Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(15):3086-3098. doi:10.7150/jca.14835 This issue Cite
Research Paper
Dihydroorotate dehydrogenase Inhibitors Target c-Myc and Arrest Melanoma, Myeloma and Lymphoma cells at S-phase
Mathura Subangari Dorasamy1, 2, Bhavesh Choudhary2, Kavitha Nellore3, Hosahalli Subramanya3, Pooi-Fong Wong1 ![]()
1. Department of Pharmacology, Faculty of Medicine, 50603 University of Malaya, Kuala Lumpur, Malaysia;
2. Aurigene Discovery Technologies Limited, Bollaram Road, Miyapur Hyderabad - 500049, Telangana, India;
3. Aurigene Discovery Technologies Limited, 39-40, KIADB Industrial Area, Phase II Electronic City, Hosur Road, Bangalore - 560100 Karnataka, India.
Received 2015-12-29; Accepted 2017-3-5; Published 2017-9-5
Abstract
Dihydroorotate dehydrogenase (DHODH) is a rate-limiting enzyme in the de novo biosynthesis pathway of pyrimidines. Inhibition of this enzyme impedes cancer cell proliferation but the exact mechanisms of action of these inhibitors in cancer cells are poorly understood. In this study, we showed that cancer cells, namely melanoma, myeloma and lymphoma overexpressed DHODH protein and treatment with A771726 and Brequinar sodium resulted in cell cycle arrest at S-phase. Transfection with DHODH shRNA depleted DHODH protein expression and impeded the proliferation of melanoma cells. shRNA knockdown of DHODH in combination with DHODH inhibitors further reduced the cancer cell proliferation, suggesting that knockdown of DHODH had sensitized the cells to DHODH inhibitors. Cell cycle regulatory proteins, c-Myc and its transcriptional target, p21 were found down- and up-regulated, respectively, following treatment with DHODH inhibitors in melanoma, myeloma and lymphoma cells. Interestingly, knockdown of DHODH by shRNA had also similarly affected the expression of c-Myc and p21 proteins. Our findings suggest that DHODH inhibitors induce cell cycle arrest in cancer cells via additional DHODH-independent pathway that is associated with p21 up-regulation and c-Myc down-regulation. Hence, DHODH inhibitors can be explored as potential therapeutic agents in cancer therapy.
Keywords: A771726, Brequinar, Ramos, DHODH shRNA, p21.
Introduction
Proliferating cancer cells show substantially different metabolic needs compared to normal differentiated cells as they require additional nutrients to support their high rates of proliferation (1). Success in targeting cancer cell metabolism will materialize from an improved understanding of exactly how cells control and consume nutrients into pathways that are essential for biosynthesis (2). As all cancer cells rely on this alteration in metabolism, these altered pathways represent strong therapeutic targets (3, 4). However, discovering a 'therapeutic window' between normal proliferating and cancer cells remains a major challenge as the metabolic requirements of these cells are similar. Thus, only a few molecules which target metabolic pathways have been established as a form of cancer treatment (2).
The de novo biosynthesis of pyrimidine is an essential metabolic pathway for nucleic acid synthesis (5). Although most cells meet their needs for nucleotides by reutilizing current ones through the salvage pathway, activated T cells and other rapidly proliferating cells, namely cancer cells are highly dependent on de novo nucleotide synthesis (6, 7). DHODH is the fourth sequential and rate-limiting enzyme in the de novo biosynthesis pathway of pyrimidines and it is the only enzyme found within the mitochondrial inner membrane of eukaryotes (6, 8). Inhibition of this enzyme leads to intense reductions in cellular pyrimidine pools and eventually results in the failure of cells to proliferate (9). This protein is considered to be of great interest to the scientific community as it is one of the key enzymes in sustaining the proliferation of transformed cells and a potentially good target for cancer chemotherapy. The therapeutic potential of hindering de novo pyrimidine biosynthesis at the DHODH oxidation phase was shown by the anti-proliferative agents namely A771726, an active metabolite of Leflunomide (LFM) and Brequinar sodium salt (BQR) (10, 11). Leflunomide is an immunomodulatory and anti-inflammatory drug approved by FDA for the cure of rheumatoid arthritis (RA) patients in 1998. It was later determined that LFM works via the inhibition of DHODH in activated lymphocytes (12, 13). Apart from DHODH inhibition, LFM, at higher doses is also known to inhibit tyrosine kinases responsible for B and T cell signaling (14). On the other hand, BQR was designed to be a specific DHODH inhibitor and is known to disrupt DHODH activity with much higher potency than LFM (11, 15, 16). Earlier studies revealed that the inhibition of proliferation of some tumor cells such as melanoma (17), neuroblastoma (18), glioblastoma and breast cancer (19-21) was effective through LFM. In addition, BQR was also found effective against colon cancer in vivo (22).
To date, extensive research has been performed to study the effectiveness of targeting DHODH enzyme in cancer therapy. Nevertheless, the extent to which the enzyme inhibition affects cancer cell proliferation is still not well understood. The aim of the present work is to elucidate the mechanism of action of DHODH inhibitors in melanoma, myeloma and lymphoma cells. Our results suggest that the active metabolite, A771726 and BQR exert their anti-proliferative effects on the cancer cells. These inhibitors arrested cancer cells growth in S-phase, decreased c-Myc and increased p21 protein expression. Furthermore, DHODH suppression by shRNA affected cell proliferation, c-Myc and its target protein, p21. The current data suggest that these effects are possibly associated with an additional DHODH-independent pathway comprising of c-Myc inhibition.
Materials and Methods
Cells
Human melanoma (A375), human myeloma (H929), and human Burkitt's lymphoma (Ramos) cell lines were obtained from American Type Cell Collections. A375 cells were grown in DMEM (Sigma, USA), H929 and Ramos cells were grown in RPMI 1640 (Sigma, USA). The medium was supplemented with 10% heat-inactivated fetal bovine serum (Sigma, USA), 100 units/mL penicillin and 100 µg/mL streptomycin (Gibco, USA). An additional 0.05 mM/L 2-mercaptoethanol (Sigma, USA) was supplemented for the growth of H929 cells. Cell lines were maintained in a 5% CO2 atmosphere at 37 °C.
Antibodies and chemicals
A771726 and BQR were synthesized at Aurigene Discovery Technologies Limited, Bangalore, India. Monoclonal primary antibodies against human DHODH were purchased from Santa Cruz Biotechnology, Inc, USA. Antibodies for c-Myc, p21, phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), NFkB and Bcl-xL were obtained from Cell Signaling Technology, USA. Loading controls - β actin, GAPDH and tubulin β antibodies were procured from SignalChem, Canada. Secondary antibody, anti-rabbit polyclonal antibody was secured from Sigma, USA.
