Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(10):1809-1817. doi:10.7150/jca.16486 This issue Cite
Research Paper
HIF is not essential for suppression of experimental tumor growth by mTOR inhibition
Karl X. Knaup ![]() , Regina Guenther, Johanna Stoeckert, Juliana M. Monti, Kai-Uwe Eckardt, Michael S. Wiesener
, Regina Guenther, Johanna Stoeckert, Juliana M. Monti, Kai-Uwe Eckardt, Michael S. Wiesener
Department of Nephrology and Hypertension, Friedrich-Alexander-Universität Erlangen-Nürnberg, 91054 Erlangen, Germany.
Received 2016-6-15; Accepted 2017-3-23; Published 2017-7-1
Abstract
The Hypoxia Inducible Transcription Factor (HIF) is the master regulator of cellular response to hypoxic adaptation. Solid tumors inevitably harbour hypoxic regions with subsequent stabilization and activation of HIF and HIF target genes due to poor vascularization and rapid growth. The mammalian target of rapamycin (mTOR) is a global regulator of cellular growth and proliferation, which can also regulate HIF expression independantly of hypoxia via specific activation of cellular translation and transcription. An effective blockade of mTOR results in attenuation of HIF under hypoxic conditions in vitro. This mechanism could enable a simultaneous inhibition of both the mTOR- and the HIF-pathway, resulting in an effective tool for cancer targeting. We set out to analyze the effect of mTOR inhibition and the involvement of mTOR regulation on HIF in vivo in a subcutaneous xenograft model in nude mice. Our results demonstrate that mTOR inhibition in our model leads to a clear reduction in tumor growth of various cellular origins, most likely due to inhibition of cellular proliferation. Moreover, these effects can also be achieved independently of the HIF status of the tumor cells. The HIF levels per se seem to remain unaffected by mTOR inhibition, probably due to the profound hypoxic environment in these threedimensional structures, consequently leading to a strong HIF stabillization. Therefore, treatment of these experimental tumors with mTOR inhibitors is an effective tool to achieve size regression. The involvement of and the effect on HIF in this in vivo setting is nevertheless negligible.
Keywords: cancer, hypoxia, immunosuppression, mTOR, HIF, xenograft
Introduction
Cancer is one of the leading causes of death worldwide, resulting in 8.2 million deaths in the year 2012 (WHO, World Cancer Report 2014). Defining individual pharmacological therapies for the wide variety of cancer is one of the most broadly investigated and competitive fields.
The mammalian target of rapamycin (mTOR) is one therapeutic target which has moved into the center of attention over the last years in tumor treatment.1, 2 Functionally it is a Ser/Thr kinase which integrates mitogenic signals and nutritional status and regulates important cellular functions such as cellular growth, proliferation, cell cycle progression and transcriptional and translational processes.3, 4 The best defined mTOR-downstream effectors are the p70S6Kinase (p70S6K) and the eukaryotic initiation Factor 4E binding protein (eIF4E-BP). In many cancers the mTOR pathway is constitutively active due to mutation of negative upstream regulators such as the phosphatase and tensin homolog (PTEN)5, 6 or the tuberous sclerosis complex (TSC).7, 8 Moreover, mutation of PTEN has also been shown to lead to enhanced angiogenesis via upregulation of the hypoxia inducible transcription factor (HIF).9
HIF is a heterodimeric transcription factor which is widely recognized as the master regulator to a hypoxic environment. It mediates an adaptive transcriptional response and ensures cellular survival at low oxygen levels via transactivation of a variety of target genes. On a molecular level, HIF consists of a constitutive ß-subunit (HIF-1ß) and at least two oxygen regulated α-subunits (HIF-1α and HIF-2α). The oxygen dependent degradation of the α-subunits is initiated by hydroxylation of certain prolyl residues by specific prolyl hydroxylases (PHDs). This leads to recognition by an E3 ubiquitin ligase complex harbouring the von Hippel-Lindau protein and subsequent polyubiquitination and degradation via the proteasome. To regulate recruitment of the necessary transcriptional cofactor p300/CBP an additional asparagyl residue of HIF is also hydroxylated. Besides this undisputed dominant oxygen-dependent degradation, there have been a number of reports showing that there is also a relevant regulation of HIF via the PI3K/AKT/mTOR pathway.10 Amongst others, we could demonstrate in a previous study that this regulation is mTOR dependent.11, 12 Nevertheless the exact molecular mechanisms underlying this regulation and its biological relevance are still unclear.
