Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(14):2729-2739. doi:10.7150/jca.18396 This issue Cite
Research Paper
Discovering the miR-26a-5p Targetome in Prostate Cancer Cells
Milena Rizzo1, 2 ![]() , Gabriele Berti1, Francesco Russo3, 4, Sofia Fazio1, Monica Evangelista1, Romina D'Aurizio3, Marco Pellegrini3, Giuseppe Rainaldi1, 2
, Gabriele Berti1, Francesco Russo3, 4, Sofia Fazio1, Monica Evangelista1, Romina D'Aurizio3, Marco Pellegrini3, Giuseppe Rainaldi1, 2
1. Non-coding RNA Laboratory, Institute of Clinical Physiology (IFC), CNR, Pisa, Italy;
2. Tuscan Tumor Institute (ITT), Firenze, Italy;
3. Laboratory of Integrative Systems Medicine (LISM), Institute of Informatics and Telematics (IIT) and Institute of Clinical Physiology (IFC), CNR, Pisa, Italy;
4. Novo Nordisk Foundation Center for Protein Research, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark.
Received 2016-11-17; Accepted 2017-2-27; Published 2017-8-22
Abstract
Purpose. miR-26a-5p is a tumor suppressor (TS) miRNA often downregulated in several tumor tissues and tumor cell lines. In this work, we performed the re-expression of the miR-26a-5p in DU-145 prostate cancer cells to collect genes interacting with miR-26a-5p and analyzed their integration in the tumorigenesis related pathways.
Methods. The transfection of DU-145 prostate cancer cells with miR-26a-5p was done using nucleofection. The biological effects induced by miR-26a-5p re-expression were detected with routine assays for cell proliferation, cell cycle, survival, apoptosis and cell migration. The miRNA pull out technique was used to collect and next generation sequencing to identify the complete repertoire of the miR-26a-5p targets (miR-26a-5p/targetome). TargetScan 7, PITA and RNA22 were used to find the predicted miR-26a-5p targets in the miR-26a-5p/targetome. Gene set enrichment analysis were used to integrate target genes in KEGG pathways and Protein-Protein Interaction networks (PPINs) and modules were built.
Results. miR-26a-5p exerted an anti-proliferative effect acting at several levels, by decreasing survival and migration and inducing both cell cycle block and apoptosis. The analysis of the miR-26a-5p/targetome showed that 1423 (1352 coding and 71 non-coding) transcripts interacted with miR-26a-5p. Filtering the miR-26a-5p/targetome with prediction algorithms, 628 out of 1353 transcripts were miR-26a-5p predicted targets and 73 of them were already validated miR-26a-5p targets. Finally, miR-26a-5p targets were involved in 22 KEGG pathways and 20 significant protein-protein interaction modules
Conclusion. The TS-miR-26a-5p/targetome is a platform that shows both unknown and known miRNA/target interactions thus offering the possibility to validate genes and discover pathways in which these genes could be involved.
Keywords: re-expression of miR-26a-5p, DU-145 prostate cancer cells, miRNA pull out assay, next generation sequencing, miR-26a-5p/targetome, pathway in cancer.
Introduction
The miR-26a-5p is a tumor suppressor (TS) miRNA often downregulated in tumor tissues as well as in tumor cell lines [1-5]. In prostate cancer cells, the loss of the miR-26a-5p increased proliferation, migratory capacity and invasiveness [6]. Conversely, the re-expression of miR-26a-5p in the androgen-independent PC3 tumor cell line rescued the TS activity by directly repressing wnt5a and inhibiting cell proliferation [7]. Similarly, the miR-26a-5p re-expression in the PC3 cells repressed the LARP protein-1 by inhibiting the invasive capability of the cells [3] or blocking the G1/S transition by targeting the cell cycle regulating network [8]. Interestingly, the re-expression of miR-26a-5p (miRNA replacement) in a murine model of hepatocellular carcinoma reduced the tumor mass formation in the liver [9]. Altogether, these data strongly support the hypothesis that the tumor suppressor activity of miR-26a-5p is linked to its capability to repress genes involved in cancer. Since a single miRNA targets hundreds of genes simultaneously, it is conceivable that as more interactions miR-26a-5p/mRNAs are uncovered the easier it will be to identify the pathways affected by its tumor suppressor activity. In this work, we proposed a way to accelerate the 'knowledge' of miR-26a-5p/mRNAs interactions. Several protocols based on Ago immunoprecipitation (Ago-HITS-CLIP [10], Ago-PAR-CLIP [11], Ago-iPAR-CLIP [12], CLASH [13]) and the miRNA pull out assay [14] are appropriate to capture the miRNA/targets interactions. We exploited the miRNA pull out assay to capture and next generation sequencing (NGS) to identify the RNA transcripts specifically interacting with miR-26a-5p. We identified the transcripts specifically captured by miR-26a-5p (miR-26a-5p/targetome) and verified which of them were miR-26a-5p putative targets based on the prediction of specific algorithms. We also reported the procedures adopted to validate the miR-26a-5p/targetome and the bioinformatics analysis performed to highlight the pathways in which the target genes could be involved.
Materials and Methods
Cells and culture conditions
DU-145, 22Rv1, LNCaP prostate cancer cells were grew in RPMI Medium 1640 (EuroClone). PC3 prostate cancer cells grown in HAM's medium (Euroclone). The medium was added with 10% FBS (Fetal Bovine Serum, EuroClone), 1% penicillin/streptomycin (2mm, EuroClone) and 1% L-glutamine (2mM, Sigma-Aldrich). The cells were incubated at 37°C in a humidified atmosphere containing 5% CO2. The HCT116 Dicer-/- cell line, which grown in McCOY's medium (Euroclone), was used to perform the luciferase assay.
