Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(14):2774-2784. doi:10.7150/jca.18731 This issue Cite
Research Paper
CT-721, a Potent Bcr-Abl Inhibitor, Exhibits Excellent In Vitro and In Vivo Efficacy in the Treatment of Chronic Myeloid Leukemia
Yinghui Sun1, Na Zhao1, Huan Wang1, Qiong Wu1, Yunqi Han1, Qichao Liu1, Mangang Wu1, Yuliang Liu2, Fansheng Kong3, He Wang3, Ying Sun3, Deguang Sun2, Lutao Jing2, Guojing Tang2, Yuandong Hu2, Dengming Xiao2, Hong Luo3, Yongxin Han2, Yong Peng1 ![]()
1. Department of Biology, Centaurus BioPharma Co., Ltd.;
2. Department of Medical Chemistry, Centaurus BioPharma Co., Ltd.;
3. Department of DMPK, Centaurus BioPharma Co., Ltd.
Received 2016-12-13; Accepted 2017-5-18; Published 2017-8-23
Abstract
Kinase inhibitors that target Bcr-Abl are highly effective in the treatment of chronic myeloid leukemia (CML). However, these inhibitors are often invalidated due to the drug resistance. Therefore, the discovery and development of novel Bcr-Abl inhibitors is required to overwhelm the drug resistance in the treatment of CML resistant to the currently used first-line Bcr-Abl inhibitors. Herein we have described a newly developed Bcr-Abl inhibitor CT-721, which displayed potent inhibitory effects on wild-type and T315I mutant Bcr-Abl. It functioned as a typically ATP-competitive inhibitor, superior to other existing Bcr-Abl inhibitors. CT-721 also demonstrated time-dependent inhibition of Bcr-Abl activation and the resultant downstream signaling transduction pathways in Bcr-Abl positive cells. Furthermore, CT-721 induced cell apoptosis and cell cycle arrest, and efficaciously inhibited tumor growth in Bcr-Abl-expressed K562 and KU812 xenograft models in a mechanism-based manner. Further PK/PD studies revealed a positive in vivo correlation between the compound concentration and inhibition of Bcr-Abl activity. Taken together, CT-721 is a potent and time-dependent Bcr-Abl kinase inhibitor, and has shown strong in vitro and in vivo anti-CML activities with a favorable pharmacokinetic profile, differentiating it from other Bcr-Abl kinase inhibitors already approved and current in development for the treatment of CML.
Keywords: CT-721, Bcr-Abl, anti-tumor, leukemia.
Introduction
Chronic myeloid leukemia (CML), a myeloproliferative disease, is characterized by the presence of a fused Bcr-Abl protein resulting from Philadelphia chromosome (a rearrangement between chromosome 9 and 22) [1, 2]. This fusion protein leads to the constitutive activation of Abl and downstream kinases on several signaling pathways, and consequently makes the cells escape from the normal regulation of cellular signaling possible. In the past two decades, studies showed that the fused Bcr-Abl protein was the main pathogenetic cause of CML, since the constitutive activation of Bcr-Abl played a critical role in cell transformation and leukemia oncogenesis [3]. The activation of multiple signaling pathways resulted in increased cell proliferation, reduced dependence on growth-factors, decreased apoptosis and accelerated cell division [4, 5]. Taken the in vitro and in vivo studies together, Bcr-Abl has been a new drug target for the treatment of CML.
Imatinib (Gleevec/STI-571) [6, 7], a first-line clinical drug for CML patients based on its excellent efficacy and safety, is a small molecule Bcr-Abl inhibitor [7]. However, as the first generation Bcr-Abl inhibitor, imatinib was often invalidated due to the development of cellular resistance to the drug during the treatment [8]. Now it has been demonstrated that the resistance is associated with Abl kinase mutations, especially T315I mutation in the gatekeeper region [9]. The second generation inhibitors, such as nilotinib [10], dasatinib [11, 12] and bafetinib [13], have been developed to overcome the imatinib resistance, and they are generally more potent and have achieved higher and faster responses than imatinib. Unfortunately, both T315I mutated Bcr-Abl and leukemia-initiating cells remained insensitive to them. Such a resistance occurs via several cell signaling pathways and/or is mediated by multiple protein kinases since the single T315I mutation in Bcr-Abl has accounted for only 15-20 % of the drug resistant cases [14, 15]. Therefore, tremendous efforts are being focused on development of the third-generation Bcr-Abl inhibitors targeting T315I mutation for the treatment of drug-resistant CML [16]. Some of those inhibitors, such as ponatinib/AP24534 [17-19] and DCC-2036 [20, 21], have shown potent efficacy on the mutant resistance. In the current study, we reported an imidazopyridazine-based compound, CT-721, as a novel Bcr-Abl inhibitor based on computational modeling and Structure-Activity Relationship (SAR) analysis. CT-721 has a novel chemical structure with excellent inhibition on wild-type and T315 mutant Bcr-Abl kinase activity. We systemically studied the inhibitory mechanism and in vitro and in vivo efficacy of CT-721. Due to its excellent efficacy and favorable pharmacokinetic profile, CT-721 has been selected for further development for the treatment of Leukemia.
Materials and Methods
Compound synthesis
CT-721, [(S)-N-(3-((6-chloroimidazo[1,2-b]pyridazin-3-yl)ethynyl)-4-methylphenyl)-1-(4-methylpiperazin-1-yl)-2,3-dihydro-1H-indene-5-carboxamide] was designed and synthesized by Centaurus BioPharma Co., Ltd. Reference compound, Ponatinib [3-[2-(Imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)-methyl]-3-(trifluoromethyl)phenyl}benzamide] was synthesized in house. Compounds were stocked at 10 mM concentration in 100% DMSO and diluted for biological test prior to use in each experiment.