HIF is also closely related to cancer development13 and its expression in tumors is correlated with increased resistance to chemotherapy14. There have been a number of reports showing that mTOR activity can be closely correlated to HIF expression levels.9, 12, 14 Since both systems, mTOR and HIF, exhibit pro-tumor attributes, it would be of special interest to simultaneously block both pathways to achieve a maximum inhibition of tumor growth, in the best scenario, a regression of tumor size. This could be achieved, at least in part, by applying mTOR inhibitors to reduce mTOR activity and HIF expression. If true, this could be particularly effective in tumors with high levels of HIF expression, such as renal cell carcinomas.15 There are a number of very potent mTOR inhibitors available (rapamycin, RAD001, Temsirolimus etc.) which are licensed for treatment of different tumor entities. The route of application is mostly oral and the tolerance rate of side effects is generally lower as compared to conventional cytotoxic regimens.
In this context, we set out to elucidate the impact of mTOR inhibition on tumor growth in a subcutaneous xenograft mouse model in vivo. In particular we aimed to analyze the effect on HIF expression and the influence of mTOR inhibition on HIF dependent tumor growth.
Results
Prior to establishing the xenograft tumor model in BalbC nu/nu mice the response of the different tumor cell lines intended for injection (Fig.1 HeLa (A); Caki-1 (B); Hepa-1 C1C7 and Hepa-1 C4 cells, (C)) in response to hypoxia, a hypoxia mimicking agent (Dipyridyl, DP) and mTOR inhibition was analyzed. These cell lines were chosen either due to their broad application in tumor biology (HeLa, human cervical cancer), their unique HIF-response to mTOR inhibition (Caki-1, human renal clear cell carcinoma) or their variability of functional HIF-status (Hepa-1, mouse hepatoma). In contrast to the Hepa-1 wildtype cells (C1C7), their genetical counterpart (C4) was subjected to chemical mutagenesis resulting in defective HIF-1ß. This cell line is therefore able to stabilize HIF-1α, yet it cannot form dimers with HIF-1ß to generate a functional HIF-1 transcriptional complex.16
Immunoblot analysis of (A) Hela, (B) Caki-1 and (C) Hepa-1 cells. Cells were exposed to either normoxia (N, 21% O2), hypoxia (H, 1% O2) or DP (Dipyridyl, 100µM) treatment for 6 or 24 hours. 30µg of total cell lysate was loaded and western blotting was performed for the following proteins: HIF-1α, HIF-2α, p70S6K-P (Thr 389), p70S6K, rpS6-P (Ser 235/236), rpS6 and ß-actin. Hepa-1 cells are either wildtype cells (Hepa-1 C1C7) or HIF-1ß deficient (Hepa-1 C4). Therefore, these cells were additionally analyzed for the expression of HIF-1ß. The mTOR inhibitor rapamycin was applied 45 minutes prior to hypoxic/pharmacological exposure at a concentration of 100nM.
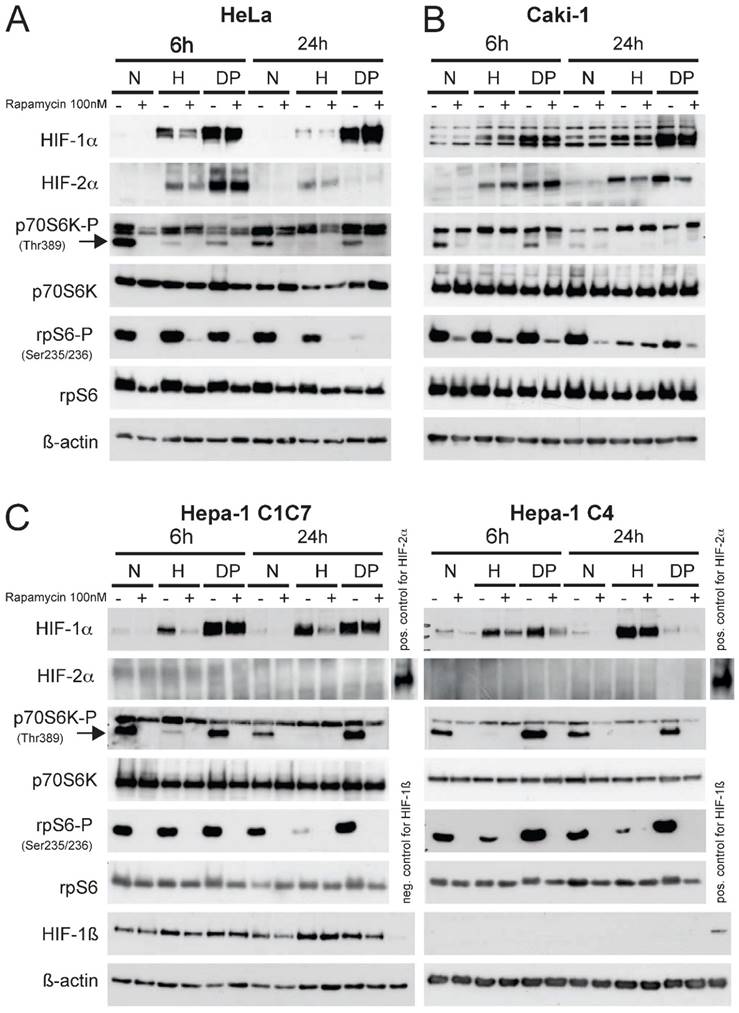