Transfection
Transient transfections of double stranded miRNAs mimics (miR-26a-5p) or negative control (miR-CT) (GenePharma) and siRNA pool against BIRC5 (siR-BIRC5) or negative control pool (siR-CT) (OriGen) of DU-145 cells were carried out using Amaxa Nucleofector®Device program 0-006 and Nucleofector®Solution L (Lonza). For each transfection, 1x106 of exponentially growing DU-145 cells were suspended in 100 µl of (solution L + supplement) mix and then the miRNA mimics/siRNA was added to a final concentration of 60 nM. After electroporation, 400 µl of RPMI medium were added and samples of 1x105 cells were seeded in culture dishes of 30 mm diameter (P30). The lipofectamine-mediated transfection was performed as following: 1.5x105 cells were seeded in P30 dishes and 48 hours after seeding cells were transfected with miRNA mimic/siRNA using 10µl of Lipofectamine 2000 (Thermo Fisher), according to the protocol provided by the manufacturer. The suspension of transfected cells was used for cellular and molecular assays.
Cell proliferation
1×105 cells were seeded in a series of 30 mm diameter dish and grown for 96 hours. At 24 hours intervals cells were collected and counted.
Cell cycle analysis
Cell cycle analysis was performed as following: 5x105 cells were fixed with 95% cold ethanol and marked with 300 µl of a solution 50 µl/ml propidium iodide (Sigma-Aldrich). After overnight incubation at 4°C, the cell cycle analysis was performed with Accuri C6 flow cytometer (BD Biosciences). Using specific software supplied with the instrument, the percentage of cells in each phase of the cell cycle was determined for each sample, considering the parameters SSC-H/FL2-A.
Survival assay
Cell survival was measured as following: cells were collected and seeded at cell density of 200 cells/60 mm diameter culture dish to allow colony formation. After 10-12 days, dishes were stained with 0.1 % CV and the ratio (number colonies/number seeded cells) was used to calculate the fraction of surviving cells.
Apoptosis assay
The apoptosis was measured as follows: 1x106 cells were suspended in 300 µl Binding Buffer 1X and left at room temperature for 15 minutes. Thereafter, cell labelling was done according to the kit Annexin V-FITC (Sigma). Cells were then passed through flow cytometer Accuri C6 (BD Bioscences) and analyzed using FL3-H/FL1-H parameters.
Cell migration assay
Cell migration was measured as follows: 2x105 cells were seeded on a plate using culture insert (IBIDI, Martinsried, Germany). The septum was removed when 80% of cell confluence was reached (0 hour), leaving the cells free to migrate according to their ability and to fill the empty space. The cells were observed with 10x and 20x objective lenses and images were captured using Leica DM IL LED microscope at 6, 9 and 13 hours after the removal of the septum and analyzed with ImageJ. The migratory rate of each sample was determined as the relative percentage of gap closure at 13 hour compared to 0 hour.
miRNA pull out assay
The miRNA pull out assay was performed modifying the protocol described by Orom et al. [14]. DU-145 cells were seeded into the wells of a 6-well plate at the density of 1.5 x105. After 24 hours after seeding, cells were transfected using Lipofectamine 2000 (Thermo Fisher) with 60nM of either miR-26a-5p duplex (ds-miR-26aCT) or a mix of 3' biotin-tagged miR-26a-5p 7tU (nucleotide 7 was a thiouridine) and miR-26a-5p 17tU duplexes (ds-miR-26aBIO). The day after transfection, the cells were washed with PBS and irradiated with UV (365nm, 2J/cm2) using the Bio-Link crosslinking (BLX) (Ambrose Lourmat) to induce cross-linking of tU nucleotides to RNA. Total RNA was extracted adding TRIzol reagent (Thermo Fisher) directly on adherent cells and following the instructions provided by the manufacturer. After DNase treatment, 15 μg of RNA was incubated for 4 hours at 4°C with 100 μl of streptavidin-conjugated beads (200 μl of Streptavidin Sepharose high performance, GE Healthcare) previously suspended in PO buffer (1M Tris pH8, 5M NaCl, 1M MgCl2, NP40 50 μl in 100 ml buffer). After 2 washes with PO buffer and 2 washes with DEPC-treated water, the RNA complexed with beads was recovered by adding 1 ml Trizol directly on the beads followed by the TRIzol RNA extraction protocol. We performed two biological replicates obtaining two miR-26aCT (control) and two miR-26aBIO (miR-26a-5p) pull out samples
Target Identification by Next Generation Sequencings (NGS)
The RNA isolated after the miRNA pullout procedure from both miR-26aCT and miR-26aBIO samples was used for the construction of the cDNA libraries using the TruSeq Stranded Total RNA Sample Preparation kit (Illumina) according to the manufacturer's suggestions. cDNA libraries were sequenced using the HiSeq2000 (Illumina) in single-reads mode (50bp) by IGA Technology Service, Udine, Italy, obtaining about 20 million reads for each sample.
NGS data analysis
NGS data (GEO accession number GSE85306) were analyzed using the Galaxy platform (https://usegalaxy.org/). Briefly, after quality control analysis (using FastQC) we made use of TopHat2 to align the reads on the human genome (version hg38). Then, the Cufflinks suite of tools was used to assemble transcripts and estimate their abundances. The final transcriptome assembly was obtained by using Cuffmerge. The differential expression analysis was performed using Cuffdiff. The differentially expressed transcripts with q-value < 0.05 were selected for downstream analyses.
Bioinformatics analyses
The available normalized log2 miRNA expression data of PCa patients were retrieved from cBioPortal (http://www.cbioportal.org/; GEO accession number: GSE21032). This dataset consists of 99 primary and 14 metastatic tumor tissues and 27 normal controls.
