Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(5):716-729. doi:10.7150/jca.17779 This issue Cite
Review
The controversial role of phospholipase C epsilon (PLCε) in cancer development and progression
Anna Tyutyunnykova1, Gennady Telegeev2, Anna Dubrovska1, 3, 4 ![]()
1. OncoRay - National Center for Radiation Research in Oncology, Faculty of Medicine and University Hospital Carl Gustav Carus, Technische Universität Dresden and Helmholtz-Zentrum Dresden-Rossendorf, Fetscherstrasse 74, 01307 Dresden, Germany.
2. The Institute of Molecular Biology and Genetics of NASU, Kyiv, Ukraine.
3. German Cancer Consortium (DKTK), Dresden, Germany.
4. Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, Germany.
Received 2016-9-30; Accepted 2016-12-23; Published 2017-2-25
Abstract
The phospholipase C (PLC) enzymes are important regulators of membrane phospholipid metabolism. PLC proteins can be activated by the receptor tyrosine kinases (RTK) or G-protein coupled receptors (GPCR) in response to the different extracellular stimuli including hormones and growth factors. Activated PLC enzymes hydrolyze phosphoinositides to increase the intracellular level of Ca2+ and produce diacylglycerol, which are important mediators of the intracellular signaling transduction. PLC family includes 13 isozymes belonging to 6 subfamilies according to their domain structures and functions. Although importance of PLC enzymes for key cellular functions is well established, the PLC proteins belonging to the ε, ζ and η subfamilies were identified and characterized only during the last decade. As a largest known PLC protein, PLCε is involved in a variety of signaling pathways and controls different cellular properties. Nevertheless, its role in carcinogenesis remains elusive.
The aim of this review is to provide a comprehensive and up-to-date overview of the experimental and clinical data about the role of PLCε in the development and progression of the different types of human and experimental tumors.
Keywords: Phospholipase Cε, cancer development, intracellular signaling, oncogene, tumor suppressor
Introduction
Phospholipase C (PLC) family, consisting of 6 subgroups - PLCβ, γ, δ, ε, η, ξ, is a group of proteins able to hydrolyze membrane phosphoinositol 4,5-bisphosphates (PIP2) to inositol-1,4,5-phosphate (IP3) and diacylglycerol (DAG) - both important second messengers - in response to extracellular stimuli including hormones and growth factors through activation of the different receptor tyrosine kinases (RTK) or G-protein-coupled receptors (GPCR) [1]. IP3 stimulates Ca2+ signaling and DAG acts through protein kinase C (PKC) leading to different cellular events, such as proliferation, growth and migration. Phosphoinositide signaling is important for normal functions of cells as well as for development of different pathological conditions including cancer [2].
PLCε homologue in C. elegans, PLC210 protein was first discovered in 1998 by Kataoka and coworkers by using a yeast two-hybrid system with Ras homologous protein LET-60 as the bait [3]. Shortly after, mammalian homologues of PLC210 have been independently identified by three groups by screening of rat and human expressed sequence tag databases [4-6].
Since then, PLCε has been further extensively studied to reveal its role in the regulation of different cellular functions. PLCε protein has the biggest size among all PLC family members - 230 kDa and consists of several functional domains including core domain (consisting of EF, X and Y subdomains) which possesses an ability to hydrolyze PIP2, pleckstrin-homology PH and C2 domains, GTP-exchanging (CDC25-like) domain at the C-terminus and two Ras-associating domains at N-terminus [4-6]. Two splice variants have been described for PLCε: PLCε1a and PLCε1b, which are expressed differently in different tissues [7]. However, the distinct roles which these splice variants might play are not described yet.