An equal number of cells were seeded one day prior to the hypoxic (1% O2) exposure or DP treatment (100µM). Cells were then pre-treated with 100nM of rapamycin for 45 minutes prior to 6 or 24 hours of control, hypoxia or DP treatment, respectively. Total cell extracts were made and immunoblotting was performed (Fig. 1). In all cell lines the application of 100nM rapamycin for 6 hours was sufficient to block the mTOR pathway, as the levels of phospho-p70S6K and phospho-rpS6 as downstream effectors were very effectively inhibited at this timepoint. Hypoxia displays a similar effect on the activity of mTOR, yet this physiological blockage kinetically clearly differs from pharmacological mTOR inhibition. Hypoxia itself has a profound inhibitory effect on the phosphorylation status of p70S6K at 6 hours of exposure. Phospho-levels of its downstream substrate rpS6 initially begin to decline at 24 hours of hypoxic insult in all cell lines. This may be in part due to a negative feedback loop of HIF-1α and rpS6.11 Additionally, HIF-1 target genes REDD1 and BNIP3 actively enhance suppression of mTOR activity.17, 18 HIF-1α is readily induced by hypoxia or DP-treatment in all cell lines, whereas HIF-2α is not expressed in both Hepa-1 cell clones. In contrast to HeLa and the two Hepa-1 cell lines, Caki-1 cells do not show a HIF-1α response to mTOR inhibition after hypoxic stimulation. HIF-2α is similarly regulated in both HeLa- and Caki-1 cells. HIF-2α nevertheless shows a downregulation after 24 hours of stimulation and mTOR inhibition. Nevertheless, Caki-1 cells are a very useful tool to evaluate the influence of mTOR inhibition on tumor growth without the influence of HIF-1α. The reason for this cell type-specific behaviour nevertheless remains unclear, yet the individual characteristics of the different cell lines implied can provide an insight into the regulatory influences of mTOR on HIF and tumor growth in general.
Interestingly, a differential behaviour is visible when comparing hypoxic to pharmacological HIF stabilization in response to mTOR inhibition. DP treatment consistantly leads to a stronger accumulation of HIF-1α in HeLa, Caki-1 and Hepa-1 C1C7 cells and this induction cannot be reduced by the application of rapamycin. Hepa-1 C4 cells only show a weak inducibility of HIF-1α by DP. As the response of mTOR inhibition is obviously linked to the intensity of the stress signal 11, DP-induction of HIF-1α can consequently be reduced by mTOR inhibition only in Hepa-1 C4 cells (Fig. 1C).
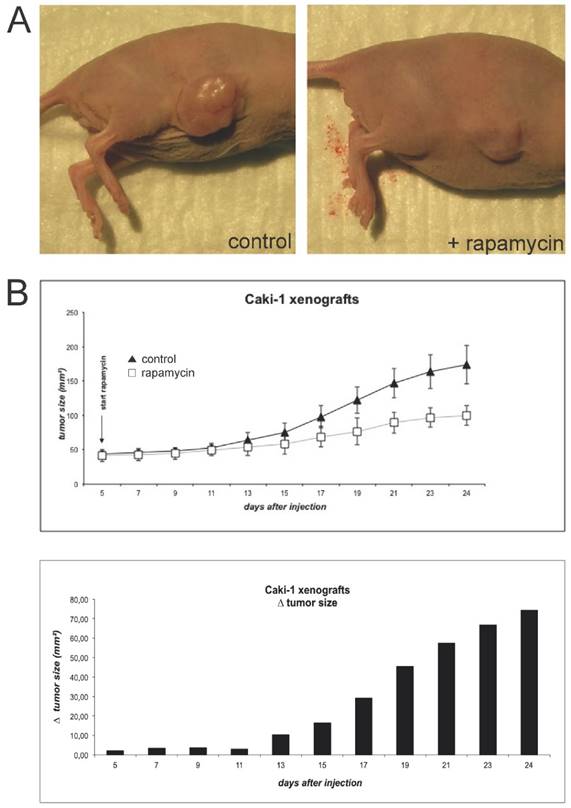
To induce Caki-1 xenograft tumors, BalbC nu/nu mice were subcutaneously injected with 5x106 cells (Fig. 2A). Tumor growth was monitored until it reached an established measurable size. At this time, rapamycin treatment was initiated. Animals were given a total of 1.5mg/kg bodyweight via daily oral gavage and further tumor growth was monitored every 2 days (Fig. 2B). Tumors of animals treated with the mTOR inhibitor quickly showed growth retardation in comparison to controls.