In order to identify the miR-26a-5p predicted targets in the miR-26a-5p/targetome we used the following prediction algorithms: TargetScan 7 [15], PITA [16] and RNA22 [17]. The analyses were performed using the script versions of these programs. We considered for PITA and RNA22 a maximum of one mismatch and one G:U in the seed match. Moreover, RNA22 returned also a p-value and the folding energy for each miRNA-target interaction. We selected as cut-off a folding energy <= -10 and a p-value <= 0.05. For PITA we selected a score (i.e. the ddG score based on the folding energy) <= -10. A transcript was considered as putative target if at least one target prediction algorithm predicted the interaction. For these analyses, we predicted the miRNA binding sites in the 3'-UTR retrieving the sequences by UCSC Table Browser (http://genome.ucsc.edu/).
The Gene Set Enrichment Analysis was performed using the GSEA online tool developed by the Broad Institute (http://software.broadinstitute.org/gsea/msigdb/index.jsp) using the MSigDB (the Molecular Signatures Database) as annotated gene sets. Significant terms (e.g. pathways) were considered using q < 0.05.
The Protein-Protein Interaction Network (PPIN) was constructed using the StringApp, a Cytoscape app (http://www.cytoscape.org/) that is able to query the String database [18], a protein-protein interaction database. A medium confidence score of 0.4 was used as minimum required interaction score. All the disconnected nodes in the network were removed, maintaining only the proteins with at least one interaction.
The ClusterOne algorithm implemented in Cytoscape was used to find functional modules. The default parameters were used considering a minimum size of three proteins and the String confidence score was used as edge weights for the clustering.
Quantification of miRNAs and mRNAs (qRT-PCR)
Total RNA was extracted from 1x106 cells using the miRNeasy mini kit following the manufacturer's recommendations. 1µg of total RNA were retro transcribed using either the miScript II RT kit (Qiagen) or the QuantiTect reverse Transcription kit (Qiagen) for the miRNA or the mRNA quantification, respectively. The reverse transcription was made following the instructions given by the manufacturer. MiRNAs and the mRNAs were quantified with Rotor-Gene Q 2plex (Qiagen), using the miScript SYBR Green PCR Kit (Qiagen) or the SsoAdvanced ™ SYBR ® Green Supermix (Bio-Rad) respectively, according to the protocol indicated by the manufacturer. The relative quantification was performed using the Rotor-Gene Q Software, normalizing to the internal controls (U6, SNORD55 and SNORD110 for miRNA and GAPDH, ACTB and HPRT for mRNA). The relative miR-26a-5p expression level in PCa tumor cell lines was evaluated respect to the normal prostate cells RNA (FirstChoice human prostate total RNA, Ambion). All reactions were performed in triplicate and the results are the mean of three biological replicates.
Western Blot analysis
Proteins were extracted from cell pellets with Lysis Buffer (1 M Tris HCl pH 8, Triton X 100 1%, Na deoxycholate 0.25%) with the addition of PMSF 1mM. The proteins were then quantified with colorimetric method using the Biorad protein Assay Reagent (Biorad). The absorbance was measured at 595 nm with ChroMate microplate reader (Awareness Technology). The proteins were separated on polyacrylamide gels SDS-PAGE (10% and 4-15 %, gel precast MINI-PROTEAN ® TGX Stain-FreeTM, BIO-RAD), and transferred to 0.2 μm nitrocellulose membranes by electro blotting using the Trans-Blot Turbo Blotting System (BIO-RAD). The resulting blots were blocked with 5% nonfat dry milk or BSA solution in TBST. Anti GAPDH (Cell Signaling) (1:10000) and anti BIRC5 (Santa Cruz Biotechnology) (1:100) primary antibodies were used. Incubation was performed overnight at 4°C and bands were revealed after incubation with the recommended secondary antibody coupled to peroxidase using ECL (GE Healthcare). Scanned images were quantified using ImageJ software and normalized to GAPDH.
Luciferase Reporter Assay
To verify the interaction of miR-26a-5p with BIRC5 gene, the sequence of the 3'UTR-BIRC5 was cloned downstream the luciferase coding sequence into the pmiR vector (pMIR-Report Luciferase, Ambion). HCT116 Dicer-/- colon cancer cells were seeded at density of 1,5x105 cells /well in a 12 well multiplate. The day after, cells were co-transfected using the Lipofectamine 2000 (Thermo Fisher) according to the manufacturer's instructions, with 150 ng of pmiR-BIRC5 3'UTR, 100 ng of pRL control vector-TK (Renilla, Promega) and with 60 nM of ds-miR-26a-5p. The luciferase assay was performed 24 hours after transfection, using Dual-Luciferase Reporter Assay System (Promega) and luminescence was evaluated by the luminometer GloMax-Multi Detection System, Promega). The pRL-TK (Renilla) was used to normalize the luminescence arising from pmiR-BIRC5 3' UTR construct.
Statistical Analyses
Results are expressed as mean SD of at least three independent experiments and data analysed by Student's t-test (*P<0.05, **P<0.01, ***P<0.001).
Results
The re-expression of miR-26a-5p in prostate cancer cells
A characteristic of TS-miRNAs is their under expression in either tumor tissues or tumor cell lines. Using the miRNA expression data from PCa patients of the cBioPortal database, we evaluated the miR-26a-5p expression in tumor tissues of patients with either primary or metastatic PCa. We found that miR-26a-5p tended to be under expressed in primary PCa and even more in metastatic PCa (Figure 1A), suggesting a positive relationship between the under expression and PCa tumor development. These data are in line with the recent literature in which the miR-26a-5p reduced expression was associated with metastasis and poor overall prognosis in PCa [6, 19].