Cell culture
K562, KU812, Kasumi-1, MV-4-11, HL-60, Colo-205, A549 and MCF-7 cell lines were purchased from the cell bank of Chinese Academy of Science (Shanghai, China). MKN-45 human gastric carcinoma cell line was obtained from Health Science Research Resource Bank (Osaka, Japan). Ba/F3-Bcr-AblWT/T315I stable cell line was generated as described [27, 28]. Cells were cultured in DMEM or RPMI-1640 supplemented with 10% FBS, and 50 IU penicillin/streptomycin in a humidified atmosphere with 5% CO2 at 37 ℃. Human PBMC cells were collected with Ficoll-Paque PREMIUM (GE Healthcare) according to manufacturer's guide. The medium, serum and antibiotics were purchased from Invitrogen.
Kinase enzyme assay
Abl, c-Kit, VEGFR2, PDGFRβ, c-Met, Alk enzymes were purchased from Millipore (Billerica, MA), and HTRF® KinEASETM-TK was obtained from Cisbio Bioassays. HTRF (Homogeneous Time-Resolved Fluorescence) assay was used to analyze kinase enzyme activity and compound potency. Compounds were serially diluted in 3 folds from 1 mM stock solution in DMSO for 11 points. 4 μL of the diluted solution of the compounds was added to 96 μL of reaction buffer (50 mM HEPES pH7.0, 0.1 mM Na3VO4, 0.01% BAS, 5 mM MgCl2, 1 mM DTT), then 2.5 μL of 4×compound solution and 5 μL of 2×kinase solution were added to a 384-well plate(OptiPlate-384, PerkinElmer). The resulted solutions were briefly mixed, centrifuged and incubated for 5min. Finally, 2.5 μL of 4×ATP/TK peptide (ATP at Km for each kinase) solution was added to the reaction system to initiate the reaction. The enzymatic reactions were carried out for 60-120 min at 23 oC, then terminated by adding 5 μL of detection solution containing TK antibody-cryptate and 5 μL of Streptravidin-XL-665. The mixtures were incubated for additional 1 hour at 23 oC. The fluorescent signals were read using EnVision multilable plate reader (PerkinElmer). IC50 values of the compounds were generated using GraFit software (Version 6.0) and presented at the mean of three independent experiments performed in duplicate. The selective inhibition of CT-721 against site-mutated Abl variants E225K, G250E, Y253F, H369P, M351T and Q252H was performed by Thermo Fisher SCIENTIFIC with service of Broad Assay Panel.
Cell proliferation assay
The effects of CT-721 on cell proliferation were measured using Promega's kits (CellTiter-Glo® or CellTiter-Blue® Cell Viability Assay). Cells were seeded in 96-well plate at low destiny with 195 µL medium per well. The stock solutions of compounds in 100% DMSO were 3-fold serially diluted from 10 mM to 0.3 nM, and 4 µL of solution at each concentration was transfered to 96 µL of serum free medium (SFM), then 5 µL of the resulted solution was added to each well. After treatment for 72 hours, 25 or 35 µL of CellTiter-Glo® (for adherent cells) or CellTiter-Blue® (for suspension cells) reagent was added to each well, and the resulted mixture was further incubated at RT for 10 minutes or 4 hours. The luminescence/fluorescent signals were measured using EnVision® multi-labelled plate reader (PerkinElmer) or FlexStation 3 (Molecular Devices) and IC50 values were calculated using Prism® software (Version 5.0) and reported at the average of three independent experiments performed in duplicate.
Western blot
K562 or KU812 cells were plated in 6-well plates at the density of 1x106 cells per well in 2 mL of the culture medium and cultured at 37 oC overnight under 5% CO2. The various concentrations of compound solutions were added to each well. Cells were collected and lysed after 60 minutes, and total protein concentrations were determined with a Bio-Rad BCA® kit. Equal amounts of cell lysates were loaded onto 10% SDS gel and separated by electrophoresis. Separated proteins were then electro-transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA). After being blocked with 1 X Tris-buffered saline (TBS) containing 0.1% Tween-20 and 5% bovine serum albumin (BSA), the membranes were incubated with primary antibodies at room temperature for 2 hours or at 4 oC O/N, then washed with 1X TBS containing 0.1% Tween-20 and followed by treatment with the horseradish peroxidase (HRP)-conjugated secondary antibody at room temperature for another 1 hour. The targeted proteins were visualized using an enhanced chemiluminescence (ECL) plus system (Thermo Fisher Scientific, Waltham, MA). Primary antibodies against phospho-Abl, phospho-Crkl, phospho-ERK, phospho-STAT3, phospho-STAT5, phospho-AKT, AKT and β-Actin and the horseradish peroxidase (HRP) conjugated goat anti-rabbit IgG were purchased from Cell Signaling Technology.
Flow cytometry
K562 cells were treated with 0.1 % DMSO (control) or with CT-721 solutions at indicated concentrations for 24 hours at 37 oC, then the cells were harvested by centrifugation and washed twice with PBS. Cell apoptosis was determined with a PE Annexin V Apoptosis Detection Kit (BD Biosciences, San Jose, CA) following the manufacture's guides. Breifly, cells were resuspended with 100 µL of binding buffer containing with Annexin V and 7-AAD. After 15 minutes incubation at room tempreture in the dark, 400 µL of binding buffer was added to the mixture and the cells were analyzed with a Guava flow cytometer (Millipore, Bedford, MA). Cell cycle arrest was evaluated by incorporation of propidium (PI) (Sigma-Aldrich, St.Louis, MO) into the permeablized cells. After being treated with DMSO or compounds for 24 hours, cells were harvested, washed twice with cold PBS, fixed with ice-cold 70% ethanol O/N followed by a secondary staining step with staining buffer (0.25 mg/mL Rnase, 0.025 mg/mL PI, in PBS) for 1 hour at 37 oC. Compound regulation of cell-cycle was analyzed using a Guava flow cytometer (Millipore, Bedford, MA). Data were analyzed with FlowJo software.