(A) For the initiation of xenograft tumors, BalbC nu/nu mice were injected subcutaneously with 5x106 Caki-1 cells (diluted in 200µl PBS). Animals were sacrificed when tumors reached a maximal size conforming to animal rights. (B) When the tumors initiated sufficient growth, rapamycin was applied via oral gavage daily at a concentration of 1.5 mg/kg bodyweight. Tumor size was monitored by measurement of length and width every two days.
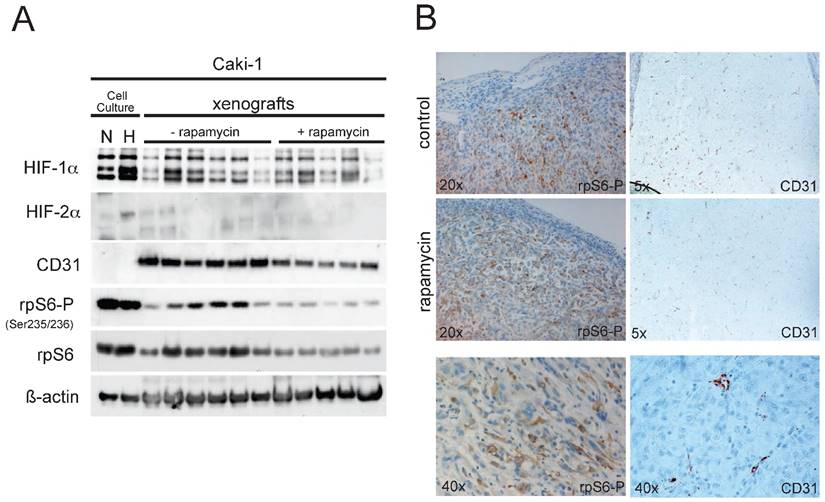
When tumors reached a limiting size according to the animal rights law, animals were sacrificed and tumors were excised. Caki-1 xenografts were allowed to grow for 24 days after injection. Total protein extracts from the Caki-1 tumors were analyzed via immunoblotting. Phosphorylation of rpS6 was clearly reduced in xenografts of animals receiving rapamycin, compared to the control group (Fig. 3A), implying effective mTOR inhibition within the xenografts. Also, vascularisation in the xenografts of the rapamycin-treated animal seems to be slightly reduced as western blotting for the endothelial marker CD31 (PECAM-1) shows, nevertheless failing statistical significance. HIF-1α levels were well detectable in the tumor extracts, yet the expression levels showed an enormous variation, which makes it difficult to perform legitimate quantitative comparison of the two groups. Compared to the cell culture experiments shown in Fig.1, HIF-2α cannot be detected in the xenograft tumor lysates. Expression of HIF-1α and HIF-2α can nevertheless be confirmed in immunohistological staining of Hela- and Caki-1 xenografts (Suppl. Fig. 4). These stainings show that both subunits have overlapping and distinct expression patterns, emphasizing the distinct cellular expression and function of HIF-1α and HIF-2α in general. Further, immunohistochemical staining for both phospho-rpS6 and CD31 of the tumors (Fig. 3B) seem to emphasize our findings from immunoblotting. Yet this form of analysis is probably not the best suitable for a quantitative evaluation of gene expression and must be considered very carefully. Regions of the tumors which harbour high mTOR activity (P-rpS6 staining) co-localize well with highly proliferative sections stained for PCNA (supplementary Figure 3A). The application of mTOR inhibitors seem to reduce overall proliferation in Caki-1 xenografts (supplementary Figure 3B and 3C).
(A) 30µg of total xenograft tumor lysates were immunoblotted for the following proteins: HIF-1α, HIF-2α, CD31, p70S6K-P (Thr389), p70S6K, rpS6-P (Ser235/236), rpS6 and ß-actin. As controls, total cell lysates from Caki-1 cell culture experiments (N, normoxia, 21%O2; H, hypoxia, 1%O2) were added. (B) Immunohistochemical staining of paraffin embedded sections of the Caki-1 xenograft tumors were analyzed for the expression of CD31 and rpS6-P (Ser 235/236). Magnifications as indicated.
Xenograft tumor growth was monitored for (A) Hepa-1 C1C7 (wildtype) and (B) Hepa-1 C4 (HIF-1ß deficient) cells. Rapamycin was administered to the animals in a concentration of 1.5mg/kg bodyweight by daily oral gavage. Tumor size was monitored by measurement of length and width every two days.