Relative quantification of miR-26a-5p expression level in tumor cell lines. (A) Analysis of the miR-26a-5p expression level data retrieved from the cBioPortal database. (B) qRT-PCR of miR-26a-5p in prostate (PC3, DU-145, VCaP, 22Rv1) cancer cell lines respect to the normal prostate cells RNA. Data are reported as mean of at least three independent experiments (*P<0.05, **P<0.01, ***P<0.001, unpaired t-test).
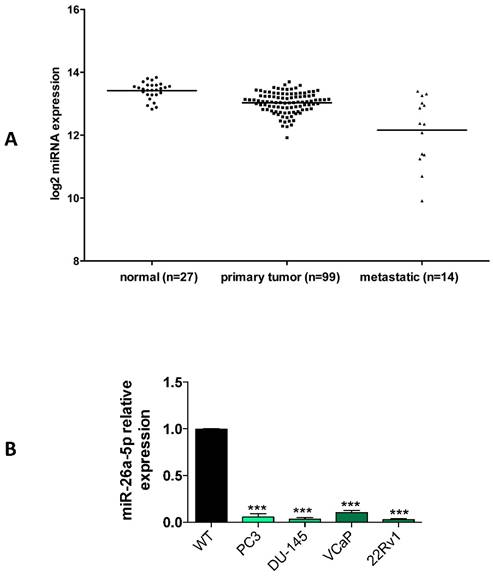
We then quantified the expression of miR-26a-5p in both androgen independent (DU-145, PC3) and androgen dependent (22Rv1 and VCaP) prostate cancer (PCa) cell lines. Data clearly demonstrated that the miR-26a-5p was downregulated in all the analyzed tumor cell lines (Figure 1B).
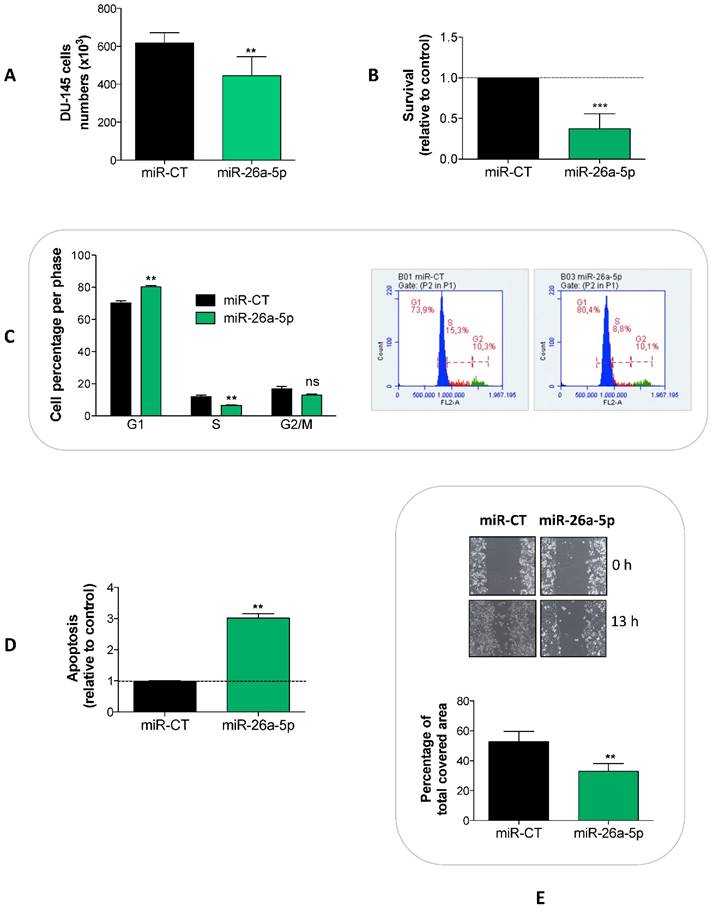
To detect the effects induced by the miR-26a-5p re-expression, we transfected miR-26a-5p in DU-145 cells and analyzed a series of cell proliferation-related events. We showed that the miR-26a-5p exerted an anti-proliferative effect (Figure 2A) acting at several levels, by decreasing survival (Figure 2B) and inducing both cell cycle block (Figure 2C) and apoptosis (Figure 2D). Moreover, the miR-26a-5p re-expression impaired the capability of tumor cell to migrate (Figure 2E).
Effects of miR-26a-5p re-expression. The re-expression of miR-26a-5p reduced cell proliferation (A), decreased survival (B), induced a cell cycle block (C) and apoptosis (D) and impaired the cell migration (E). Data are reported as mean of at least three independent experiments (*P<0.05, **P<0.01, ***P<0.001, unpaired t-test).
Identification and validation of the miR-26a-5p/targetome
Since a single miRNA targets hundreds of mRNAs, we searched for the genes which were affected by miR-26a-5p re-expression. We transfected DU-145 cells with miR-26a-5p and then performed the miRNA pull out protocol [14, 20, 21] adequately revised. The repertoire of the mRNAs linked with miR-26a-5p was collected and then the transcripts of the miR-26a-5p pull out sample identified using NGS. The miR-26a-5p/targetome consisting of 1423 transcripts (1352 protein coding and 71 non-coding RNA) was obtained (Supplementary Table S1). To evaluate whether the miR-26a-5p/targetome contained miR-26a-5p targets, the 1352 protein coding transcripts were examined using three different miRNA-target prediction algorithms (TargetScan 7, PITA and RNA22). We found that 628 out of 1352 transcripts (46%) were miR-26a-5p predicted targets (Supplementary Table S2) and 73 of them were also miR-26a-5p validated targets (based on miRTarbase, the database of experimentally validated miRNA targets [22] closure (Supplementary Table S3). These results suggested that the miRNA pullout assay was a fruitful method to capture reliable mRNA targets. Finally, we ranked the 628 transcripts according to the fold change of enrichment and 9 transcripts with different fold change value were selected. The qRT-PCRs confirmed the NGS expression data (Figure 3A) and this led to be confident about the reliability of the miR-26a-5p targets identification.
miR-26a-5p/targetome analysis. (A) Relative quantification with qRT-PCR of selected transcripts of miR-26a-5p/targetome in miR-26a-5p versus control pull out samples. Data are reported as mean of at least three independent experiments (*P<0.05, **P<0.01, ***P<0.001, unpaired t-test). (B) Result of the GSEA tool reporting the most significant KEGG pathways. In the Y axis we report each pathway with the q-value and in the X axis we report the number of genes in each pathway.