Cell proliferation assay
A375, H929 and Ramos cells were seeded in 96-well plates at different densities, 1000 to 20000 cells per well. The DHODH inhibitors - A771726 and BQR were dissolved in Hybrimax DMSO (Sigma, USA). Cells were treated with inhibitors and incubated for 72 hours. In control wells, cells were treated with 0.5% of DMSO per well. Cell viability of suspension cells was determined using XTT assay. Each well was incubated with 100 µl of 1 mg/ml XTT supplemented with 25 μM PMS (Sigma, USA) for 2 hours. The amount of formazon produced was determined by reading the absorbance of the plate at 450 nm using a microplate reader (Victor X5, PerkinElmer, USA). For adherent cells, 50 µl of 1 mg/ml Resazurin (Sigma, USA) dissolved in PBS (Sigma, USA) was added to each well. The fluorescence reading was measured at Ex/Em of 531/595 nm. Cell viability was calculated as a percentage of fluorescence or absorbance measured in the treated wells relative to the DMSO control wells. EC50 values were determined as concentrations that reduced cell viability by 50% and the curve was plotted with GraphPad Prism 6.0 (San Diego, USA). Experiments were repeated three times and mean ± SE was calculated.
DHODH biochemical assay
The DHODH biochemical assay is based on the oxidation of L-dihydroorotic acid (L-DHO) aided by reduction of 2,6-dichloroindophenol (DCIP) and decylubiquinone (DUQ) resulting in the decrease in absorbance at 610 nm (23). DHODH inhibitors, A771726 and BQR were assessed for their ability to inhibit DHODH protein in a coupled enzymatic spectrophotometric assay. The drop in absorbance at 610 nm is relative to the reduction of DCIP. The assay buffer is composed of 50 mM Tris HCl (pH 8), 150 mM KCl and 0.8 % Triton X-100 (Sigma, USA). DHODH protein (25 ng) (Origene, USA) was dissolved in assay buffer and added to the 96-well plate containing DHODH inhibitors. The mixture was pre-incubated for 30 minutes at room temperature. Upon pre-incubation, substrate mixture (20 mM of L-DHO, 2 mM of DuQ and 2 mM of DCIP) was added to activate the reaction. Lastly, the plate was read every 10 minutes on Victor X5 at 610 nm. The IC50 values of the DHODH inhibitors were calculated by GraphPad Prism 6.0. Experiments were repeated three times and mean ± SE was calculated.
Trypan blue exclusion assay
A375 cells were treated with various concentrations of A771726 and BQR for 48 hours. Cell viability was assessed with Trypan blue (Sigma, USA) exclusion method. Viable cells (unstained cells) indicate metabolically intact cells whereas blue cells are considered as dead cells. The total number of viable cells was plotted against time and experiments were repeated three times and mean ± SE was calculated.
Western blotting
A375, H929 and Ramos cells were seeded in 6-well plates. Cells were harvested and lysed with RIPA buffer (Sigma, USA) supplemented with protease and phosphatase inhibitor cocktails (Sigma, USA). Extracted protein was quantified with Pierce BCA Protein Assay Kit (Thermo Scientific, USA). Equal amount of protein was subjected to a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membrane (GE Healthcare, USA). The membrane was blocked with Tris-buffer saline with 0.1% Tween-20 (TBST) containing 5% dry milk (Cell Signaling, USA). After blocking, the membrane was probed with appropriate primary antibodies overnight at 4 °C. On the next day, the binding of the primary antibodies was detected with anti-rabbit polyclonal secondary antibody labeled with horseradish peroxidase. The blot was developed with ECL Western blotting detection system (Pierce, USA). Experiments were repeated three times.
Knockdown using DHODH siRNA
siRNA against DHODH and siRNA controls (Non-silencing siRNA and AllStar Cell Death siRNA) were procured from Qiagen, USA. The following four 21-bp double-stranded DHODH siRNA were used during transfection: siRNA 1 - CAGGTATGGATTTAACAGTCA, siRNA 2 -AGCCGTGGACGGACTTTATAA, siRNA 3 - CTCCGGGATTTATCAACTCAA and siRNA 4 - CAGGGCTTTGGCGGAGTCACA. A375 cells were transfected with DHODH siRNA using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer's instructions (Qiagen, USA). Briefly, 0.15 x 106 cells were seeded in a 6-well plate. On the next day, 5 µl of Lipofectamine 2000 was diluted in 250 µl with OPTI-MEM I Medium (Invitrogen, USA) and incubated for 10 minutes at room temperature. DHODH siRNA 1,2,3 and 4 were pooled and diluted with OPTI-MEM I Medium to a final concentration of 4 nM per well. siRNA and lipofectamine were mixed gently and added to the cells after 30 minutes incubation at room temperature. After 5 hours of incubation, fresh DMEM media was changed and plate was incubated for 48 hours. Cell lysates were prepared and DHODH expression was ascertained through SDS-PAGE and Western blotting experiments. Experiments were repeated three times.
Stable shRNA transfection
shRNA against DHODH and control plasmids were purchased from Qiagen, USA. The following two gene-specific shRNA plasmids and one negative control plasmid were used during transfection: shRNA 1 - CAAGGCCAGTTCCCATAATT and shRNA 2 -TGAGAGTTCTGGGCCATAAAT and negative control plasmid GGAATCTCATTCGATGCATAC. Each plasmid carries shRNA under the control of the U1 promoter and a marker gene (hygromycin resistance gene). The plasmids were first transformed into competent E. coli cells. Following DNA amplification, shRNA plasmid construct was extracted and purified by GenEluteTM HP Plasmid Miniprep Kit by Sigma, USA.
One day prior to transfection of plasmid shRNA construct, 0.15 x 106 per well A375 cells were seeded in a 6-well tissue culture plate. 2 µg per well of plasmid DHODH and negative control shRNA was added with Lipofectamine 2000 (Invitrogen, USA) to each well in a ratio of 1:2. The lipofectamine/DNA complexes were removed 5 hours after transfection and fresh medium was added to the cells. To produce stably transfected cells, 100 µg/ml Hygromycin was added to the media 48 hours after transfection to select for clones containing insert. The cells were left in selective medium for 10 days after which they were trypsinized and cultured in selective media for propagation. The silencing effect was verified by Western blot analysis
Cell cycle analysis by FACS
A375, H929 and Ramos cells were treated with DHODH inhibitors for 24, 48 and 72 hours. Following treatment, the quantitative cell cycle analysis was performed using a commercial kit (BD, Cycletest Plus-DNA reagent kit, USA). Samples were prepared according to the kit's instructions. Cells incorporated propidium iodide and total DNA content in cells was analyzed with FACS Calibur flow cytometer (Becton Dickinson, USA). At least 20,000 events were collected for each sample. The data was analyzed using FlowJo V10.1. Experiments were repeated three times and mean ± SE was calculated.