Due to its complex structure, PLCε is involved in the different signaling pathways (Figure 1). Core domain of PLCε is responsible for the hydrolysis of membrane phosphoinositides into second messengers inositol-1,4,5-phosphate and diacylglycerol (DAG) [4-6, 8]. This leads to the activation of protein kinase C and D and subsequent downstream signaling: PKC induces phosphorylation of multiple transcription factors and along with DAG takes part in Ca2+ signaling (Figure 1). The first studies of the biological functions of PLCε demonstrated that H-Ras directly binds to the RA domains of PLCε in a GTP-dependent manner leading to PLCε association with the plasma membrane and activation of its PIP2 hydrolyzing activity [4]-6]. According to Bunney et al., PLCε is localized in cytoplasm in self-inhibited mode, while binding of Ras stimulates it to change its conformation and to translocate to the membrane [8]. However, targeting of PLCε to the membrane is not the only mechanism regulating its activity. Overexpressing the mutant form of PLCε with a C-terminal CAAX sequence led to the constitutive membrane localization and increased activity of PLCε as compared to the wild type of PLCε. Nevertheless, even this membrane targeted PLCε still can be further activated by the EGF stimulation suggesting additional mechanisms regulating PLCε enzymatic activity beside interaction with Ras proteins and other activators [9].
A schematic overview of the PLCε signaling pathways.
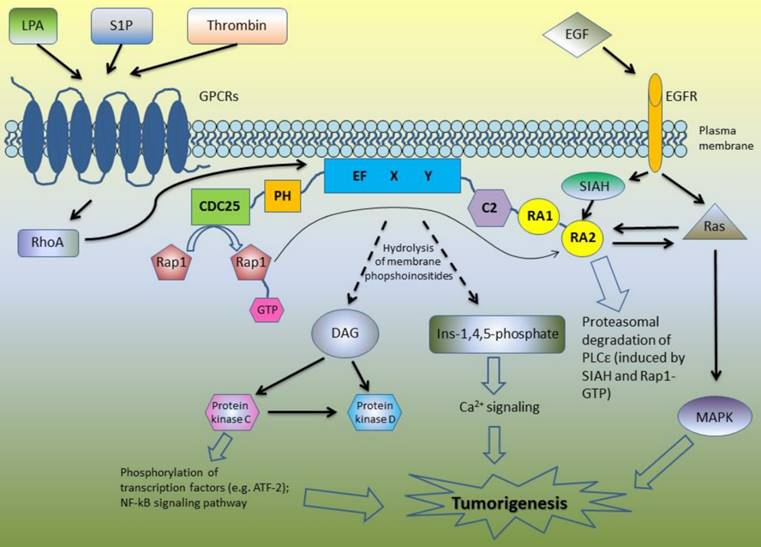
Lopez et al. revealed that N terminus of PLCε has a RasGEF domain. They demonstrated that mutant form of PLCε that lost its PIP2 hydrolyzing properties still can activate Ras-MAPK kinase pathway suggesting that this activation is due to Ras binding and GEF domain rather than to PIP2 hydrolysis [5]. Song and co-workers used cell stimulation with epidermal growth factor (EGF) as an example of the physiological stimulus which may trigger PLCε activation. This study revealed that EGFR activation can direct PLCε to the different subcellular regions depending on the activated upstream regulators. In the cells overexpressing H-Ras, EGF stimulation led to the PLCε translocation to the plasma membrane, although upon EGF-dependent Rap1A activation PLCε was localized in the perinuclear region [6]. The same group described similar mechanisms of PLCε activation in response to PDGF stimulation of hematopoietic BaF3 cells expressing PDGF receptor mutant that can activates Ras and Rap1 but not other types of PLC such as PLCγ . The activity of PLCε was induced by PDGF treatment and abrogated by disruption of the Rap and Ras pathways with overexpression of the Rap GAP, Spa1 protein and dominant negative Ras, respectively. Notably, PDGF stimulation led to the proliferation of these cells only if they express PLCε [10].
Since then, some other members of Ras family were described as the upstream regulators of PLCε including Rap2 and TC21, which activate PLCε in RA2-dependent manner in response to cell stimulation with EGF and activation of EGFR tyrosine kinase [11]. In addition to the RTK-mediated activation, PLC PIP2 hydrolyzing activity can be also induced by the transmembrane spanning GPCR receptors [12]. Mutation of RA2 domain of PLCε only partially inhibited this stimulation suggesting that GPCR can stimulate PLCε in RA2-dependent and RA2-independent way [11]. GPCR agonists such as lysophosphatidic acid (LPA), sphingosine-1-phosphate (S1P) and thrombin enhance PLCε activity through the GPCR-associated Gα12/13 and βγ subunits of the heterotrimeric G proteins [13]. This activation is at least in part mediated by the RhoGEF proteins which stimulate activation of Rho GTPase that consequently binds and activates PLCε independently on RA2 binding [11, 14]. PLCε has no specific domain to bind RhoA, but this GTPase can be bound directly to the small site within core domain of PLCε [15]. In the experiments with stimulation of PIP2 hydrolysis in the presence of RA2 mutant forms of PLCε, Kelley and coworkers identified additional GTPases that stimulate PLCε independently on RA2 binding including RalA and Rac [11]. Interestingly, that not only Ras family proteins have the ability to bind RA domains of PLCε: Siah proteins (E3 ubiquitine protein ligase) also can bind RA2 domain, but the region is distinct from that binding Ras, leading to the proteasomal degradation of PLCε after epidermal growth factor (EGF) stimulation [16].