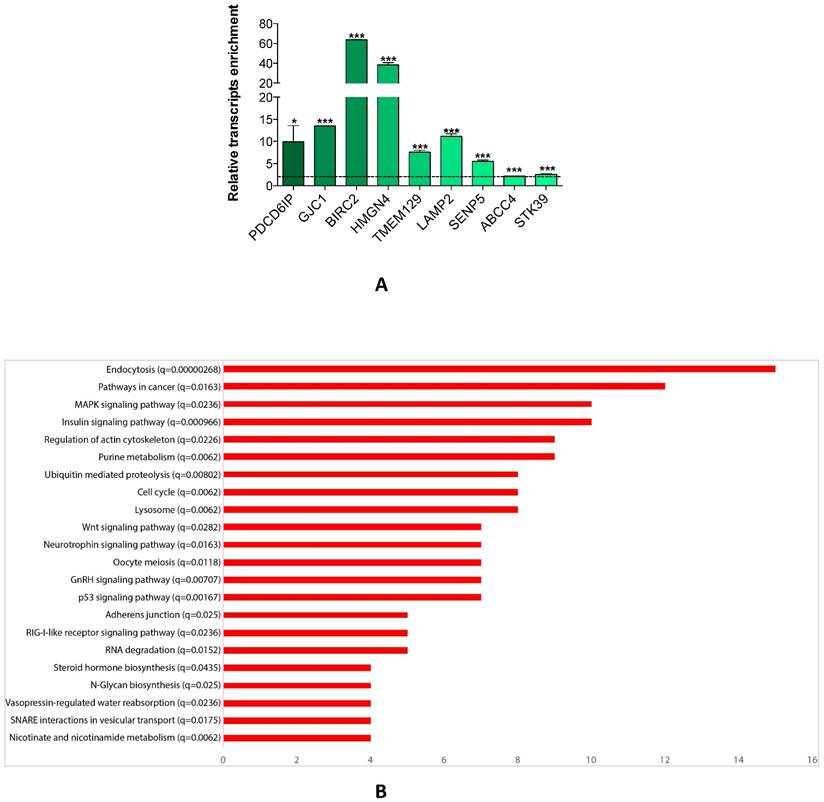
The miR-26a-5p regulatory networks
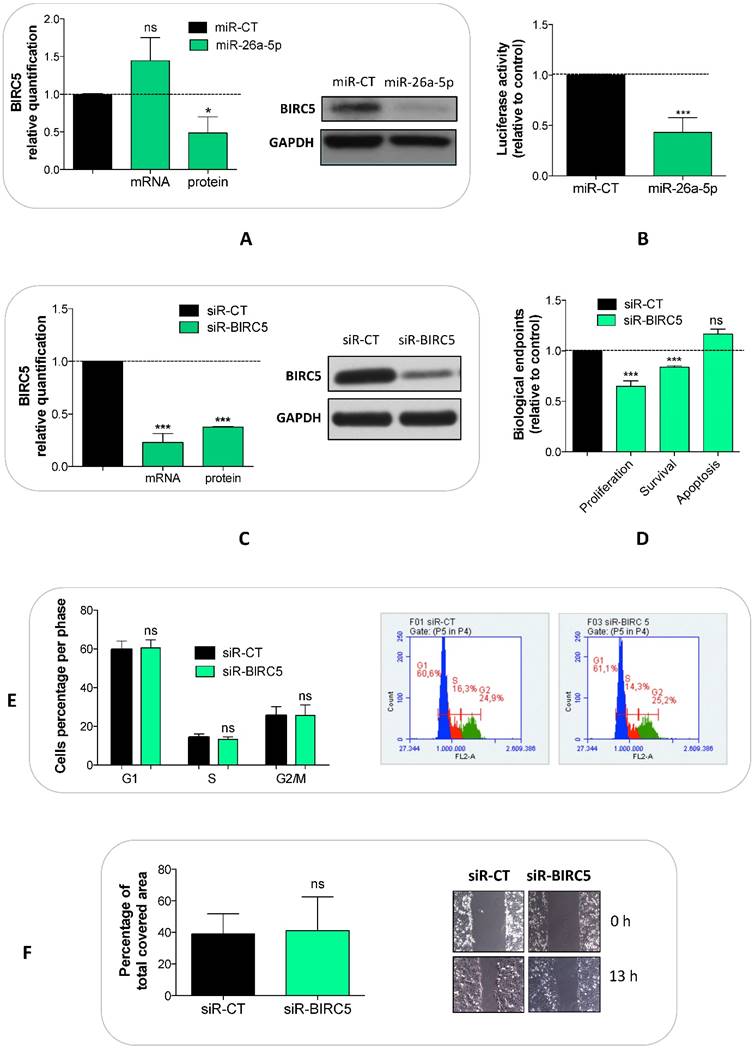
We explored whether the miR-26a-5p/targetome represents a useful platform to discover the miR-26a-5p-dependent gene regulation networks. We used the GSEA tool on the 628 miR-26a-5p targets and 22 significantly enriched pathways (q<0.05) were identified (Figure 3B). The majority of the pathways identified with this method were biological processes fundamental for cancer development (e.g. cell cycle) or pathways strictly correlated with cancer biology (e.g. MAPK, p53 and wnt signaling). Excluding endocytosis, the pathway in which the highest number of miR-26a-5p targets were involved was the “Pathway in cancer”. Among these targets, there were EGFR and BIRC5 (Supplementary Table S4), which are involved in several aspects of cancer biology [23, 24]. Although the validation of the pathways was outside of the aims of this work, we made an attempt in this direction by investigating the role played by BIRC5 (survivin) in prostate tumor cells. We verified that BIRC5 was downregulated (only at post-transcriptional level) in DU-145 cells transfected with miR-26a-5p (Figure 4A) and then demonstrated that BIRC5 was a miR-26a-5p direct target (Figure 4B). We then investigated if BIRC5 per se had a role in prostate cancer cells. We silenced BIRC5 in DU-145 (Figure 4C) and we observed the reduction of cell proliferation and cell survival (Figure 4D) whereas apoptosis (Figure 4D), cell cycle (Figure 4E) and cell migration (Figure 4F) were unmodified.