Statistical Analysis
Cell proliferation assay and DHODH biochemical assay were performed in triplicates and each experiment was repeated three times. Trypan blue exclusion assay, siRNA transfection, flow cytometry analysis and Western blot experiments were performed singly and each experiment was repeated three times. Statistical significance of the differences between control and treatment was analyzed with Student's t-test and ANOVA in GraphPad Prism 6.0. P values less than 0.05 (*), 0.01 (**), 0.001(***) and 0.0001 (****) were considered statistically significant.
Results
Cancer cells overexpressed DHODH protein
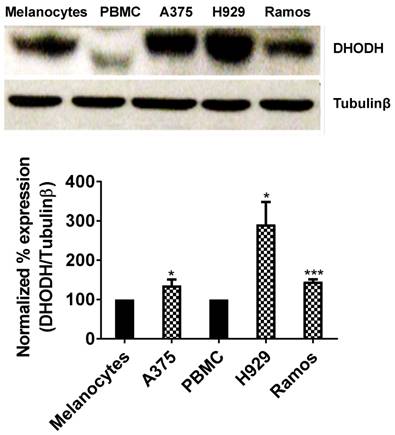
It has been well established that DHODH enzyme plays a pivotal role in highly proliferating cells. In the present study, we determined the expression of DHODH in normal melanocytes, peripheral blood mononuclear cells (PBMCs) and cancer cells - human melanoma (A375), multiple myeloma (H929) and Burkitt's lymphoma (Ramos). We found that DHODH is overexpressed in A375, H929 and Ramos cancer cells compared to their corresponding normal cells, melanocytes and PBMCs (Figure 1), suggesting that DHODH protein is overexpressed in transformed cells.
DHODH expression in melanocytes, PBMCs (normal cells) and A375, H929, Ramos (cancer cells). Equal amount of protein lysate was used for Western blot analysis with human primary DHODH antibody. Tubulinβ served as protein loading control. Data represents mean ± SE of 3 independent experiments. *, P<0.05 and ***, P < 0.001.
DHODH inhibitors reduce cancer cell proliferation
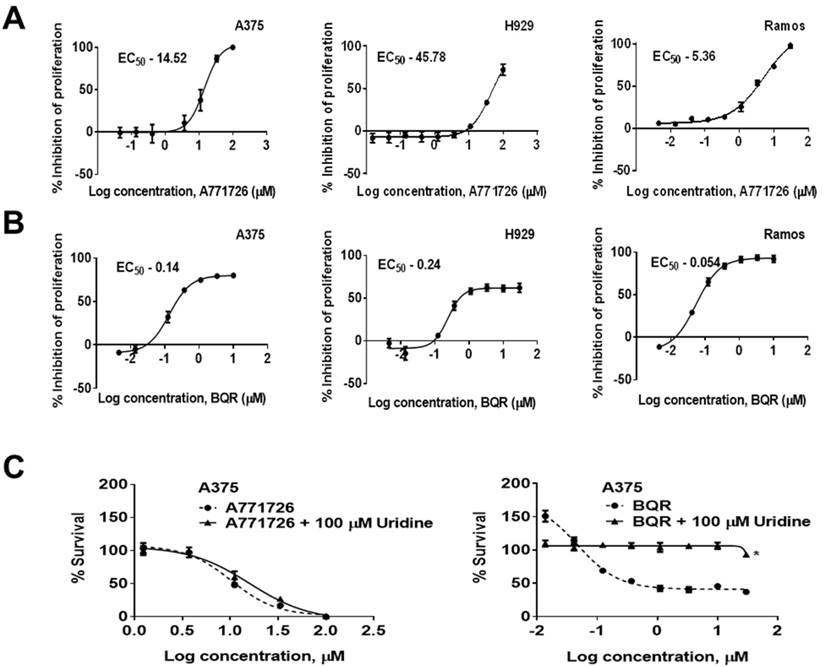
A771726 has been reported to inhibit the proliferation of melanoma and myeloma cells (17, 24, 25). In agreement to these reports, we found that A771726 caused a dose-dependent decrease in the proliferation of melanoma and myeloma cells. A771726 reduced the viability of A375 and H929 cell growth by 50% at concentrations of 14.52 µM and 45.78 µM, respectively, relative to the vehicle control treated cells whereas BQR inhibited cell proliferation of these cells at an EC50 of 0.14 µM and 0.24 µM, respectively. In addition, we extended the investigation on Burkitt's lymphoma cells, Ramos. Cell proliferation was significantly reduced by A771726 and the EC50 achieved was 5.36 µM (Figure 2A). Ramos cells were more sensitive to the treatment with BQR compared to A375 and H929 cells with EC50 of 0.054 µM (Figure 2B). In order to examine if inhibition of proliferation by A771726 and BQR is completely mediated via DHODH, we performed uridine rescue experiments in A375 cells (Figure 2C). Exogenous uridine used at 100 µM did not rescue the inhibitory effect of A771726. However, the inhibitory effect of BQR in A375 cells was completely reversed with uridine at 100 µM.
DHODH inhibitors cause a dose-dependent decrease in the proliferation of human melanoma, myeloma and lymphoma cell lines. A375, H929 and Ramos cells were treated with increasing concentrations of A771726 and BQR for 72 hours. Inhibition of proliferation was measured by XTT assay. Values shown are mean ± SE of 3 independent experiments. A, EC50 of A771726, A375 - 14.52 μM; H929 - 45.78 μM; Ramos - 5.36 μM. B, EC50 of BQR, A375 - 0.14 μM; H929 - 0.24 μM; Ramos - 0.054 μM. C, Uridine rescue with DHODH inhibitors. *, P<0.05.
DHODH inhibitors affect DHODH enzyme activity but not the DHODH expression level
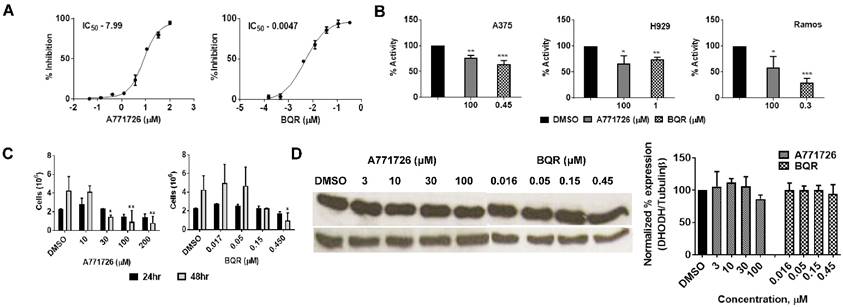
We performed human DHODH enzymatic assay with A771726 and BQR using the purified enzyme. In agreement with earlier reports, A771726 and BQR inhibited DHODH enzyme activity in a dose-dependent manner at IC50 of 7.99 μM and 0.0047 μM, respectively (Figure 3A). To quantitate the enzymatic activity in vitro, we treated A375, H929 and Ramos cells with A71726 and BQR for 48 hours and the DHODH enzymatic assay was performed with these cell lysates. As hypothesized, the DHODH inhibitors reduced in vitro DHODH enzymatic activity by 30 - 36% in A375, 25 -27% in H929 and 42 -71% in Ramos cells (Figure 3B). Having verified the ability to affect the enzyme activity, we further assessed whether these inhibitors affect the protein expression of DHODH in A375 cancer cells. We therefore, treated A375 cells with A771726 and BQR for 24 and 48 hours and the DHODH expression was examined. Both inhibitors reduced cell growth in a dose-dependent manner (Figure 3C) but not the protein expression of DHODH (Figure 3D). Our data indicate that DHODH inhibitors affect the enzyme activity but not the DHODH protein expression.
