic defect in Ataxia-telangiectasia. Annu Rev Immunol. 1997;15:177-202
16. Nicolas N, Moshous D, Cavazzana-Calvo M. et al. A human severe combined immunodeficiency (SCID) condition with increased sensitivity to ionizing radiations and impaired V(D)J rearrangements defines a new DNA recombination/repair deficiency. J Exp Med. 1998;188(4):627-34
17. Abbaszadeh F, Clingen PH, Arlett CF. et al. A novel splice variant of the DNA-PKcs gene is associated with clinical and cellular radiosensitivity in a patient with Xeroderma pigmentosum. J Med Genet. 2010;47(3):176-81
18. Buchholz TA, Wu X, Hussain A. et al. Evidence of haplotype insufficiency in human cells containing a germline mutation on BRCA1 or BRCA2. Int J Cancer. 2002;97:557-61
19. Leong T, Whitty J, Keilar M. et al. Mutation analysis of BRCA1 and BRCA2 cancer predisposition genes in radiation hypersensitive cancer patients. Int J Radiat Oncol Biol Phys. 2000;48(4):959-65
20. Bourton EC, Foster HA, Plowman PN. et al. Hypersensitivity of BRCA1 heterozygote lymphoblastoid cells to gamma radiation and PARP inhibitors. J Genet Syndr Gene Ther. 2013:4 (5). doi: 10.4172/2157-7412.1000146
21. Bourton EC, Plowman PN, Harvey AJ. et al. The PARP-1 inhibitor Olaparib causes retention of γ-H2AX foci in BRCA1 heterozygote cells following exposure to gamma radiation. J Cancer Ther. 2013;4:44-52
22. Underhill C, Toulmonde M, Bonnefoi H. A review of PARP inhibitors: from bench to bedside. Ann Oncol. 2010;22(2):268-79
23. Bourton EC, Plowman PN, Adam Zahir S. et al. Multispectral imaging flow cytometry reveals distinct frequencies of γ-H2AX foci induction in DNA double strand break repair defective human cell lines. Cytometry A. 2012;81A:130-37
24. Weinberg RA. The rational treatment of cancer. In: (ed.) Weinberg RA. The biology of cancer. New York: Garland Science. 2013:797-876
25. Delaney G, Jacob S, Featherstone C. et al. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer. 2005;104(6):1129-37
26. Bourton EC, Plowman PN, Smith D. et al. Prolonged expression of the γ-H2AX DNA repair biomarker correlates with excess acute and chronic toxicity from radiotherapy treatment. Int J Cancer. 2011;129(12):2928-34
27. Hoeller U, Tribius S, Kuhlmey A. et al. Increasing the rate of late toxicity by changing the score? A comparison of RTOG/EORTC and LENTA/SOMA scores. Int J Radiat Oncol Biol Phys. 2003;55:1013-18
28. Valdiglesias V, Giunta S, Fenech M. et al. γ-H2AX as a marker of DNA double strand breaks and genomic instability in human population studies. Mutat Res. 2013;753:24-40
29. Malkin D. Li Fraumeni syndrome. Genes Cancer. 2011;2(4):475-484
30. Parris CN, Adam Zahir S, Al-Ali H. et al. Enhanced γ-H2AX DNA damage foci detection using multimagnification and extended depth of field in imaging flow cytometry. Cytometry A. 2015;87(8):717-23 doi: 10.1002/cyto.a.22697
31. Ulus-Senguloglu G, Arlett CF, Plowman PN. et al. Elevated expression of artemis in human fibroblast cells is associated with cellular radiosensitivity and increased apoptosis. Br J Cancer. 2012;107(9):1506-13
32. Chehade R, Pettapiece-Phillips R, Salmena L. et al. Reduced BRCA1 transcript levels in freshly isolated blood leukocytes from BRCA1 mutation carriers is mutation specific. Breast Cancer Res. 2016;18:87. doi: 10.1186/s13058-016-0739-8
33. Kim H, George E, Ragland RL. et al. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin Cancer Res. 2016 Dec 19. doi: 10.1158/1078-0432.CCR-16-2273
34. Shi Y, Zhou F, Jiang F. PARP inhibitor reduces proliferation and increases apoptosis in breast cancer cells. Chin J Cancer Res. 2014;26(2):142-47
Author contact
![]() Corresponding author: christopher.parrisac.uk
Corresponding author: christopher.parrisac.uk