Validation of miR-26a-5p/BIRC5 interaction and BIRC5 function in prostate cancer. Relative quantification of BIRC5 expression level at both transcriptional (by qRT-PCR) and translational (by western blot) levels after BIRC5 silencing (A) and miR-26a-5p overexpression (C) in DU-145 cells. (B) Relative Luciferase activity after the cotransfection of pmiR-BIRC5 3' UTR and either miR-26a-5p or miR-CT. Cell proliferation, survival, apoptosis (D), cell cycle (E) and cell migration (F) in DU-145 cells after BIRC5 silencing. Data are reported as mean of at least three independent experiments (*P<0.05, **P<0.01, ***P<0.001, unpaired t-test).
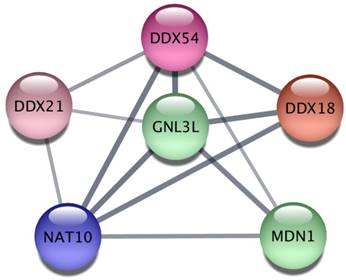
Finally, we explored the Protein-Protein Interaction Network (PPIN) of the 628 miR-26a-5p targets and then extracted subnetworks of highly connected proteins in order to understand whether miR-26a-5p was able to regulate functional modules. We obtained a large network of 450 proteins and 956 interactions where at least one protein was connected to another one (Supplementary Table S5). Then, using a clustering algorithm we extracted 20 significant modules (p<0.05, Table 1), suggesting that miR-26a-5p can interact with several targets within the same module regulating multiple functions. The obtained modules involved proteins that may play a role in cancer. In particular, the most significant module (p=0.003) included six proteins (Figure 5) some of which have already been studied in the context of tumor biology. For instance, DDX, an RNA helicase whose family is often deregulated in cancer [25] and GNL3L, which is involved in cell cycle regulation [26] and overexpressed in tumor cells that exhibit markers and tumorigenic properties of the tumor initiating cells (TICs) [27].
Network analysis of miR-26a-5p targets.
| Clusters | Number of proteins | Proteins in modules | p-value |
|---|---|---|---|
| C1 | 6 | DDX21, DDX54, MDN1, GNL3L, NAT10, DDX18 | 0.002537434 |
| C2 | 7 | DDX23, HNRNPA3, SRSF11, NUP214, CPSF2, ZC3H4, NUP210 | 0.00364518 |
| C3 | 11 | CARM1, TBL1XR1, ABCA1, SREBF2, CEBPA, CROT, SOAT1, MED20, MED29, MED28, MED6 | 0.005176176 |
| C4 | 9 | RAB3IP, STX6, RAB3B, SEPT11, STX2, RAB35, RAB3D, STX3, YKT6 | 0.008559119 |
| C5 | 5 | AGPAT6, AGPAT3, MBOAT1, PISD, ADPRM | 0.010785874 |
| C6 | 9 | RAB3IP, STX6, RAB3B, STX2, RAB35, RAB3D, STX3, YKT6, TXLNA | 0.010808197 |
| C7 | 9 | DDX23, HNRNPA3, SRSF11, NUP214, CPSF2, HDAC4, IFRD1, ZC3H4, NUP210 | 0.013479035 |
| C8 | 4 | GDAP1, MFN2, ZCCHC11, SMCR7L | 0.014700525 |
| C9 | 4 | EXOSC6, EXOSC2, NAA38, PATL1 | 0.014700525 |
| C10 | 4 | GALNT7, B3GNT2, B4GALT1, SLC35D1 | 0.015191412 |
| C11 | 5 | TMEM43, WNK3, SLC12A4, STK39, DSC2 | 0.018356928 |
| C12 | 4 | CYLD, TRIM25, DDX58, TRIM56 | 0.027257239 |
| C13 | 4 | WNK3, SLC12A4, FAM120C, STK39 | 0.030300985 |
| C14 | 3 | C19orf54, ZDHHC8, SLC25A30 | 0.031801285 |
| C15 | 6 | TAB2, CYLD, TRIM25, DDX58, BIRC2, TRAF5 | 0.03277608 |
| C16 | 3 | ARSJ, ARSI, ARSA | 0.03826125 |
| C17 | 3 | RAB11FIP5, EHD1, RAB11FIP1 | 0.03826125 |
| C18 | 3 | MTHFR, AHCYL2, ENO2 | 0.040427799 |
| C19 | 5 | CELSR2, VANGL2, RYK, CELSR1, WNT7B | 0.04684631 |
| C20 | 12 | INPP5B, ARHGEF7, PARD6B, CYFIP1, RND2, PLEKHG2, RHOBTB3, PLEKHG4B, RHOQ, OPHN1, CDC42EP3, RHOB | 0.049791258 |
Protein-protein interaction analysis. The most significant cluster obtained by using the ClusterOne algorithm on the Protein-Protein Interaction network of miR-26a-5p targets.