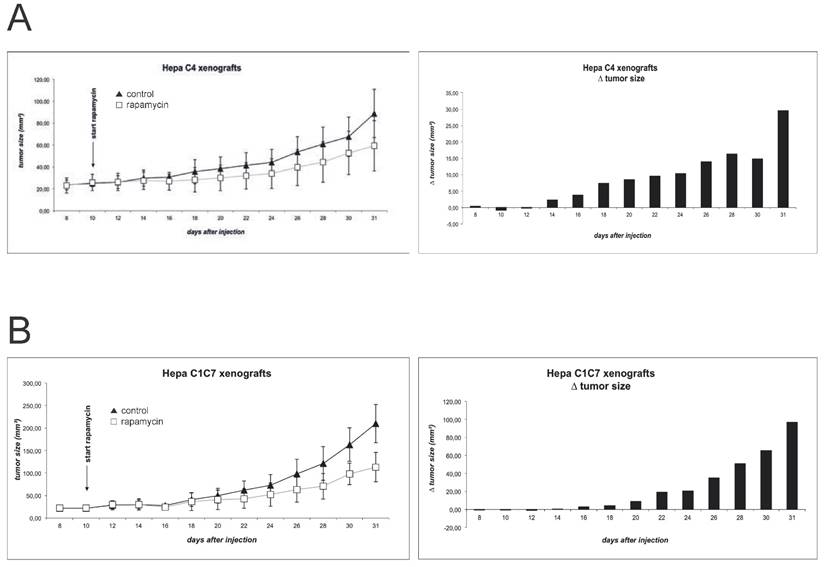
Figure 4 shows the growth curves of the Hepa-1 xenografts. As published previously and confirmed in a pilot experiment, Hepa-1 C4 derived tumors show a delayed initiation and growth, as compared to the wildtype cells 19. In an attempt to harmonize growth kinetics, we chose to increase the number of injected Hepa-1 C4 cells to 5x106, compared to 3x106 cells Hepa-1 C1C7. Indeed, this manipulation allows a better comparison of these xenografts, as tumor formation and growth now show more comparable kinetics. Nevertheless, Hepa-1 C4 cells, lacking the HIF dimerization partner form clearly smaller tumors than the wildtype cells with an intact HIF system. Immunoblot evaluation of tumor extracts reveals a detectable mTOR inhibition in both Hepa-1 systems, when comparing phospho-rpS6 levels. Yet, CD31 and HIF-1α levels either show no difference or evaluation of the two groups is difficult due to poor detection of the protein (suppl. Fig. 1).
When establishing the HeLa xenografts we encountered several unexpected difficulties. The initiation of tumor growth was difficult. A total of 4x106 cells were injected subcutaneously and it took about three weeks until reliable stable tumors were established. In addition, the growth of these xenografts was very slow and they tended to ulcerate and bleed (suppl. Fig. 2A), making it difficult to perform accurate size measurements. After the initiation of rapamycin treatment, the growth curves of control and rapamycin-treated groups did begin to differentiate (suppl. Fig. 2B), yet progressive ulceration of these tumors only allowed imprecise measurements and furthermore forced us to prematurely terminate this experiment in compliance with animal rights law. Furthermore, immunoblot analysis of HeLa xenografts proved to be very difficult as the tumor material appeared mostly necrotic and yielded only little and often degraded protein extracts of very poor quality (data not shown). Of note, mTOR inhibition in HeLa xenografts obviously could not prevent ulceration of the tumors, even though the pathway is efficiently blocked.
Discussion
The main goal of our study was to elucidate the involvement of HIF expression mediated by mTOR in experimental tumor growth. Numerous reports describe HIF regulation via the mTOR pathway 12, 11 and clinical studies clearly demonstrate the powerful anti-tumor effects of mTOR inhibitors. 20 Although HIF and mTOR are seperately positively correlated to tumor growth, cell motility and tumor metastatsis 13, 21, 22, it has never clearly been shown if the positive effects of mTOR inhibition are dependent on HIF reduction. If and to which extent this mTOR mediated HIF-expression is involved or present in solid tumor growth was the focus of our attention.