Tumor xenograft studies in nude mice
Female athymic nude mice (5-6 week) were used for all the in vivo studies. 5x106 cells in 100 µL of serum-free medium were planted subcutaneously on the right flank of mice. Tumor volumes were monitored by caliper measurement using the formula: tumor volume (mm3) = (w2 x l)/2, where w = width and l = length. When the tumor volumes reached 150~200 mm3, mice were randomized to groups (8-10 mice per group) and treated with compounds or vehicle by oral gavage every day for 2-4 weeks. The tumor volumes and body weight were measured every 2-3 days. Tumor growth inhibition (TGI) and body weight changes were calculated compared to the control groups. Statistical significance of the treatment groups compared to vehicle group was evaluated with t-test. All the experiments were carried out in accordance with the guidelines of institutional animal care and use committee (IACUC) of Centaurus BioPharma and approved by IACUC of Centaurus BioPharma.
Pharmacokinetic studies in rats
Female SD rats (5-6 week) were administrated with CT-721 at a single dose of 5 mg/kg and blood samples were taken sequentially from 2 minutes to 24 hours after compound administration. The serum samples were collected by centrifugation and stored at -80 oC. Compound concentrations in the sample serums were further analyzed by LC-MS/MS (API-4000, Applied Biosystem Inc.), and pharmacokinetic parameters (T1/2, Tmax, Cmax, AUC0-INF) were generated using WinNonlin software (WinNonlin Professional, version 6.2).
Results
CT-721 is a potent inhibitor of wild-type and T315I mutant Brc-Abl kinases
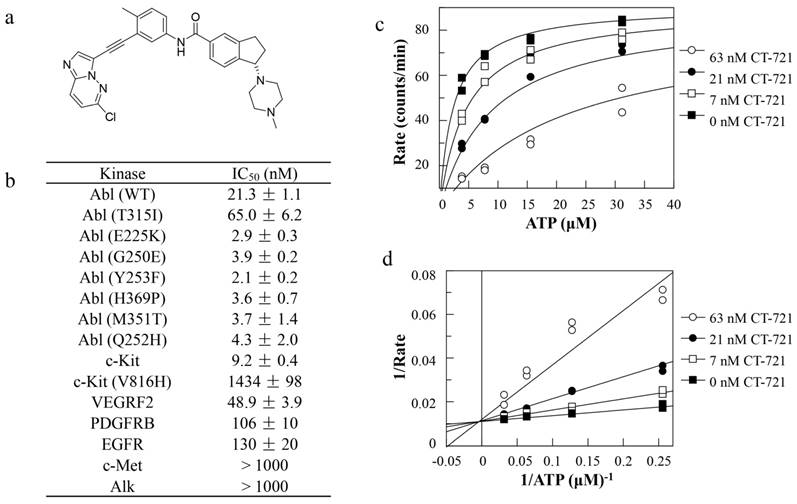
In an effort to develop T315I mutant Bcr-Abl sensitive inhibitor to overcome CML drug resistance, we had designed and synthesized CT-721 (Fig. 1a), a novel small molecular inhibitor of wild-type and T315I mutant Bcr-Abl kinases. The efficacious inhibition of CT-721 on the Bcr-Abl kinase and T315I mutant Bcr-Abl kinase was similar to that of Ponatinib (data not shown) with IC50 values of 21.3 ± 1.1 nM and 65.0 ± 6.2 nM, respectively (Fig. 1b). Potent inhibition on other 6 imatinib-resistant mutants (E225K, G250E, Y253F, H369P, M351T and Q252H) was also observed. Furthermore, the selectivity of CT-721 was investigated by determining the inhibition profile over other human kinases involved in the progress of CML as well as AML, including c-Kit, VEGFRF2, PDGFRβ, EGFR, c-Met and Alk. As shown in Figure 1b, CT-721 also markedly inhibited c-Kit, VEGFR2, PDGFRβ and EGFR with IC50 values of 9.2 nM, 48.9 nM, 106 nM and 130 nM, respectively, implicating CT-721 could exert additional effects on CML other than targeting Bcr-Abl kinase. The kinetic experiments were performed to explore the inhibitory mechanism of CT-721 against Bcr-Abl. As shown in Fig. 1c, CT-721-mediated inhibition on Abl kinase activity can be significantly attenuated with increasing concentrations of ATP. Moreover, CT-721 had higher affinity to free Abl enzyme with a dissociation constant (Ki) value of 7.3 nM than that to the enzyme-ATP complex with a Ki' value of 775 nM. Lineweaver-Burk plots (Double-reciprocal plots) (Fig. 1d) illustrated that CT-721 inhibited Bcr-Abl as an ATP-competitive inhibitor.
In vitro enzymatic inhibition, selectivity and its mechanism of CT-721. (a) Chemical structure of CT-721. (b) Kinase inhibition profile of CT-721. (c) Mechanism of inhibition of CT-721 to Abl enzyme. Abl enzyme activities were evaluated at various concentrations of ATP (0, 0.5, 1, 2, 4, 8, 16, 32 μM) in the presence of CT-721 (63 (○), 21 (●), 7 (□) and 0 (■) nM respectively). (d) Double-reciprocal plots (Lineweaver-Burk plots) of velocity versus ATP concentrations was generated at various concentrations of CT-721
CT-721 has demonstrated time-dependent inhibition on Bcr-Abl kinase
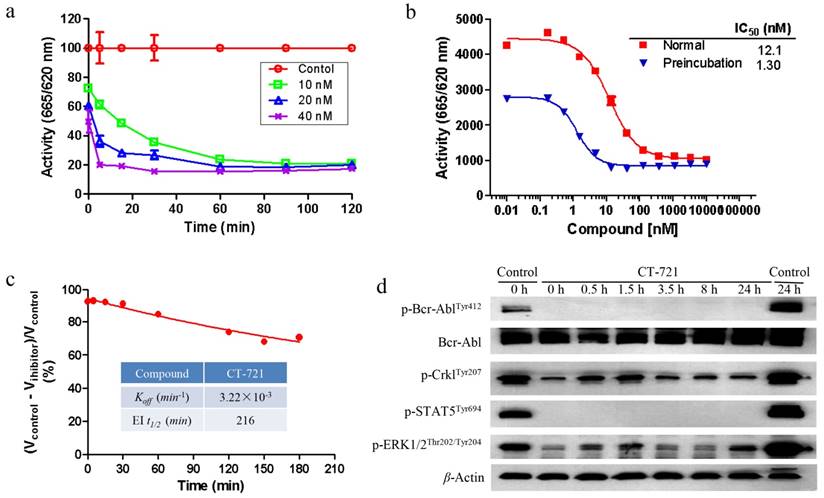
In order to further characterize the inhibitory mechanism of CT-721 on Bcr-Abl, both biochemical assay and cellular assay were carried out. As shown in Fig. 2a, CT-721 demonstrated dose-dependent and time-dependent inhibitions on Bcr-Abl kinase activity in in vitro enzymatic studies. The IC50 value of CT-721 for Abl kinase was decreased by 9-fold from 12.1 nM to 1.3 nM after the kinase was pre-incubated with CT-721 for two hours, implicating that CT-721 inhibited Abl enzyme in a time-dependent manner (Fig. 2b). In the further enzymatic kinetics study, CT-721 was characterized as a slow- and tight-binding inhibitor of Bcr-Abl kinase with Koff and EI t1/2 values at 3.22 × 10-3 min-1 and 216 minutes, respectively (Fig. 2c). The main advantages of slow- and tight-binding inhibitors result from their high affinity and long residence time on their target enzymes due to the slow dissociation rates of typical tight binding inhibitors. Therefore, the slow- and tight-binding inhibitors usually lead to persistent and sustained inhibition of targeted enzyme activity. Indeed, the pre-treatment of K562 leukemia cells with 100 nM of CT-721 for one hour led to a persistent inhibition on phosphorylation of Bcr-Abl and its downstream targets, such as Crkl kinase and Stat3, up to at least 24 hours even after the compound was washed out (Fig. 2d). Collectively, these results suggest that CT-721 is a time-dependent and slow-binding Abl kinase inhibitor.