Discussion
MiRNAs are involved in a variety of biological processes and their deregulation may be associated with the genesis of various diseases, including cancer [28]. Many studies have shown that some miRNAs, defined onco-miRNAs, inhibit TS gene expression while other miRNAs, defined TS-miRNAs, are capable to inhibit oncogenes. TS-miRNAs are often under-expressed in tumors [29] thus presuming a causal association between the under expression of TS-miRNAs and tumor development. The current interest in studying the replacement of TS-miRNA derives from two observations: miRNAs are globally under-expressed in cancer [30] and the replacement of TS-miRNAs to physiological levels was evaluated as an effective therapeutic approach in vivo [31]. The identification of the genes affected by the re-expression of miR-26a-5p could allow deciphering the TS activity of miR-26a-5p. We have isolated miR-26a-5p targets using the miRNA pull out assay, a technique that, unlike other computational and experimental approaches, allowed the isolation of mRNA targets of a specific miRNA in a determined cellular context. The expression profiles of the captured transcripts performed with NGS technology allowed the identification of 1423 differentially expressed transcripts, 1352 transcripts coding and 71 transcripts non-coding sequences. Most of the 71 non-coding transcripts belong to categories of RNA with known regulatory function (lincRNAs, pseudogenes, antisense RNAs and miRNA precursors). In addition, for some of them an involvement in the tumor biology has been already demonstrated: e.g. SNHG16 (Small Nucleolar Host RNA Gene 16) is a long non-coding RNA that has been associated with the development and the invasiveness of bladder cancer [5]. This observation suggests that the regulation of the expression of the non-coding RNAs may play a role in the anti-proliferative activity of miR-26a-5p. Therefore, the identified non-coding RNAs may represent novel candidates suitable for investigation as molecules with a potential role in cancer development.
We used three algorithms based on different approaches to evaluate the enrichment of the coding portion of the miR-26a-5p/targetome of miR-26a-5p predicted targets. Particularly, we selected TargetScan 7, PITA and RNA22 to take into consideration both canonical and non-canonical miRNA/targets interaction because a large number of miRNAs interact with their targets through this type of interaction [13]. The result of our analysis was that about 46% of the identified transcripts were miR-26a-5p predicted targets indicating that the miR-26a-5p/targetome was significantly enriched of miR-26a-5p targets.
We also showed that the predicted targets were part of pathways relevant to cancer. Using GSEA we identified 22 KEGG pathways, the majority of which are fundamental or directly associated with cancer progression and/or biology. This result underlines the fact that the availability of miR-26a-5p/targetome is a useful platform to discover the gene networks that play a relevant role in the miRNAs function. For instance, among the KEGG pathways comprising the largest number of targets, we selected “Pathway in cancer”. The genes belonging to this pathway would be relevant for tumorigenesis and, as proof of principle, we focused on BIRC5, a multifunctional protein that participates in the cell cycle control, apoptosis and response to DNA damage in tumor cells [24, 32]. Data showing that the silencing of BIRC5 causes a decrease of cell proliferation both in vitro and in vivo have been also reported [33, 34]. Moreover, high BIRC5 expression is related to a higher Gleason score [35] as well as with clinic-pathological features of aggressive PCa [36, 37].
We demonstrated that miR-26a-5p directly regulated BIRC5 expression in DU-145 cells and that the BIRC5 silencing reduced cell proliferation and survival without affecting cell cycle, apoptosis and migration. As BIRC5 alone influenced only some of the biological effects induced by miR-26a-5p, the validation of more elements of each pathway rather than a single gene must be pursued.
Finally, by using the Protein-Protein Interaction Networks we discovered that a large number of the miR-26a-5p targets coding for proteins functional connected each other, suggesting that miR-26a-5p regulation is mediated by the repression of more than one gene involved in the same process. It is of note that some members of the most significant modules of functional related proteins have been already associated to cancer [25, 27].
In conclusion, we demonstrated that the re-expression of miR-26a-5p in DU-145 prostate tumor cells reduced the proliferation indicating that miR-26a-5p tumor suppressor activity was restored. Moreover, we showed that the analysis of the transcripts interacting with miR-26a-5p (miR-26a-5p/targetome) with specific bioinformatics tools allowed the identification of genes and gene expression networks regulated by miR-26a-5p. In particular, the most relevant output of this work was the identification of several genes and pathways affected by miR-26a-5p regulation with a possible role in PCa. Therefore, an increasing availability of miRNA-specific and tumor-specific targetomes will increase the probability to find out genes and pathways related to cancer development/progression regulated by miRNAs. In the future, we will investigate more in detail the effect of miR-26a-5p regulation on pathway/s relevant for cancer and in which more than one miR-26a-5p targets are involved both in vitro and in a xenograft model. Moreover, to evaluate the clinical relevance of the above-mentioned target/s in PCa, we will investigate their expression, together with miR-26a-5p expression, in biopsy and normal tissues from prostate cancer patients with primary or metastatic tumor, as well as their possible correlation with parameters of tumor aggressiveness.
Supplementary Material
Supplementary tables.
Acknowledgements
We would like to thank Dr. Lorena Tedeschi (Institute of Clinical Physiology, CNR, Pisa) for giving us siRNA pool against BIRC5 and for technical suggestions.