DHODH inhibitors affect DHODH enzyme activity but not the DHODH protein expression in A375 cells. A, DHODH enzymatic assay with A771726 and BQR using pure human DHODH enzyme. B, DHODH enzymatic assay with A771726 and BQR using A375, H929 and Ramos cells lysates. C, A375 cells were incubated with 10, 30, 100 and 200 µM A771726 and 0.016, 0.05, 0.15 and 0.45 µM of BQR for 24 and 48 hours and cell viability was determined by Trypan blue stain. D, Western blot of A375 cell lysate after treatment with A771726 and BQR. Cells were harvested at 48 hours after treatment for Western blot analysis. Tubulinβ served as protein loading control. Values shown are mean ± SE of 3 independent experiments. *, P < 0.05, **, P < 0.01 and ***, P <0.001.
DHODH inhibitors arrest cancer cells primarily at S-phase
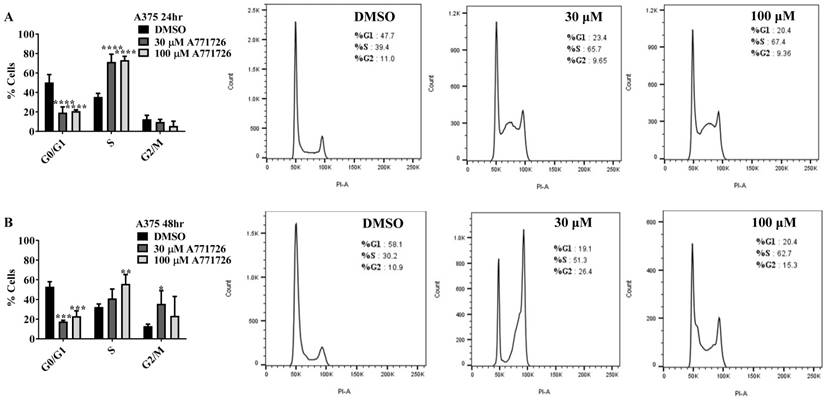
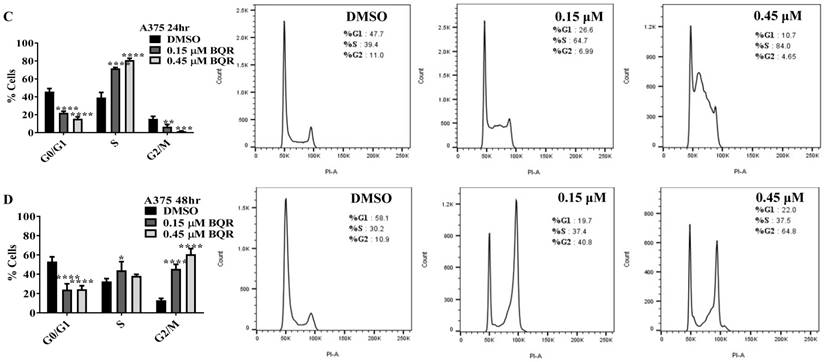
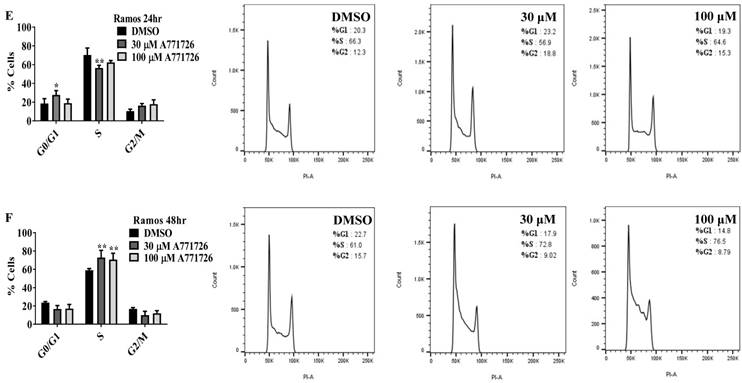
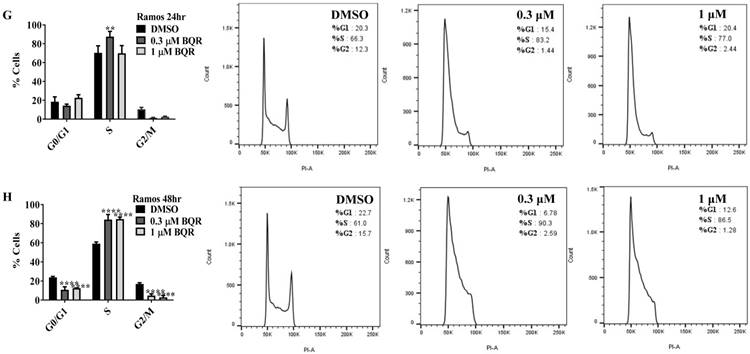
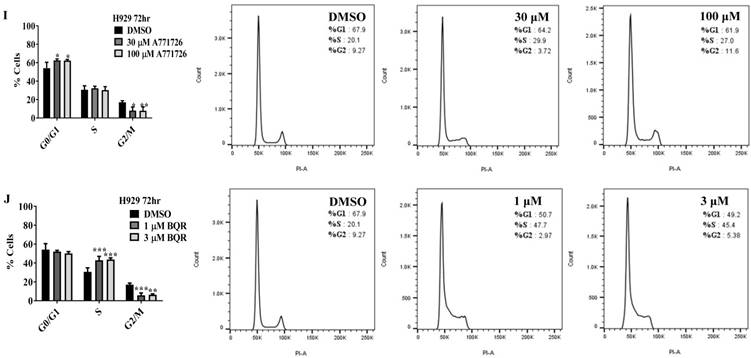
Since A771726 and BQR impede proliferation of A375, H929 and Ramos cells, we further investigated the cell cycle phase that was targeted by DHODH inhibitors. Figure 4 shows the percentage of cell population in each cell cycle phase following 24, 48 and 72 hours of exposure to A771726 or BQR at two different concentrations. As illustrated in Figure 4A and 4C, A375 cells treated with 30 µM or 100 µM A771726 and 0.15 µM or 0.45 µM BQR resulted in significant arrest at S-phase after 24 hours of treatment. A significant G2/M accumulation was observed after 48 hours of treatment with both compounds in these cells (Figure 4B and 4D). Ramos cells showed significant S-phase arrest following 48 hours of exposure to A771726 and BQR at two different concentrations compared to the non-treated cells (Figure 4F and 4H) and marginal increase in S-phase population at 24 hours of treatment (Figure 4E and 4G). Since H929 cells are slow growing cells, we analyzed cell cycle progression after 72 hours of treatment. These cells showed S-phase arrest after treatment with BQR but not with A771726. As illustrated in Figure 4J, the S-phase population of BQR increased from 20.1% in controls to 47.7% and 45.4% in both 1 µM and 3 µM concentrations and in turn significantly reduced G2/M population in these cells. Interestingly, treatment with A771726 at both concentrations slightly increased the percentage of G0/G1 phase (Figure 4I), showing a different response in these two inhibitors. Our data clearly indicate that A771726 and BQR primarily target S-phase in all three cancer cell lines. These inhibitors block the cell cycle in all three cell lines upon DHODH depletion thus providing a clear association between cell proliferation inhibition and DHODH protein.