Development and characterization of PLCε knockout mice models revealed an important physiological role of this protein in the maintaining of cardiac and pancreatic functions by regulation of Ca2+ mobilization in β cells and cardiomyocytes [17-19].
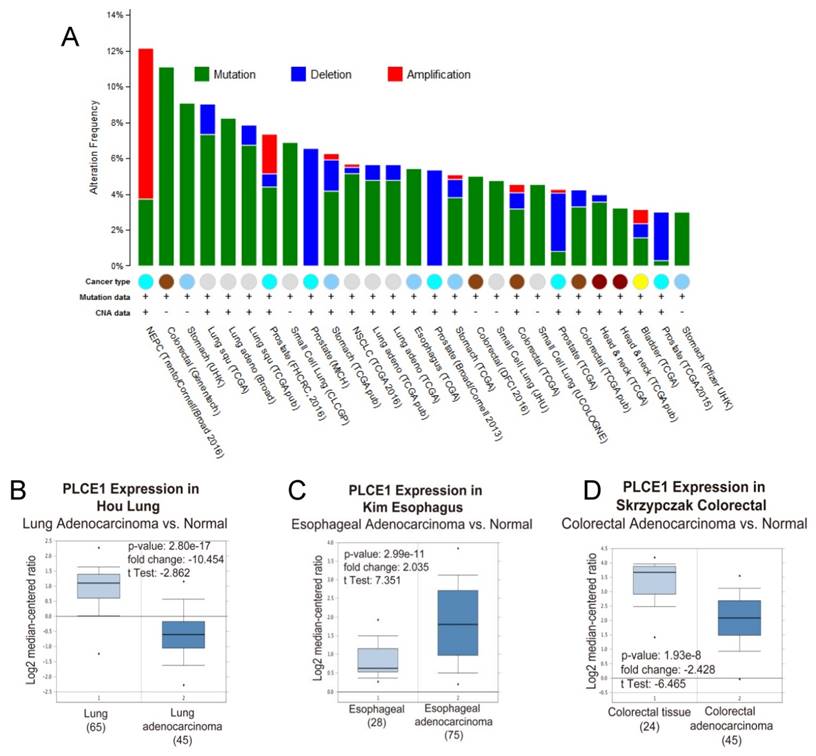
Because of its involvement in the different signaling pathways, PLCε plays pivotal role in development of many human diseases including childhood nephrotic syndrome [20, 21] and different types of cancers. Analysis of the PLCE1 gene alterations using The Cancer Genome Atlas (TCGA) database demonstrates that PLCE1 is frequently mutated gene in the different types of tumors (Figure 2A). Nevertheless, the contribution of PLCE1 in the carcinogenesis remains controversial, and it can swap the role from tumor suppressor to oncogene depending on the type of cancer (Figure 2BCD) that will be discusses in detail below.
PLCε expression and mutations in different types of human cancer. (A) Frequency of PLCE1 genetic alterations in different types of tumors. NEPC - neuroendocrine prostate cancer. (B-D) Analysis of PLCE1 mRNA expression from Oncomine data sets for lung, esophagus and colorectal normal and tumor tissues, correspondingly [73-75].
Skin cancer
One of the most interesting examples of PLCε involvement in cancer development is skin cancer. Because of the fact that PLCε has the ability to interact with Ras family proteins it has been postulated that it can also play a role in Ras-triggered cancers, one of which is skin cancer.