Time-dependent inhibition studies of CT-721. (a) Concentration-response analysis of CT-721 at the indicated concentrations, 10 (□), 20 (△) and 40 (╳) nM respectively. Following preincubation with CT-721 for the different time as indicated, the enzyme reaction was initiated by adding the substrates at different time points, and the reaction rates were determined. (b) Effects of preincubation time with CT-721 on the steady state velocity of the Abl catalytic reaction. Abl enzyme was pre-incubated with CT-721 at indicated concentrations for 0 min or 40 min, then inhibitory activities (IC50) was measured as described in Methods and Materials. (c) The inhibitory reversibility study of CT-721. Abl enzyme (100 nM, 100×) was pre-incubated with CT-721 (100 nM) for 2 hours, then the enzyme reaction was initiated by adding the substrates to a 100-fold diluted pre-incubation mixture. Enzyme rates in the presence of compound were determined at different times after a 100-fold dilution of pre-incubation mixture, and the rate of uninhibited reaction was normalized by using the enzyme activity in absence of compound as 100%. The kinetics parameter Koff and EI t1/2 was obtained from a one-phase exponential decay equation using the fitted data points. (d) Time-dependent and tight-binding characteristic of CT-721 inhibition on Abl mediated signal transduction. K562 cells were treated with 100 nM of CT-721 for 1 hour, then the medium containing compound was washed out and replaced with fresh medium for another 24 hours incubation (CT-721/0-24 h labeling in Figure 2d), DMSO-untreated cells was used as control (control/0 h and control/24 h in Figure 2d) .
CT-721 selectively inhibits proliferation of Bcr-Abl-expressing leukemia cells
The cell proliferation studies were executed to evaluate the inhibitory effects of CT-721 on several cell lines including solid tumor and leukemia cells abnormally expressing Bcr-abl or other oncogenic kinases. As shown in Table 1, CT-721 not only significantly inhibited the growth of CML cell lines with Philadelphia chromosome, K562 and KU812, with IC50 values of around 1 nM, but also showed remarkable inhibition on proliferation of Ba/F3 stable cell lines harboring wild-type Bcr-Abl or T315I mutated Bcr-Abl. In contrast, CT-721 had very poor potency on parental Ba/F3 cells (IC50 > 1000 nM), and was significantly less potent against growth of Bcr-Abl-negative MV-4-11 cells with an IC50 value more than 200 nM. On normal human PBMC cells and several solid cancer cell lines including lung cancer (A549, H460), colorectal adenocarcinoma (Colo-205), breast carcinoma (MCF-7), gastric carcinoma (MKN-45), prostate adenocarcinoma (PC-3) and hepatocellular carcinoma (SMMC-7721). CT-721 presented very weak inhibitory potency with IC50 values between 1000 to 10000 nM. These data directly supported that CT-721 mediated a target-dependent inhibition of cell growth by selectively targeting Bcr-Abl, and indirectly implicated well accepted tolerance of immuno-protective PBMC function (IC50 > 1000 nM).