Cell cycle distribution of melanoma, myeloma and lymphoma cell lines after treatment with DHODH inhibitors. A771726 and BQR: Induce S and G2/M phase arrest in A375 cells (A, B, C and D); Induce S-phase arrest in Ramos cells (E, F, G and H); Induce G0/G1 and S-phase arrest in H929 cells (I and J). Values shown are mean ± SE of 3 independent experiments. *, P <0.05, **, P <0.01, ***, P <0.001 and ****, P <0.0001.
DHODH knockdown potentiates melanoma cells sensitivity to DHODH inhibitors
To investigate whether the anti-proliferative effect of A771726 and BQR in A375 cells is dependent on DHODH protein, we performed a transient knockdown of DHODH using siRNA 1, 2, 3 and 4. The expression of DHODH decreased significantly by almost 85% (Figure 5A). Interestingly, DHODH suppression in A375 cells did not affect its growth in comparison to non-suppressed cells. Both showed almost 100% of cell viability, suggesting that knockdown of DHODH does not significantly affect cell proliferation, probably due to the transient effects of siRNA. Due to the limitations with siRNA transfection, we performed DHODH knockdown with shRNA transfection. Two independent DHODH shRNA knockdown stable cell line clones were generated in which DHODH shRNA clone 2 induced robust gene knockdown, 89% compared to negative control shRNA (Figure 5B). shRNA knockdown of DHODH resulted in impaired cell growth by 35% in comparison to cells transfected with control shRNA at 72 hours (Figure 5C). With the stable knockdown of DHODH, we next examined whether significant suppression of DHODH will sensitize the melanoma cells to DHODH inhibitors. DHODH shRNA suppressed cells were treated with A771726 and BQR for 24, 48 and 72 hours and the cell viability was examined. As hypothesized, treatment with A771726 and BQR further reduced the viability of stable DHODH shRNA cells by 36% and 53%, respectively (Figure 5D).
DHODH silencing potentiates cancer cell sensitivity to DHODH inhibitors in A375 cells. A, Cells were transfected with DHODH siRNA (4 nM) or control siRNA (4 nM) and harvested after 48 hours. B, Cells were transfected with DHODH shRNA (2 μg) or control shRNA (2 µg) and harvested after 48 hours. Equal amounts of protein lysates were used for Western blot analysis with human primary DHODH antibody. β-actin and anti-GAPDH served as protein loading controls. C, Prolonged depletion of DHODH caused growth retardation. D and E, Growth curve of DHODH suppressed and non-suppressed cells (prolonged and transient) after 72 hours of treatment with A771726 and BQR. Cell count was determined by Trypan blue exclusion assay as described in Materials and Methods. Values shown are mean ± SE of 3 independent experiments. *, P <0.05, **, P <0.01 and ***, P <0.001.
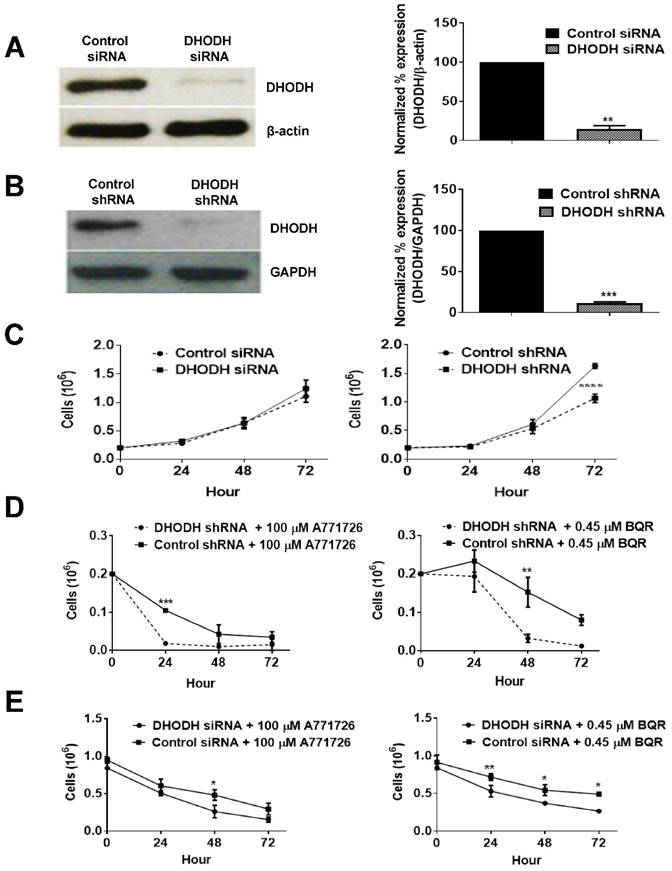
Although DHODH siRNA-transfected cells did not show reduced cell proliferation, these cells showed a dose-dependent reduction of cell viability after the treatment with both DHODH inhibitors (Figure 5E). Hence, both transient and prolonged knockdown of DHODH sensitized the cells to the inhibitory effect of DHODH inhibitors.