To address this, we specifically chose different cell lines and characterized their response to mTOR inhibition in respect to HIF expression levels in vitro. In a previous study11 we were already able to show that under standard conditions of 10% FCS not all cells seem to have an identical behaviour in this context. Caki-1 cells do not show a lower induction rate of HIF-1α by hypoxia or DP treatment after mTOR inhibition. In contrast, HIF-2α follows the inhibitory effects on HIF observed in other cells, yet only after 24 hours of stimulation and mTOR inhibition. Therefore, these cells nevertheless provide a tool to determine tumor regression by mTOR inhibition regardless of HIF-1α expression levels, as this is unaffected by rapamycin (Figure 2). Developing xenografts of Caki-1 cells clearly showed a retarded growth curve when mice were treated with rapamycin, implying a HIF-1α-independent mechanism for this effect. In these cells the inhibition of global cellular proliferation by mTOR inhibition is obviously sufficient to slow tumor growth. In analyzing the xenografts we nevertheless did find lower levels of CD31, a well-known endothelial marker which could in turn be a readout of less vascularization in the treated tumors. Even though HIF is well described as a player of (neo)vascularization23, 24, we could not find any evidence for the involvement of HIF in reduced vascularization of the xenografts, as quantitative realtime PCR of the tumors didn't show a regulation of VEGF in response to rapamycin treatment (data not shown). Although there seems to be a regulatory effect of mTOR inhibition on HIF-2α in the in vitro assay (Fig 1B), we were not able to confirm this in the xenografts. Combined with the fact that there is growth retardation in Hepa-1 cells, which clearly lack HIF-2α expression, this supports the notion that the inhibition of cellular proliferation by mTOR inhibitors is the major reason for this phenomenon, possibly by inducing G1-cell cycle arrest.25 Immunohistochemical staining for P-rpS6 and PCNA could support this as cells with an active mTOR pathway co-localize with well proliferating regions of the tumors (suppl. Fig 3A). Moreover, immunohistochemical (suppl. Fig. 3B) and immunoblot (suppl. Fig. 3C) analysis of PCNA in the xenograft tumors show reduction PCNA levels in rapamycin-treated tumors.
A very useful tool to investigate our hypothesis are the Hepa-1 cells. Interestingly both developing xenografts show a similar response to mTOR inhibiton, especially when comparing the mean delta of the tumor size of these two groups, resulting in a clear reduction of tumor growth after mTOR inhibition (Figure 4) in both cases. These data support our findings in Caki-1 cells, suggesting that HIF is not an essential player in tumor size reduction achieved by mTOR inhibition.
Our results demonstrate that even though there can be a clear mTOR-related regulation of HIF expression in vitro, the influence of this is obviously not critical in our implemented xenograft models. This is most probably due to the far more complex environment of threedimensional tumor structures, when compared to in vitro studies. In xenografts there are per se many hypoxic regions due to poor vascularization and a rapid increase in tumor size, which clearly leads to HIF induction via the classical prolyl hydroxylase inhibition due to oxygen deprivation. This regulation may be so dominant that the effect of mTOR on HIF expression levels is either hard to see or impossible to measure in these tumors. This is in line with our previous findings that the effect of HIF regulation via mTOR is dependent on the surrounding oxygen levels.11 A strong hypoxic stimulus, as it can develop in solid tumor structures such as xenografts seems to clearly override the mTOR directed HIF regulation. Moreover, mTOR influence on HIF is probably more prominent and important in normoxic surroundings or regions of mild hypoxia in terms of regulating basal HIF levels. It is widely accepted that HIF is profoundly regulated via post-translational modification and degradation via the von Hippel-Lindau complex. Nevertheless, there are a number of non-hypoxic stimulators such as lipoloysaccharides (LPS), thrombin or angiotensin II (Ang II) of HIF which have been shown to enhance HIF protein levels independently of hypoxia.26
To further elucidate the involvement of mTOR regulation on HIF in tumor growth it would be important to analyze the early developmental stages of solid tumor development, where hypoxia only plays a minor or intitiating role in HIF stabilitzation. If subcutaneous xenografts are an ideal model for these described investigations is now indeed questionable. Of note, not all cells used for these experiments developed into proper tumors (suppl. Fig. 2). The cervical cancer cell line Hela only slowly started to grow into established solid tumors, yet these quickly began to ulcerate and bleed. This made it very difficult to get a reliable scientific readout in terms of tumor size and immunhistochemical and protein analysis was also difficult due to the limited quality of the samples.
It may be more efficient to analyze an heterotopic tumor model, as tumors can arise in their “physiological“ surroundings, grow slower and are therefore less dependent on rapid neovascularization as xenograft models. In these scenarios it is indeed possible that basal regulatory elements of HIF, such as mTOR play a much more fundamental role in tumor development and growth. As we described in a previous study11, mTOR regulatory features on HIF seem to be more active in surroundings which are not subjected to massive hypoxia, such as xenografts.
Our data clearly implicates that treatment of solid tumors with mTOR inhibitors is an effective tool to reduce experimental tumor growth. Moreover, we show that this effect can be achieved independently of the HIF status and the HIF responsivity of the cells.