Cell growth inhibition of CT-721
| Cell line | Cancer type | IC50 (nM) |
|---|---|---|
| Parental Ba/F3 | Murine pro B cell | 1000~10000 |
| Ba/F3-Bcr-ablWT | Stable cell line | 1.6 ± 0.3 |
| Ba/F3-Bcr-ablT315I | Stable cell line | 18.5 ± 12 |
| K562 | Chronic myeloid leukemia | 0.38 ± 0.07 |
| KU812 | Chronic myeloid leukemia | 1.0 ± 0.13 |
| Kasumi-1 | Acute myeloid leukemia | 53 ± 6 |
| MV-4-11 | Acute myeloid leukemia | 232 ± 55 |
| HL-60 | Myeloid Leukemia | 1000~10000 |
| A549 | NSCLC | 1434 ± 98 |
| H460 | Large lung cell | 48.9 ± 3.9 |
| Colo-205 | Colorectical adenocarcinoma | 48.9 ± 3.9 |
| MCF-7 | Breast carcinoma | 106 ± 10 |
| MKN-45 | Gastric carcinoma | 1000~10000 |
| PC-3 | Prostate adenocarcinoma | 1000~10000 |
| SMMC-7721 | Hepatocellular carcinoma | 1000~10000 |
| Human PBMC | Human lymphocyte | 1000~10000 |
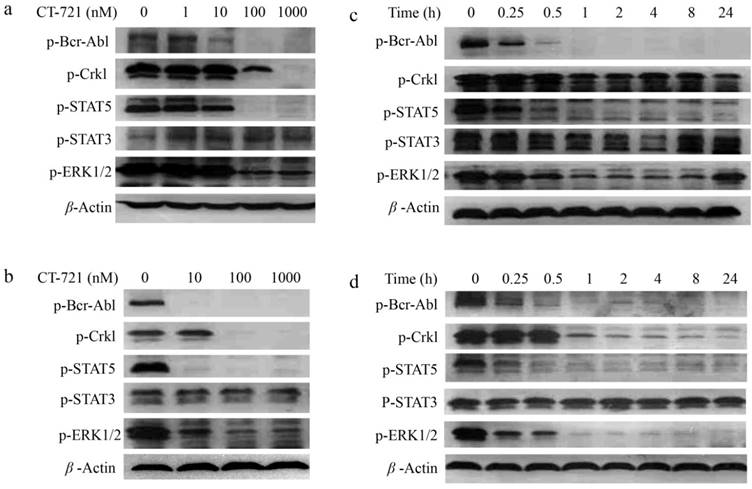
To investigate the underlying mechanisms of CT-721 inhibition on leukemia cells abnormally expressing Bcr-Abl, we evaluated the effects of CT-721 on cell signaling pathways that played important roles in proliferation of K562 and KU812 cells. CT-721 dose-dependently suppressed the phosphorylation of Bcr-Abl and its downstream protein Crkl in K562 and KU812 cell lines after these cells were treated with CT-721 for 1 hour (Fig. 3a and 3b). CT-721 significantly suppressed phosphorylation of Bcr-Abl kinase at 10 nM, and totally blocked the phosphorylation at 100 nM. In the meantime, the phosphorylation of Crkl was also significantly inhibited by CT-721 at 100 nM. In the time course of inhibition studies, CT-721 demonstrated time-dependent inhibition on the phosphorylation of Bcr-Abl and Crkl in both K562 and KU812 cell lines (Fig. 3c and 3d). The phosphorylation of these proteins underwent substantial and sustained blockage up to at least 24 hours (Fig. 3c and 3d). Multiple signal transduction pathways play key roles in Bcr-Abl mediated cell growth and tumorgenesis. The further studies showed that CT-721 only dose-dependently repressed the levels of phosphorylated ERK1/2 and STAT5 (Fig. 3a-3d), and had no meaningful inhibition on the phosphorylation of STAT3 and AKT (data not shown). These results have mechanized that proliferation-associated MAPK and STAT5 signaling pathways are keys of CT-721 inhibition of the growth of K562 and KU812 cells.
Inhibitory effects of CT-721 on the phosphorylation of kinases in K562 and KU812 cells. (a and b) Concentration-dependent inhibition of CT-721 on Bcr-Abl mediated protein phosphorylation. K562 (a) and KU812 (b) cells were cultured in 6-well plate and treated with the indicated concentrations of CT-721 for 1 hour. Control cells received the drug vehicle with 0.1% DMSO. Cell lysates were analyzed by Western blot with specific antibodies against p-Bcr-Abl, p-Crkl, p-AKT and p-ERK respectively. (c and d) Time-dependent inhibition of CT-721 on Bcr-Abl mediated protein phosphorylation. K562 (c) cells and KU812 (d) cells were treated with 100 nM of CT-721 for 0, 0.25, 0.5, 1, 2, 4, 8, 24 hours respectively, cells were then collected and cell lysates were analyzed by Western blot with special antibodies against p-Bcr-Abl, p-Crkl, p-AKT and p-ERK respectively.
CT-721 induces apoptosis and cell-cycle arrest in leukemia cells
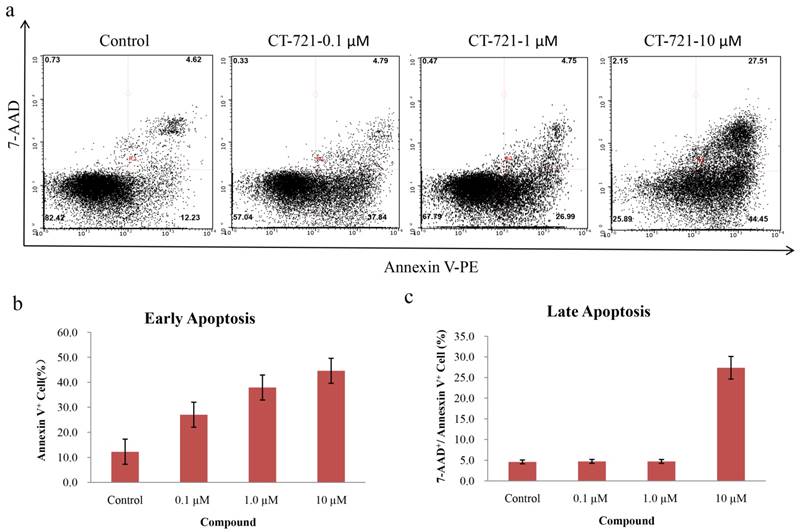
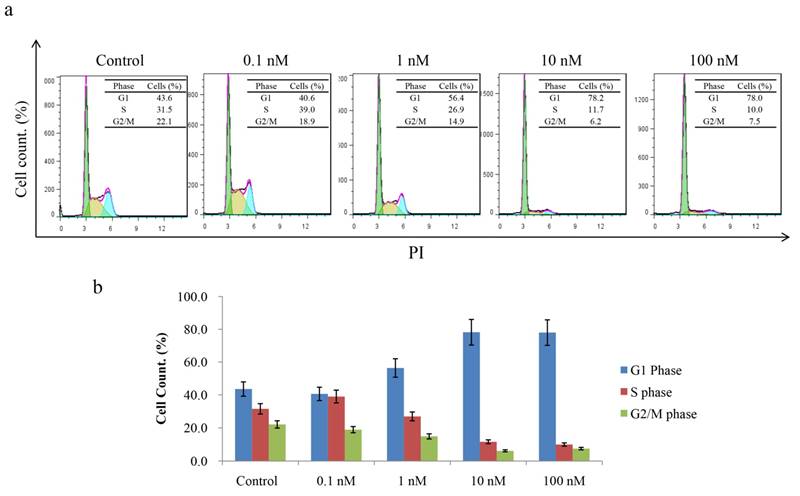
Since CT-721 demonstrated significant inhibition on K562 cell growth, we further assessed its effects of on cell-cycle progression and apoptosis in K562 cells and Ba/F3-Bcr-AblWT/T315I cells. CT-721 dose-dependently induced significant early-phase apoptosis in a dose-dependent manner after 24 hour treatment. Meanwhile, late-phase apoptosis was also observed in presence of 10 μM of CT-721 in the cells for 24 hours (Fig. 4a). Similar results were obtained in Ba/F3-Bcr-AblWT/T315I cells (data was not shown). These results supported that the observed anti-proliferation potency of CT-721 was correlated with its induction action of cell death. In the cell cycle analysis of K562 cells after cells were exposed to CT-721 for 24 hours (Fig. 5), CT-721 was able to induce pronounced G0/G1 phase arrest at 1 nM with a dose-dependent increase in the percentage of cells in G1 phase, and a decrease in the percentage of cells in S phase and G2/M phase (Fig. 5). This was in agreement with the growth curves of K562 cells treated with CT-721, implicating that CT-721 suppressed the entrance of cells from G1 to S phase, interfered with the cell cycle progression and resulted in growth inhibition. Overall, there was a generally close agreement between CT-721-induced growth inhibition, apoptosis and cell-cycle arrest in Bcr-Abl-expressing cells.