In 2004 Kataoka's group generated transgenic mice lacking a part of the catalytic domain and EF subdomain of PLCε (PLCε Δx/Δx) to study the role of PLCε in development of two-stage chemically induced carcinogenesis [22]. For this carcinogenesis model, single application of dimethylbenzanthracene (DMBA) led to the initiating of the oncogenic mutation of the HRAS gene. Subsequent weekly application of 12-O-tetradecanoyl-phorbor-13-acetate (TPA) for 20 weeks results in the clonal expansion of the initiated cells in the form of benign squamous tumors. Study of Bai and coworkers showed that PLCε Δx/Δx mice developed much less tumors (mostly papillomas) compared to PLCε+/+ and PLCε+/- (mostly adenocarcinomas) mice. According to authors' conclusions, PLCε may act as an oncogene for Ras-triggered skin cancer. Later the same group showed that PLCε plays crucial role in skin inflammation induced by phorbol ester, linking the initial inflammation to the subsequent skin cancer development [23]. Interestingly that more recent study of Martins et al., which is also based on the characterization of the PLCε knockout mice demonstrated completely opposite results [19]. In contrast to the Kataoka's mice model where PLCε was expressed in a shortened and catalytically inactive form, the knockout models described by Martins and coauthors either have a complete loss of the PLCε expression (PLCε-/-) or have expression of the mutant form of PLCε with mutant RA domains enable to bind to Ras (PLCεRAm/RAm). According to their findings, PLCε cannot be considered as an oncogene for Ras-triggered skin cancers, but rather as tumor suppressor, because this study revealed that PLCε-/- and to a lesser degree PLCεRAm/RAm mice possessed increased susceptibility to tumor development as compared to the mice with PLCε+/+ and PLCε+/- genotype. Difference in the observed data shows that PLCε may be involved in the process of skin tumor development but the exact mechanisms determining the role of PLCε in these mechanisms should be investigated more deeply.
Lung cancer
The knockout models described by Martins et al. which have a complete loss of the PLCε expression (PLCε -/-) or express the mutant form of PLCε (PLCε RAm/RAm) were also used to investigate potential role of PLCε in KRAS-driven lung tumor development. For this study, Martins and coworkers used conditional LSL-KrasG12DNSCLC mouse model where lung tumor development is induced by a single infection with an AdCre virus which results in the removal of the transcriptional termination Stop element and activation of the KrasG12D oncogene expression [19]. PLCε -/- and PLCε RAm/RAm mice were crossed with LSL-KrasG12D mice and expression of KrasG12D was induced by AdCre infection. Analysis of the tumor burden revealed no significant differences in the mice with different phenotypes. Interestingly, analysis of the LSL-KrasG12D MEF cells revealed a rapid reduction of PLCε expression after KrasG12D induction with AdCre. This KrasG12D mediated converting wild type cells into the cells lacking PLCε can explain similar features of KrasG12 mice models with or without PLCε knockout [19]. The tumor suppressor role of PLCε for human lung tumor development was confirmed by comparative analysis of cDNA level in the 21 pairs of tumor and normal tissues derived from the same patients. The results of this study demonstrated that PLCε was decreased in about 73% of tumors [19]. The same study also documented downregulation of PLCε expression in several non-small lung cancer cell lines. Notably that PLCε expression in these cells can be induced by the histone deacetylase inhibitor (TSA) and DNA methylation inhibitor [24] suggesting epigenetic mechanism of PLCε regulation in human tumors [19].
In contrast to the results of Martins and coworkers, the study conducted by Luo and colleagues showed that the expression of PLCε at mRNA level was higher in non-small cell lung carcinoma (NSCLC) cells derived from 36 patients than in non-cancerous cells obtained from adjacent lung tissues [25]. Treatment of NSCLC cells with PLC inhibitor U-73122 resulted in upregulation of p53 level and induced cell apoptosis. According to these results, authors hypothesized that the high levels of PLCε protein decrease the expression of p53 in NSCLC and thus inhibit apoptosis, but the exact mechanism warrants further investigation.
Interestingly, analysis of the datasets from Oncomine cancer microarray database confirmed a lower PLCε expression in the lung adenocarcinoma tissues as compared to the respective normal tissues suggesting rather tumor suppressor role of this protein for lung tumor development [26](Figure 2B).