Materials and Methods
Cell Culture
HeLa, CAKI-1, Hepa-1 C1C7 and Hepa-1 C4 cells were cultured in DMEM (PAN) containing 1.0 g/L glucose, 10% FCS, 2 mmol/L L-glutamine, 100 units/mL penicillin, and 100 µg/mL streptomycin. Cells were incubated at 37°C at 5% CO2 in humidified air. Hypoxic exposure took place in an “invivo400” hypoxic workbench (Ruskinn Technology Ltd.) with 1% oxygen, 5% CO2, and balanced N2.
Protein Extraction and Immunoblotting
For protein extraction, cells were seeded 24 h before experiments and then pretreated for 45 min with the mTOR inhibitor rapamycin (100nM) (Sigma-Aldrich) and subsequently subjected to 6 h or 24 h of stimulation. Cells were washed twice with PBS, removed by scraping and homogenized into extraction buffer [8 mol/L urea, 10% glycerol, 1% SDS, 10 mmol/L Tris-HCL (pH 6.8), protease inhibitor Complete (Roche), 1 mmol/L sodium vanadate, 1 mmol/L AEBSF] using a T8 Ultra Turrax homogenizer (IKA). Liquid nitrogen frozen xenograft tumors were homogenized into extraction buffer as well. Extracts were quantified using the DC protein assay (BioRad). Equal amounts of protein were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Milipore) overnight in 10 mmol/L Tris, 100 mmol/L glycine, 10% methanol, and 0.05% sodium dodecyl sulphate. Membranes were blocked with PBS and 5% fat-free dried milk and stained with antibodies detailed in Supplementary Table S1. Signals were visualized using enhanced chemoluminescence systems from GE Healthcare. Unless otherwise stated, chemicals and reagents were purchased from Sigma-Aldrich.
Immunohistochemistry
Paraffin-embedded specimens were cut into 4µm sections, dewaxed, and rehydrated in a series of ethanol washes, and endogenous peroxidase activity was blocked. A commercial protocol was followed based on a streptavidin-biotin-peroxidase reaction (DAKO, Hamburg, Germany). For antigen retrieval, slides were cooked for 7 minutes in target retrieval solution (TRS, DAKO) by using a standardized pressure cooker (Fissler). Next, slides were blocked with TBS and 5% BSA for 30 minutes at room temperature. Sections were incubated with primary antibodies overnight at 4°C (HIF-1α, Cayman 1:20000; HIF-2α, NB100-480 1:50000; rpS6-P(Ser235/236), cell signalling, 1:200; PCNA, cell signalling 1:100; PECAM-1, Santa Cruz, 1:500). Thorough rinsing was followed by incubation with biotinylated secondary antibody for 1 hour at room temperature and streptavidin/biotinylated alkaline phosphatase for 30 minutes. Finally, AEC solution (DAKO) was used as chromogen according to the manufacturer's instructions. All incubations were performed in a humidified chamber. Between incubations, specimens were washed three times in Tris-buffered saline (50 mmol/L Tris-HCl and 136 mmol/L NaCl, pH 7.4). All samples were processed in parallel. Finally, sections were counterstained with aqueous hematoxylin (DAKO) and were analyzed with a Leica DMRB microscope (Leica, Bensheim, Germany), partly. Photographs were recorded digitally (Visitron, Puchheim, Germany).
Animal Experiments
The study was approved by the institutional review board for the care of animal subjects and was done in accordance with NIH guidelines. HeLa cells (4 x 106), CAKI-1 cells (5 x 106), Hepa C1C7 cells (3 x 106) or Hepa C4 cells (5 x 106) were injected s.c. into BALB/c nude mice. 1.5 mg/kg/d of the mTOR inhibitor rapamycin (Wyeth) was administered by oral gavage. Subcutaneous tumors were measured in the outer width and length with a sliding calliper every second day. Xenograft tumors were divided and either immersion fixed in 3% paraformaldehyde for immunohistochemical analysis or frozen in liquid nitrogen for protein analysis.
Supplementary Material
Supplementary figures and table.
Acknowledgements
The authors would like to thank the Deutsche Krebshilfe (110675) for financial support. The present work was performed in (partial) fulfillment of the requirements for obtaining the degree “Dr. med.“ for R.G.
Competing Interests
All authors believe that no financial and personal relationships exist with other people or organisations that could innapropriately influence (bias) their work.