Cell apoptosis analysis of CT-721 in K562 cells. (a) Apoptosis of K562 cells treated with CT-721. Cells were cultured and treated with CT-721 under the indicated concentrations (10, 1.0 and 0.1 μM respectively) for 24 hours, then stained with Annexin-V and 7-AAD for cell apoptosis analysis with Guava flow cytometry. (b and c) Early apoptosis (b) and late apoptosis (c).
Effects of CT-721 on cell cycle. (a) Cell cycle of K562 cells treated with CT-721. Cells were harvested and washed with PBS for three times after incubation with the specified concentration of CT-721 for 24 hours, and cells were then labeled with propidium (PI) after fixing with ice ethanol O/N. Cell cycle arrest was determined with Guava Flow cytometry. (b) Cell cycle phases (G1, S, and G2/M phase) were quantified based on the data in (a).
Pharmacokinetics studies of CT-721 in SD rats
Since CT-721 showed excellent in vitro potency in enzymatic and cellular assays, we further evaluated its pharmacokinetics profile in SD rats. As shown in Table 2, CT-721 was eliminated slowly in SD rats with a t1/2 of 8.83 hours, but it was also absorbed slowly with a Tmax of 6.67 hours. CT-721 displayed high plasma exposures in rats with mean AUC at 5424 hr*ng/mL. In addition, CT-721 did not show significant inhibition on liver CYP450 enzymes (IC50 greater than 10 μM for all tested enzymes, data not shown). PK parameters supported that CT-721 could be a strong candidate to move forward for further in vivo efficacy studies.
Pharmacokinetics parameters of CT-721 in rat
| Parameter | 5 mg/kg, PO |
|---|---|
| T1/2 (hr) | 8.83 |
| Tmax (hr) | 6.67 |
| Cmax (ng/mL) | 303 |
| AUC0-INF (hr*ng/mL) | 5424 |
CT-721 blocks tumor growth in K562 and KU812 xenograft mice
Due to its strong cell growth inhibition potency, we next performed a series of in vivo anti-cancer efficacy studies using xenograft mice bearing K562 and KU812 cells-derived tumors. In the K562 xenograft model, mice were orally administered with vehicle or CT-721 at 5, 15 or 45 mg/kg after tumors reached to 150-300 mm3. The results (Fig. 6a) showed that CT-721 suppressed K562 tumor growth in a dose-dependent manner and induced tumor regression at high dose of 45 mg/kg. After treated with CT-721 at 45 mg/kg for 9 days, 90% of mice were completely cured and the tumors disappeared completely (p < 0.01), and the others showed partial regression in this treatment group. In 15 mg/kg treatment group, CT-721 inhibited tumor growth significantly with 76% (p < 0.05) tumor growth inhibition after treatment for 12 days. In 5 mg/kg treatment group, the tumor growth inhibition was 51.4%, p < 0.05. Meanwhile, the body weights of xenograft mice were also monitored and CT-721 appeared to be well tolerated at all three doses without causing significant bodyweight loss (Fig. 6b).
Anti-tumor activities of CT-721 against human tumor xenografts in nude mice. (a and c) Xenograft tumor growth inhibition by CT-721. Xenograft tumors were established by inoculating K562 (a) cells or KU812 (c) cells at 5x107 cells in 100 µL of serum-free medium into the right flank of nude mice. Oral administration of CT-721 at 5, 15 and 45 mg/kg (PO, QD) for K562 xenograft model or at 30 mg/kg (PO, QD) for KU812 xenograft model was initiated when tumors reached to an average of approximately 150-300 mm3 in volume and continued through the experiment. Tumor volume was measured on the indicated days with the mean tumor volume ± SE indicated for each group (10 mice/group). (b and d) Body weight changes of nude mice under treatment of CT-721 K562 (b) or KU812 (d). Body weight changes were also monitored on the indicated days. Each treatment groups was compared to vehicle group with t-test, statistical significance (p < 0.05 or p < 0.01) was indicated with an asterisk (*, p < 0.05; **, p < 0.01).