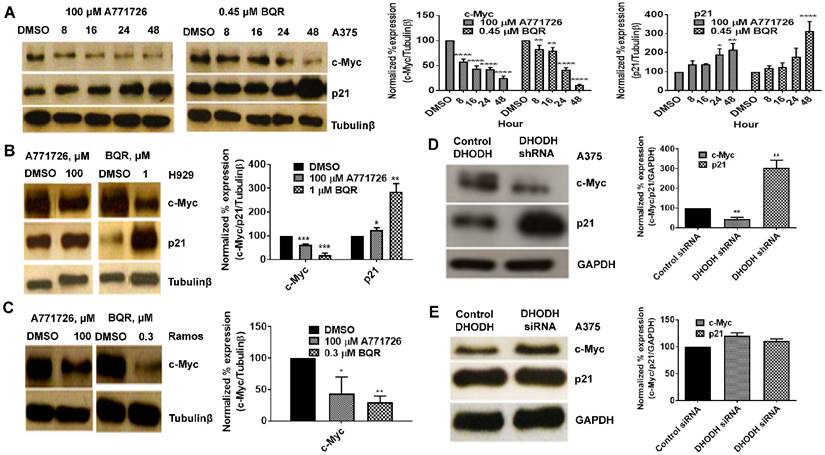
DHODH inhibitors target c-Myc and p21
Having found that DHODH inhibitors affect cancer cell proliferation and S-phase cell cycle arrest, we next evaluated the potential downstream targets of A771726 and BQR in A375, H929 and Ramos cells. Signaling proteins such as c-Myc, p21, pERK 1/2T202/204, NFkB and Bcl-xL were assessed. Treatment with A771726 and BQR down-regulated the expression of c-Myc in a time-dependent manner in A375 cells (Figure 6A). Down regulation of c-Myc was similarly achieved in H929 and Ramos cells after the treatment (Figure 6B and 6C). As expected, the c-Myc target gene p21, a cell cycle regulatory protein was up regulated in A375 and H929 cells after the treatment (Figure 6A and 6B). Intriguingly, we did not see the expression of this protein in Ramos cells. We also examined if the suppression of DHODH protein expression through DHODH shRNA would suppress c-Myc and p21 levels in A375 cells (Figure 6D). The expression of c-Myc was significantly downregulated in shRNA-transfected A375 cells but not the siRNA-transfected cells (Figure 6D and 6E). Similarly, p21 was upregulated in shRNA-transfected cells but remain unchanged in the siRNA-transfected cells. The expressions of pERK1/2T202/204, pAkt, NFkB and Bcl-xL in A375 cells were not affected by the treatment with DHODH inhibitors and cells transfected with either siRNA or shRNA (data not shown).
DHODH inhibitors target c-Myc and p21 signaling proteins in cancer cells. Equal amounts of protein lysates were used for Western blot analysis with the antibodies indicated. Tubulin-β and GAPDH served as protein loading control. A, A375 cells were treated with DMSO or DHODH inhibitors and cells were harvested at the indicated time point. B, H929 cells were treated with DMSO or DHODH inhibitors and cells were harvested at 48 hours after treatment. C, Ramos cells were treated with DMSO or DHODH inhibitors and cells were harvested at 48 hours after treatment. D, A375 cells plated in 6-well plates were transfected with DHODH shRNA (2 μg) or the control shRNA (2 μg) for 48 hours before immunoblotted with antibodies against c-Myc and p21. E, A375 cells plated in 6-well plates were transfected with DHODH siRNA (4 nM) or the control siRNA (4 nM) for 48 hours before harvesting for Western blot assays as described above. Data represents mean ± SE of 3 independent experiments. *, P <0.05, **, P <0.01, ***, P <0.001 and ****, P<0.0001.
Discussion
De novo pyrimidine synthesis is essential in fast growing cells for meeting their increased demand for nucleic acid precursors (5). Dihydroorotate dehydrogenase (DHODH) is the fourth and rate liming enzyme in the synthesis pathway of pyrimidines (8) found in the inner membrane of mitochondria (19). Depletion of this enzyme leads to reduction in cell growth, thus marking it as the best target for therapeutic intervention (9, 26). Previous studies have shown that DHODH protein was up-regulated in transformed prostate epithelial cells, PWR-1E and LNCaP cells (27) and transformed keratinocytes, NHEK, HaCaT and COLO 16 cells (28), relative to their normal counterparts. We examined the expression of DHODH in human melanoma, multiple myeloma and lymphoma and their corresponding normal cells, melanocytes and PBMCs and found that the expression level of DHODH within the cancer cells was higher than their corresponding normal cells, likely in response to a higher demand for pyrimidines in the fast growing cancer cells.
Thus far, several DHODH inhibitors namely, LFM and BQR have been demonstrated to be effective in treating immunological disorders such as multiple sclerosis and rheumatoid arthritis (29) and certain types of cancer (20). In the present study, we investigated the role of DHODH inhibitors in A375, H929 and Ramos cells and found that A771726 inhibited the cell proliferation of all three cancer cell lines. Nearly 95% inhibition of cancer cell proliferation was observed at clinically achievable drug concentrations. These results stand in line with findings from others who have seen similar cell proliferation inhibition in myeloma and melanoma cells (17, 24, 25). Likewise, treatment with BQR inhibited proliferation of these cells in nanomolar concentrations and this is also in agreement with a previous report (30). We also demonstrated that the anti-proliferative effects of A771726 and BQR in all three cancer cell lines were primarily due to S-phase arrest, as observed earlier in human prostate cancer, DU145 (27), human gastrointestinal carcinoid cells (33) and HaCaT cells (28).
It is well documented that A771726 and BQR inhibit the enzymatic activity of DHODH (10, 16, 34) and this was postulated to be the mechanism of cell growth inhibition in cancer cells (17, 24, 25, 32). Besides its role in de novo pyrimidine synthesis, Fang, et al. (2013) reported that DHODH depletion induced mitochondrial dysfunction in HeLa human cervical cancer cells (31). It has also been reported that LFM at very high concentrations can inhibit some tyrosine kinases which can further contribute to immunosuppressive effect seen in vivo. Hence, additional molecular mechanisms responsible for its anti-cancer effect remain to be further investigated.
Supplementation of exogenous uridine in cancer cells is known to be able to rescue the DHODH inhibitory effects of A771726 and BQR. To examine whether depletion of DHODH protein is largely accountable for the anti-proliferative effects of A771726 and BQR, we carried out uridine rescue experiments in A375 cells. In line with earlier reports (25, 32) our data showed that uridine supplementation did not rescue A375 cells from the anti-proliferative effects of A771726. On the contrary, exogenous uridine was able to completely reverse the anti-proliferative effect mediated by BQR at all concentrations. These results strongly suggest that two mechanisms, DHODH-independent and DHODH-dependent may possibly be involved in the anti-proliferative effect of A771726 on A375 cells. Since Brequinar sodium is a more selective inhibitor of DHODH, the anti-proliferative effect was completely reversed by uridine.