References
1. Spunt SL, Grupp SA, Vik TA, Santana VM, Greenblatt DJ, Clancy J. et al. Phase I study of temsirolimus in pediatric patients with recurrent/refractory solid tumors. J Clin Oncol. 2011;29:2933-40
2. Cabrera Lopez C, Marti T, Catala V, Torres F, Mateu S, Ballarin Castan J. et al. Effects of rapamycin on angiomyolipomas in patients with tuberous sclerosis. Nefrologia: publicacion oficial de la Sociedad Espanola Nefrologia. 2011;31:292-8
3. Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23:3151-71
4. Proud CG. The multifaceted role of mTOR in cellular stress responses. DNA Repair (Amst). 2004;3:927-34
5. Fang J, Ding M, Yang L, Liu LZ, Jiang BH. PI3K/PTEN/AKT signaling regulates prostate tumor angiogenesis. Cellular signalling. 2007;19:2487-97
6. Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E. et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000;14:391-6
7. Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345-56
8. Bissler JJ, Kingswood JC. Renal angiomyolipomata. Kidney Int. 2004;66:924-34
9. Tian T, Nan KJ, Wang SH, Liang X, Lu CX, Guo H. et al. PTEN regulates angiogenesis and VEGF expression through phosphatase-dependent and -independent mechanisms in HepG2 cells. Carcinogenesis. 2010;31:1211-9
10. Dekanty A, Lavista-Llanos S, Irisarri M, Oldham S, Wappner P. The insulin-PI3K/TOR pathway induces a HIF-dependent transcriptional response in Drosophila by promoting nuclear localization of HIF-alpha/Sima. J Cell Sci. 2005;118:5431-41
11. Knaup KX, Jozefowski K, Schmidt R, Bernhardt WM, Weidemann A, Juergensen JS. et al. Mutual regulation of hypoxia-inducible factor and mammalian target of rapamycin as a function of oxygen availability. Mol Cancer Res. 2009;7:88-98
12. Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F. et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002;22:7004-14
13. Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437-43
14. Chang Q, Qin R, Huang T, Gao J, Feng Y. Effect of antisense hypoxia-inducible factor 1alpha on progression, metastasis, and chemosensitivity of pancreatic cancer. Pancreas. 2006;32:297-305
15. Wiesener MS, Munchenhagen PM, Berger I, Morgan NV, Roigas J, Schwiertz A. et al. Constitutive activation of hypoxia-inducible genes related to overexpression of hypoxia-inducible factor-1alpha in clear cell renal carcinomas. Cancer Res. 2001;61:5215-22
16. Wood SM, Gleadle JM, Pugh CW, Hankinson O, Ratcliffe PJ. The role of the aryl hydrocarbon receptor nuclear translocator (ARNT) in hypoxic induction of gene expression. Studies in ARNT-deficient cells. J Biol Chem. 1996;271:15117-23
17. Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E. et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893-904
18. Li Y, Wang Y, Kim E, Beemiller P, Wang CY, Swanson J. et al. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J Biol Chem. 2007;282:35803-13
19. Maxwell PH, Dachs GU, Gleadle JM, Nicholls LG, Harris AL, Stratford IJ. et al. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci U S A. 1997;94:8104-9
20. Ciuffreda L, Di Sanza C, Incani UC, Milella M. The mTOR pathway: a new target in cancer therapy. Current cancer drug targets. 2010;10:484-95
21. Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4:335-48
22. Populo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. International journal of molecular sciences. 2012;13:1886-918
23. Xiang L, Gilkes DM, Chaturvedi P, Luo W, Hu H, Takano N. et al. Ganetespib blocks HIF-1 activity and inhibits tumor growth, vascularization, stem cell maintenance, invasion, and metastasis in orthotopic mouse models of triple-negative breast cancer. J Mol Med (Berl). 2014;92:151-64
24. Iwase T, Fu J, Yoshida T, Muramatsu D, Miki A, Hashida N. et al. Sustained delivery of a HIF-1 antagonist for ocular neovascularization. Journal of controlled release: official journal of the Controlled Release Society. 2013;172:625-33
25. Kawada J, Ito Y, Iwata S, Suzuki M, Kawano Y, Kanazawa T. et al. mTOR inhibitors induce cell-cycle arrest and inhibit tumor growth in Epstein-Barr virus-associated T and natural killer cell lymphoma cells. Clin Cancer Res. 2014;20:5412-22
26. Kuschel A, Simon P, Tug S. Functional regulation of HIF-1alpha under normoxia-is there more than post-translational regulation? Journal of cellular physiology. 2012;227:514-24
Author contact
![]() Corresponding author: Dr. rer. nat. Karl X. Knaup, Department of Nephrology and Hypertension, Friedrich-Alexander-Universität Erlangen-Nürnberg, Translational Research Center (TRC), Schwabachanlage 12, 91054 Erlangen, Germany. e-mail: karl.knaupde; Tel. ++49 - 9131-8539570; Fax ++49 - 9131-8539561
Corresponding author: Dr. rer. nat. Karl X. Knaup, Department of Nephrology and Hypertension, Friedrich-Alexander-Universität Erlangen-Nürnberg, Translational Research Center (TRC), Schwabachanlage 12, 91054 Erlangen, Germany. e-mail: karl.knaupde; Tel. ++49 - 9131-8539570; Fax ++49 - 9131-8539561