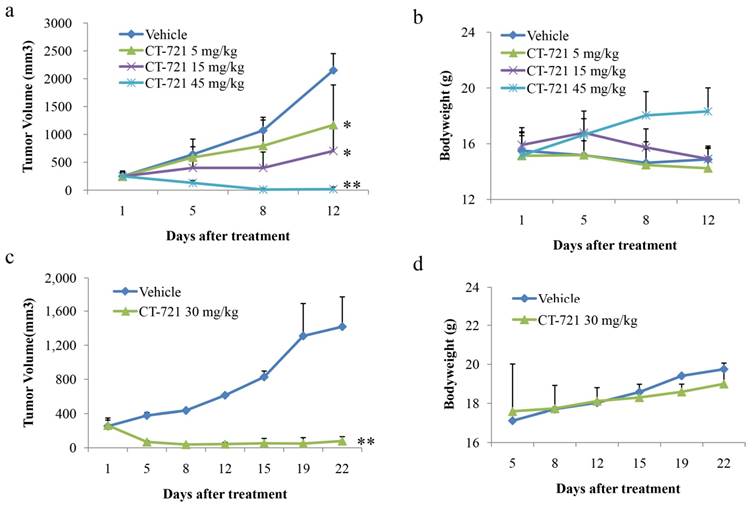
In another KU812 leukemia xenograft model, the anti-cancer efficacy of CT-721 was evaluated at the dosage of 30 mg/kg QD (Fig. 6c). In the treatment group, tumor growth was notably inhibited with partial regression observed after 5 days. After 8 days, complete tumor regression was observed in 50% of treated mice (p < 0.01). After 18 days, 80% of treated mice showed completely tumor regression and the others showed partial tumor regression (p < 0.01). Similar to studies in K562 xenograft, no significant body weight loss was observed in all treatment objects compared to the vehicle group.
PK/PD studies in K562 xenograft model
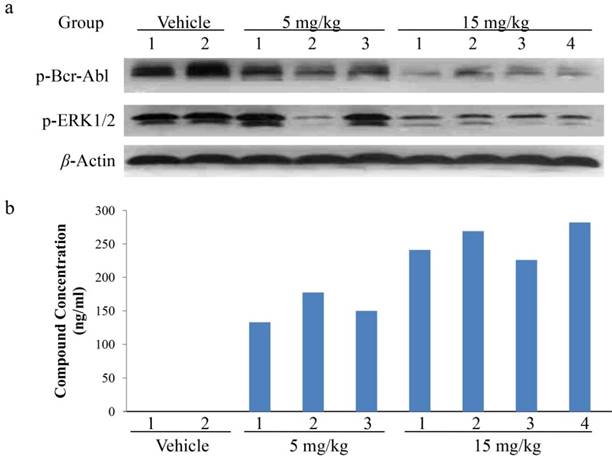
To further explore the relationship between the in vivo anti-tumor growth efficacy and the inhibition on cell signal transduction of abnormal Bcr-Abl-expressing, pharmacokinetics (PK)/pharmacodynamics (PD) studies were performed in K562 xenograft model. The tumor tissues were harvested 2 hours after a single dose of CT-721 at 5 or 15 mg/kg. CT-721 exposures in tumor tissue and the phosphorylation levels of Bcr-Abl and its down-stream signal target ERK were analyzed. Bcr-Abl phosphorylation levels in CT-721 treated groups were significantly decreased in a dose-dependent way (Fig. 7a). There existed well positively correlation between the concentration of CT-721 (Fig. 7b) and the inhibition of phosphorylation in both Bcr-Abl and ERK1/2 in tumor tissues. In the 15 mg/kg group, the compound concentration in tumor samples reached to 254 ng/mL, and almost complete inhibition of Bcr-Abl and ERK phosphrylation was observed. Collectively, these results have provided a direct relationship among compound exposures in tumor tissues, tumor growth inhibition, and the inhibition of phosphorylation of Bcr-Abl and its downstream targets.
PK/PD studies of CT-721 in K562 xenograft model. Xenograft tumor tissues were taken at 2 hours after CT-721 oral administration. (a) Tumor samples were analyzed by Western blots to detect phosphorylation of Bcr-Abl and ERK1/2 with specific antibodies. (b) CT-721 concentrations in tumor samples were quantitated by LC-MS/MS analysis.
Discussion
The Bcr-Abl fusion protein, central to the pathogenesis of CML, interacts with a variety of effector proteins, leading to deregulated cellular proliferation, decreased adherence of leukemia cells to the bone marrow stroma, and a reduced apoptotic response to mutagenic stimuli [22]. Imatinib, a Bcr-Abl selective tyrosine kinase inhibitor, is highly effective in inducing durable remission, prolonging the survival of the majority of CML patients in the chronic phase and improving the outcome in Ph+ ALL patients. The judicious use of tyrosine kinase inhibitors targeting Bcr-Abl has become a proven strategy in treatment of CML for sustained control of disease progression [6, 23]. However, the inhibitor-induced resistance and relapse develops at high incidence rate owe to mutation in Bcr-Abl kinase domain [24, 25]. All of clinically proved Bcr-Abl inhibitors are unable to inhibit T315I mutated Bcr-Abl except ponatinib, which was limited to patients who were responsive to the drug and/or had the potential benefits outweighing the risks due to adverse events of life-threatening blood clots in arteries and veins according to FDA Drug Safety Communication. Therefore, there is a need for development of new agents that can effectively treat patients with T315I mutant. We here reported that CT-721, a novel small molecular inhibitor for Bcr-Abl kinase, was able to efficiently inhibit both wild-type and T315I mutant Bcr-Abl kinase with comparable potency to Ponatinib [18], the most advanced Bcr-Abl inhibitor for leukemia treatment. More importantly, CT-721 has a more favorable PK and safety profile.
CT-721 was a potent ATP-competitive inhibitor against Bcr-Abl kinase, and it showed concentration-dependent and time-dependent inhibition on its target. In addition, CT-721 was demonstrated as a slow- and tight-binding inhibitor, which rendered it some main advantages, such as the high affinity to and the long residence time on its target. In agreement with its time-dependent and tight-binding characteristic, CT-721 showed persistent and sustained inhibition to Bcr-Abl phosphorylation and its downstream signaling molecules including Crk1 and Stat5 both in vitro and in vivo. CT-721 showed very potent and persistent inhibition on K562 and KU812 (highly-expressing Bcr-Abl fusion protein) leukemia cell growth with IC50 values around 1 nM, which was well associated with the time- and dose-dependent induction of cell apoptosis and cell cycle arrest. CT-721 sustainably prevented the phosphorylation of Bcr-Abl and its downstream protein Crk1 in a dose-dependent manner.