It is well documented that A771726 and BQR inhibit the enzymatic activity of DHODH (10, 16, 34). We showed here that treatment with DHODH inhibitors does not affect the inherent abundance or the expression of DHODH in the cancer cells although a significant decreased in its enzymatic activity was observed in all three cell lines. Similar to our finding, He, et al. (2013) reported that BQR reduced cell viability of U1690 cells but did not down regulate the DHODH enzyme (32) although Leflunomide was previously shown to drastically reduce the expression of DHODH in neuroblastoma (18). These findings suggest these inhibitors do not affect the post-transcriptional, translational and degradation regulation of DHODH in A375 cells. We employed both siRNA and shRNA knockdown of DHODH expression to validate the functional effect of DHODH in the cancer cells. Stable but not the transient knockdown of DHODH expression resulted in the reduction of cell viability. Interestingly, combination of DHODH knockdown (both transient and prolonged) with DHODH inhibitor treatment resulted in greater cell death in these cells, suggesting an additive effect with a non-DHODH mediated mechanism. Together, these results indicate inhibition of DHODH is responsible for cell cycle arrest which also sensitizes the cells to the effect of DHODH inhibitors. Since cell cycle arrest was observed, we next investigated the impact on c-Myc, pERK 1/2 T202/204, pAkt, NFkB, Bcl-xL and p21 signaling proteins.
c-Myc, a proto-oncogene and transcription factor, is one of the most commonly mutated protein in human cancers (35, 36). It plays a critical role in apoptosis, cell proliferation and differentiation and is primarily overexpressed in human tumors. It acts through regulating transcription of downstream genes (5, 37). c-Myc has been described to reduce the expression of p21 and p27 proteins and elevate the expression of cell-cycle regulators such as cyclin D1, D2 and CDK4 (38). Thus far, several molecules inhibiting the c-Myc activity are known (39, 40) and amongst them 10058-F4 is considerably the most studied compound (41). In addition, Cherwinski et al, (1995), Liu Y (2009) and White et al, (2011) reported that DHODH is a direct molecular target of c-Myc. Hence, a combination therapy with c-Myc inhibitor can be explored as a potential therapy in human cancers associated with c-Myc deregulation.
Here, we showed that DHODH inhibitors affect c-Myc expression in cancer cells. Our findings strongly demonstrate a decrease in c-Myc levels in A375, H929 and Ramos cells upon treatment with A771726 and Brequinar sodium. Correspondingly, the cell-cycle regulatory protein, p21 was observed to be up-regulated in both A375 and H929. Our data is in agreement with a previous report that has demonstrated a decline in c-Myc and increase in p21 expression in acute myeloid leukemia with 10058-F4, a small-molecule c-Myc inhibitor (42). c-Myc is up-regulated in dividing cells and down-regulated as cells exit the cell cycle. This observation supports our cell cycle data that showed DHODH inhibitors arrested A375, H929 and Ramos cells at S-phase (43). In contrast, a prior study has shown that Leflunomide did not decrease the expression of c-Myc protein in A375 cells, although a significant increase in p21 levels was observed (25). In line with compound treatment, c-Myc expression in DHODH shRNA transfected cells was lesser as opposed to the control A375 cells, which could explain the inhibition of proliferation with shRNA treatment.
Taken together, our findings provide unambiguous evidence that DHODH inhibitors, A771726 and Brequinar sodium as well as prolonged DHODH knockdown cause inhibition of cancer cells proliferation, arrest cancer cells predominantly at S-phase and decreased c-Myc protein expression. Since DHODH is vital for cell proliferation, we found that prolonged DHODH knockdown resulted in growth retardation in melanoma cells. Therefore, our studies suggest that these inhibitors can potentially be used for the treatment of melanoma, myeloma and lymphoma and DHODH enzyme can be explored as a potential drug target in cancer therapy.
Acknowledgements
The work was funded by Aurigene Discovery Technologies (M) Sdn Bhd, University of Malaya Postgraduate Research Fund (PPP) Grant Number: PG007-2014B and University of Malaya UM/MoHE High Impact Research Grant (HIR) Account Number: H-20001-E00002. The authors are thankful to Ms. Tong Kind Leng, Department of Pharmacology, University of Malaya for her assistance with flow cytometry and all Aurigene staff for their guidance and support.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029-33
2. Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nature reviews Drug discovery. 2011;10(9):671-84
3. Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136(5):823-37
4. Tennant DA, Duran RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nature reviews Cancer. 2010;10(4):267-77
5. Evans DR, Guy HI. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. The Journal of biological chemistry. 2004;279(32):33035-8
6. Jones ME. Pyrimidine nucleotide biosynthesis in animals: genes, enzymes, and regulation of UMP biosynthesis. Annual review of biochemistry. 1980;49:253-79
7. Fairbanks LD, Bofill M, Ruckemann K, Simmonds HA. Importance of ribonucleotide availability to proliferating T-lymphocytes from healthy humans. Disproportionate expansion of pyrimidine pools and contrasting effects of de novo synthesis inhibitors. The Journal of biological chemistry. 1995;270(50):29682-9
8. Cherwinski HM, Cohn RG, Cheung P, Webster DJ, Xu YZ, Caulfield JP. et al. The immunosuppressant leflunomide inhibits lymphocyte proliferation by inhibiting pyrimidine biosynthesis. The Journal of pharmacology and experimental therapeutics. 1995;275(2):1043-9
9. Copeland RA, Davis JP, Dowling RL, Lombardo D, Murphy KB, Patterson TA. Recombinant human dihydroorotate dehydrogenase: expression, purification, and characterization of a catalytically functional truncated enzyme. Archives of biochemistry and biophysics. 1995;323(1):79-86
10. Williamson RA, Yea CM, Robson PA, Curnock AP, Gadher S, Hambleton AB. et al. Dihydroorotate dehydrogenase is a high affinity binding protein for A77 1726 and mediator of a range of biological effects of the immunomodulatory compound. The Journal of biological chemistry. 1995;270(38):22467-72
11. Chen SF, Perrella FW, Behrens DL, Papp LM. Inhibition of dihydroorotate dehydrogenase activity by brequinar sodium. Cancer research. 1992;52(13):3521-7
12. Greene S, Watanabe K, Braatz-Trulson J, Lou L. Inhibition of dihydroorotate dehydrogenase by the immunosuppressive agent leflunomide. Biochemical pharmacology. 1995;50(6):861-7