This work was supported by Istituto Toscano Tumori (grant 2010, Giuseppe Rainaldi; grant 2013, Milena Rizzo).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen J, Zhang K, Xu Y, Gao Y, Li C, Wang R. et al. The role of microRNA-26a in human cancer progression and clinical application. Tumour Biol. 2016;37:7095-108
2. Fukumoto I, Hanazawa T, Kinoshita T, Kikkawa N, Koshizuka K, Goto Y. et al. MicroRNA expression signature of oral squamous cell carcinoma: functional role of microRNA-26a/b in the modulation of novel cancer pathways. British journal of cancer. 2015;112:891-900
3. Kato M, Goto Y, Matsushita R, Kurozumi A, Fukumoto I, Nishikawa R. et al. MicroRNA-26a/b directly regulate La-related protein 1 and inhibit cancer cell invasion in prostate cancer. Int J Oncol. 2015;47:710-8
4. Lin Y, Chen H, Hu Z, Mao Y, Xu X, Zhu Y. et al. miR-26a inhibits proliferation and motility in bladder cancer by targeting HMGA1. FEBS Lett. 2013;587:2467-73
5. Zhu Y, Yu M, Li Z, Kong C, Bi J, Li J. et al. ncRAN, a newly identified long noncoding RNA, enhances human bladder tumor growth, invasion, and survival. Urology. 2011;77:510 e1-5
6. Guo K, Zheng S, Xu Y, Xu A, Chen B, Wen Y. Loss of miR-26a-5p promotes proliferation, migration, and invasion in prostate cancer through negatively regulating SERBP1. Tumour Biol. 2016;37:12843-54
7. Zhao S, Ye X, Xiao L, Lian X, Feng Y, Li F. et al. MiR-26a inhibits prostate cancer progression by repression of Wnt5a. Tumour Biol. 2014;35:9725-33
8. Erdmann K, Kaulke K, Rieger C, Salomo K, Wirth MP, Fuessel S. MiR-26a and miR-138 block the G1/S transition by targeting the cell cycle regulating network in prostate cancer cells. Journal of cancer research and clinical oncology. 2016;142:2249-61
9. Kota J, Chivukula RR, O'Donnell KA, Wentzel EA, Montgomery CL, Hwang HW. et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005-17
10. Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479-86
11. Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P. et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129-41
12. Jungkamp AC, Stoeckius M, Mecenas D, Grun D, Mastrobuoni G, Kempa S. et al. In vivo and transcriptome-wide identification of RNA binding protein target sites. Mol Cell. 2011;44:828-40
13. Helwak A, Kudla G, Dudnakova T, Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153:654-65
14. Orom UA, Lund AH. Isolation of microRNA targets using biotinylated synthetic microRNAs. Methods. 2007;43:162-5
15. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015:4
16. Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39:1278-84
17. Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM. et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203-17
18. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447-52
19. Zhang J, Liang J, Huang J. Downregulated microRNA-26a modulates prostate cancer cell proliferation and apoptosis by targeting COX-2. Oncol Lett. 2016;12:3397-402
20. Orom UA, Nielsen FC, Lund AH. MicroRNA-10a binds the 5'UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell. 2008;30:460-71
21. Patella F, Leucci E, Evangelista M, Parker B, Wen J, Mercatanti A. et al. MiR-492 impairs the angiogenic potential of endothelial cells. J Cell Mol Med. 2013;17:1006-15
22. Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH. et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016;44:D239-47
23. Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR. et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2-16
24. Altieri DC. Targeting survivin in cancer. Cancer Lett. 2013;332:225-8
25. Abdelhaleem M. Over-expression of RNA helicases in cancer. Anticancer Res. 2004;24:3951-3
26. Thoompumkal IJ, Subba Rao MR, Kumaraswamy A, Krishnan R, Mahalingam S. GNL3L Is a Nucleo-Cytoplasmic Shuttling Protein: Role in Cell Cycle Regulation. PLoS One. 2015;10:e0135845
27. Okamoto N, Yasukawa M, Nguyen C, Kasim V, Maida Y, Possemato R. et al. Maintenance of tumor initiating cells of defined genetic composition by nucleostemin. Proc Natl Acad Sci U S A. 2011;108:20388-93
28. Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med. 2009;60:167-79
29. Lujambio A, Lowe SW. The microcosmos of cancer. Nature. 2012;482:347-55
30. Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20:460-9
31. Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov. 2013;12:847-65
32. Wang H, Yang M, Xu J, Zou B, Zhou Q, Bian J. et al. Survivin mRNA-circulating tumor cells are associated with prostate cancer metastasis. Tumour Biol. 2016;37:723-7
33. Cai Y, Ma W, Huang X, Cao L, Li H, Jiang Y. et al. Effect of survivin on tumor growth of colorectal cancer in vivo. Int J Clin Exp Pathol. 2015;8:13267-72
34. Zhang J, Wang S, Han F, Li J, Yu L, Zhou P. et al. MicroRNA-542-3p suppresses cellular proliferation of bladder cancer cells through post-transcriptionally regulating survivin. Gene. 2016;579:146-52
35. Eslami M, Khamechian T, Mazoochi T, Ehteram H, Sehat M, Alizargar J. Evaluation of survivin expression in prostate specimens of patients with prostate adenocarcinoma and benign prostate hyperplasia underwent transurethral resection of the prostate or prostatectomy. Springerplus. 2016;5:621
36. Danilewicz M, Stasikowska-Kanicka O, Wagrowska-Danilewicz M. Augmented immunoexpression of survivin correlates with parameters of aggressiveness in prostate cancer. Pol J Pathol. 2015;66:44-8
37. Mathieu R, Lucca I, Vartolomei MD, A MB, Klatte T, Seitz C. et al. Role of Survivin expression in predicting biochemical recurrence after radical prostatectomy: a multi-institutional study. BJU Int. 2017;119:234-8
Author contact
![]() Corresponding author: Milena Rizzo, PhD, Institute of Clinical Physiology (IFC), CNR, Via Moruzzi 1, 56124, Pisa, Italy Email: milena.rizzocnr.it
Corresponding author: Milena Rizzo, PhD, Institute of Clinical Physiology (IFC), CNR, Via Moruzzi 1, 56124, Pisa, Italy Email: milena.rizzocnr.it