An important mechanism of drug resistance associated with tyrosine kinase inhibitors is the drugs-induced compensatory activation of cellular growth signal transduction pathways that bypass the inhibitory effects of drugs. Besides Bcr-Abl, CT-721 has demonstrated potent inhibition of other tyrosine kinases such as VEGFR2, PDGFRβ, which are equally important players in hematological malignancies and the potential bypass pathways for drug resistance. CT-721 also efficaciously blocked the phosphorylation of STAT5 and MAPK in K562 and KU812 cells. Overall, our studies have strongly indicated that CT-721 suppressed cell growth possibly through targeting Bcr-Abl and cross-talking of several Bcr-Abl-associated signaling pathways.
Pharmacokinetic studies demonstrated that CT-721 was metabolized slowly and had better oral exposures than Ponatinib [26]. CT-721 did not show significant inhibitory effect against liver CYP450 enzymes (IC50 > 10 μM, data not shown). In K562 and KU812 xenograft models, CT-721 was able to induce tumor regression, manifesting excellent tumor growth inhibition. The further in vivo PK/PD studies demonstrated that the inhibition of Bcr-Abl and ERK phosphorylation in xenograft tumors by CT-721 was well correlated with its concentrations in tumor tissues, indicating that the in vivo blockage of tumor growth by CT-721 was mechanism-based.
In conclusion, our studies showed that CT-721 was a potent and time-dependent inhibitor of wild-type and T315I mutant Brc-Abl kinases, induced apoptosis and suppressed tumor growth of leukemia cells harboring Bcr-Abl fusion. Moreover, CT-721 showed the long-acting efficacy in in vitro and in vivo studies. In particular, CT-721 had better pharmacokinetics parameters and toxicity profile than Ponatinib, thus it should be much better than Ponatinib in safety profile. Therefore, CT-721 has a potential application for treatment of CML as a best-in-class candidate.
Acknowledgements
This work was supported by the National Major Scientific and Technological Special Project for “Significant New Drugs Development” during the Twelfth Five-year Plan Periods (Foundation No. 2012ZX09401007) and Beijing Nova Program (Foundation No.xx2016035).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rowley JD. Chromosomal patterns in myelocytic leukemia. N Engl J Med. 1973;289:220-1
2. Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst. 1960;25:85-109
3. Quintas-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 2009;113:1619-30
4. Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824-30
5. Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990;247:1079-82
6. Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM. et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031-7
7. Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N. et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408-17
8. Hughes T, Deininger M, Hochhaus A, Branford S, Radich J, Kaeda J. et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006;108:28-37
9. O'Hare T, Eide CA, Deininger MW. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110:2242-9
10. Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A. et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129-41
11. Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K. et al. Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658-61
12. Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399-401
13. Kimura S, Naito H, Segawa H, Kuroda J, Yuasa T, Sato K. et al. NS-187, a potent and selective dual Bcr-Abl/Lyn tyrosine kinase inhibitor, is a novel agent for imatinib-resistant leukemia. Blood. 2005;106:3948-54
14. Puttini M, Coluccia AM, Boschelli F, Cleris L, Marchesi E, Donella-Deana A. et al. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006;66:11314-22
15. O'Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J. et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65:4500-5
16. Noronha G, Cao J, Chow CP, Dneprovskaia E, Fine RM, Hood J. et al. Inhibitors of ABL and the ABL-T315I mutation. Curr Top Med Chem. 2008;8:905-21
17. O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F. et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401-12
18. Huang WS, Metcalf CA, Sundaramoorthi R, Wang Y, Zou D, Thomas RM. et al. Discovery of 3-[2-(imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-abelson (BCR-ABL) kinase including the T315I gatekeeper mutant. J Med Chem. 2010;53:4701-19
19. O'Hare T, Pollock R, Stoffregen EP, Keats JA, Abdullah OM, Moseson EM. et al. Inhibition of wild-type and mutant Bcr-Abl by AP23464, a potent ATP-based oncogenic protein kinase inhibitor: implications for CML. Blood. 2004;104:2532-9
20. Eide CA, Adrian LT, Tyner JW, Mac Partlin M, Anderson DJ, Wise SC. et al. The ABL switch control inhibitor DCC-2036 is active against the chronic myeloid leukemia mutant BCR-ABLT315I and exhibits a narrow resistance profile. Cancer Res. 2011;71:3189-95
21. Chan WW, Wise SC, Kaufman MD, Ahn YM, Ensinger CL, Haack T. et al. Conformational control inhibition of the BCR-ABL1 tyrosine kinase, including the gatekeeper T315I mutant, by the switch-control inhibitor DCC-2036. Cancer Cell. 2011;19:556-68
22. Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343-56
23. Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM. et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038-42
24. Menon H. Issues in current management of chronic myeloid leukemia: Importance of molecular monitoring on long term outcome. South Asian J Cancer. 2013;2:38-43
25. Cortes J, Goldman JM, Hughes T. Current issues in chronic myeloid leukemia: monitoring, resistance, and functional cure. J Natl Compr Canc Netw. 2012;10(Suppl 3):S1-S13
26. Wehrle J, Pahl HL, von Bubnoff N. Ponatinib: a third-generation inhibitor for the treatment of CML. Recent Results Cancer Res. 2014;201:99-107
27. Hochhaus A, Reiter A, Skladny H, Melo JV, Sick C, Berger U. et al. A novel BCR-ABL fusion gene (e6a2) in a patient with Philadelphia chromosome-negative chronic myelogenous leukemia. Blood. 1996;88:2236-40
28. La Rosee P, Corbin AS, Stoffregen EP, Deininger MW, Druker BJ. Activity of the Bcr-Abl kinase inhibitor PD180970 against clinically relevant Bcr-Abl isoforms that cause resistance to imatinib mesylate (Gleevec, STI571). Cancer Res. 2002;62:7149-53
Author contact
![]() Corresponding author: ypengcom
Corresponding author: ypengcom