13. Breedveld FC, Dayer JM. Leflunomide: mode of action in the treatment of rheumatoid arthritis. Annals of the rheumatic diseases. 2000;59(11):841-9
14. Fox RI. Mechanism of action of leflunomide in rheumatoid arthritis. The Journal of rheumatology Supplement. 1998;53:20-6
15. Chen SF, Papp LM, Ardecky RJ, Rao GV, Hesson DP, Forbes M. et al. Structure-activity relationship of quinoline carboxylic acids. A new class of inhibitors of dihydroorotate dehydrogenase. Biochemical pharmacology. 1990;40(4):709-14
16. Chen SF, Ruben RL, Dexter DL. Mechanism of action of the novel anticancer agent 6-fluoro-2-(2'-fluoro-1,1'-biphenyl-4-yl)-3-methyl-4-quinolinecarbo xylic acid sodium salt (NSC 368390): inhibition of de novo pyrimidine nucleotide biosynthesis. Cancer research. 1986;46(10):5014-9
17. White RM, Cech J, Ratanasirintrawoot S, Lin CY, Rahl PB, Burke CJ. et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011;471(7339):518-22
18. Zhu S, Yan X, Xiang Z, Ding HF, Cui H. Leflunomide reduces proliferation and induces apoptosis in neuroblastoma cells in vitro and in vivo. PloS one. 2013;8(8):e71555
19. Loffler M, Fairbanks LD, Zameitat E, Marinaki AM, Simmonds HA. Pyrimidine pathways in health and disease. Trends in molecular medicine. 2005;11(9):430-7
20. Shawver LK, Schwartz DP, Mann E, Chen H, Tsai J, Chu L. et al. Inhibition of platelet-derived growth factor-mediated signal transduction and tumor growth by N-[4-(trifluoromethyl)-phenyl]5-methylisoxazole-4-carboxamide. Clinical cancer research: an official journal of the American Association for Cancer Research. 1997;3(7):1167-77
21. van der Heijden JW, Oerlemans R, Tak PP, Assaraf YG, Kraan MC, Scheffer GL. et al. Involvement of breast cancer resistance protein expression on rheumatoid arthritis synovial tissue macrophages in resistance to methotrexate and leflunomide. Arthritis and rheumatism. 2009;60(3):669-77
22. Peters GJ, Nadal JC, Laurensse EJ, de Kant E, Pinedo HM. Retention of in vivo antipyrimidine effects of Brequinar sodium (DUP-785; NSC 368390) in murine liver, bone marrow and colon cancer. Biochemical pharmacology. 1990;39(1):135-44
23. Baldwin J, Michnoff CH, Malmquist NA, White J, Roth MG, Rathod PK. et al. High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. The Journal of biological chemistry. 2005;280(23):21847-53
24. Baumann P, Mandl-Weber S, Volkl A, Adam C, Bumeder I, Oduncu F. et al. Dihydroorotate dehydrogenase inhibitor A771726 (leflunomide) induces apoptosis and diminishes proliferation of multiple myeloma cells. Molecular cancer therapeutics. 2009;8(2):366-75
25. O'Donnell EF, Kopparapu PR, Koch DC, Jang HS, Phillips JL, Tanguay RL. et al. The aryl hydrocarbon receptor mediates leflunomide-induced growth inhibition of melanoma cells. PloS one. 2012;7(7):e40926
26. Vyas VK, Ghate M. Recent developments in the medicinal chemistry and therapeutic potential of dihydroorotate dehydrogenase (DHODH) inhibitors. Mini reviews in medicinal chemistry. 2011;11(12):1039-55
27. Hail N Jr, Chen P, Bushman LR. Teriflunomide (leflunomide) promotes cytostatic, antioxidant, and apoptotic effects in transformed prostate epithelial cells: evidence supporting a role for teriflunomide in prostate cancer chemoprevention. Neoplasia. 2010;12(6):464-75
28. Hail N Jr, Chen P, Kepa JJ, Bushman LR. Evidence supporting a role for dihydroorotate dehydrogenase, bioenergetics, and p53 in selective teriflunomide-induced apoptosis in transformed versus normal human keratinocytes. Apoptosis: an international journal on programmed cell death. 2012;17(3):258-68
29. Merrill JE, Hanak S, Pu SF, Liang J, Dang C, Iglesias-Bregna D. et al. Teriflunomide reduces behavioral, electrophysiological, and histopathological deficits in the Dark Agouti rat model of experimental autoimmune encephalomyelitis. Journal of neurology. 2009;256(1):89-103
30. Cleaveland ES, Zaharevitz DW, Kelley JA, Paull K, Cooney DA, Ford H Jr. Identification of a novel inhibitor (NSC 665564) of dihydroorotate dehydrogenase with a potency equivalent to brequinar. Biochemical and biophysical research communications. 1996;223(3):654-9
31. Fang J, Uchiumi T, Yagi M, Matsumoto S, Amamoto R, Takazaki S. et al. Dihydro-orotate dehydrogenase is physically associated with the respiratory complex and its loss leads to mitochondrial dysfunction. Bioscience reports. 2013;33(2):e00021
32. He T, Haapa-Paananen S, Kaminskyy VO, Kohonen P, Fey V, Zhivotovsky B. et al. Inhibition of the mitochondrial pyrimidine biosynthesis enzyme dihydroorotate dehydrogenase by doxorubicin and brequinar sensitizes cancer cells to TRAIL-induced apoptosis. Oncogene. 2013
33. Cook MR, Pinchot SN, Jaskula-Sztul R, Luo J, Kunnimalaiyaan M, Chen H. Identification of a novel Raf-1 pathway activator that inhibits gastrointestinal carcinoid cell growth. Molecular cancer therapeutics. 2010;9(2):429-37
34. Peters GJ, Sharma SL, Laurensse E, Pinedo HM. Inhibition of pyrimidine de novo synthesis by DUP-785 (NSC 368390). Investigational new drugs. 1987;5(3):235-44
35. Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18(19):3004-16
36. Liao DJ, Dickson RB. c-Myc in breast cancer. Endocrine-related cancer. 2000;7(3):143-64
37. Henriksson M, Luscher B. Proteins of the Myc network: essential regulators of cell growth and differentiation. Advances in cancer research. 1996;68:109-82
38. Roussel MF, Theodoras AM, Pagano M, Sherr CJ. Rescue of defective mitogenic signaling by D-type cyclins. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(15):6837-41
39. Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene. 2003;22(40):6151-9
40. Berg T, Cohen SB, Desharnais J, Sonderegger C, Maslyar DJ, Goldberg J. et al. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(6):3830-5
41. Wang H, Hammoudeh DI, Follis AV, Reese BE, Lazo JS, Metallo SJ. et al. Improved low molecular weight Myc-Max inhibitors. Molecular cancer therapeutics. 2007;6(9):2399-408
42. Huang MJ, Cheng YC, Liu CR, Lin S, Liu HE. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Experimental hematology. 2006;34(11):1480-9
43. Henriksson M, Selivanova G, Lindstrom M, Wiman KG. Inactivation of Myc-induced p53-dependent apoptosis in human tumors. Apoptosis: an international journal on programmed cell death. 2001;6(1-2):133-7
44. Liu Y. MYC regulation of nucleotide metabolism. 2009; (Order No. 3339875). (304902569).
Author contact
![]() Corresponding author: Dr Pooi-Fong Wong, Department of Pharmacology, Faculty of Medicine, 50603 University of Malaya, Kuala Lumpur, Malaysia; Phone: +603-79677022 (ext 2065); Fax: +603-79674791; Email: wongpfedu.my
Corresponding author: Dr Pooi-Fong Wong, Department of Pharmacology, Faculty of Medicine, 50603 University of Malaya, Kuala Lumpur, Malaysia; Phone: +603-79677022 (ext 2065); Fax: +603-79674791; Email: wongpfedu.my