Digestive tract cancers
Esophageal cancer
A host of recent investigations demonstrated a substantial impact of PLCε on the development of the digestive tract cancers. Meta-analysis conducted by Cui and coauthors which included 761 esophageal and gastric cancer cases and 457 controls demonstrated a strong association of PLCE1 expression with tumor progression in esophageal squamous cell carcinoma (ESCC) and gastric cancer (GC) [27].
However, not only the level of PLCE1 expression but also single nucleotide polymorphism (SNP) of PLCE1 gene is associated with ESCC and GC carcinogenesis. In 2010, Abnet and coworkers performed the genome-wide association study (GWAS) which first identified susceptibility loci for ESCC in PLCE1 gene. This study was conducted for more than 2,000 GC and ESCC cases and more than 3,000 control cases, and identified five SNPs on 10q23 that are mapped to the PLCE1 gene and have significant association to the risk of ESCC and GC development. Two of these SNPs, rs2274223 and rs3765524 result in missense mutations in the coding region of PLCE1 gene and cause the amino acid substitutions His1927Arg in the C2 domain and Thr1777Ile in the catalytic domain, respectively [28]. Interestingly, association of rs2274223 with CG was different for the different anatomical sites with a strongest association for tumors located in cardia. These results might suggest that the role of PLCε in tumor development is tissue type dependent [29].
In parallel, a large GWAS study has been conducted in China in 2010, having genotyped over 1,000 patients with ESCC and compared with DNA from more than 1,700 control individuals. The most promising SNP signatures were validated in additional large cohort study. This study confirmed association of rs2274223 with ESCC [30]. Further analysis of DNA from more than 2,700 gastric cardia adenocarcinoma (GCA) patients and over 11,000 control individuals demonstrated that this SNP is also associated with GCA susceptibility [30]. The immunohistochemical analysis also showed that GC and ESCC tissues have a higher level of PLCε expression as compared to normal gastric and esophageal epithelium, respectively, which support an idea that PLCε might contribute to the GCA and ESCC carcinogenesis [30]. Later study of Wu and coworkers for more than 2,000 ESCC patients and over 2,000 control individuals also confirmed that this rs2274223 signature in PLCE1 gene is associated with ESCC risk [29, 31].
Since then, a growing number of studies have been performed to validate association between PLCε gene polymorphism and ESCC or GC development, but the results of these studies were inconsistent. Malik and coauthors studied the polymorphisms (rs2274223A>G, rs3765524C>T and rs7922612C>T) in 135 patients with esophageal cancer [21] and 195 age and gender matched control patients from Kashmir valley, where the incidence of esophageal cancer is reported to be higher than 40% of all cancers. Researchers have showed that these SNPs did not have independent association with development of esophageal cancer, but the G2274223T3765524T7922612 haplotype was significantly associated with increased risk of EC [32]. Interestingly that similar research conducted in South Africa showed no correlation between studied PLCε SNPs and development of ESCC [33]. Recent studies of Qu et al. that included 550 patients with ESCC and 550 control individuals demonstrated that GA genotype of rs10882379 was significantly correlated with decreased ESCC risk, whereas AA genotype of rs829232 was significantly associated with a high ESCC risk in Chinese population as compared to GG genotype [34]. In attempt to obtain a more comprehensive conclusion about the possible link between PLCε rs2274223A gene polymorphism and risk of ESCC or GC development, Xue and coauthors conducted a meta-analysis of 22 published studies including 13188 cancer cased and 14666 controls [35]. This study concluded that rs2274223A>G correlates with an increased risk of both types of cancer, especially ESCC, However, the authors acknowledge that due to the retrospective character of the most of the data and high heterogeneity across the studies, which is attributed to a small number of participants and ethnic variations, their analysis might not be conclusive and needs the data from additional prospective studies for confirmation [35], One of the further reason for the discrepancy between the Chinese and African GWAS studies is a lower linkage disequilibrium (LD) which is an index of the non-random association between alleles at the different loci. According to this hypothesis, the PLCε SNPs described in the Chinese GWAS studies are not directly associated with carcinogenesis, but rather tagging with a high LD some other SNPs which are driving this association [33]
Analysis of the potential correlation between PLCε gene polymorphism and carcinogenesis fueled interest in the functional studies underlying the role of PLCε gene and described PLCε SNPs in tumor development. Bye and coauthors examined 10 polymorphic variants of PLCε which results in amino acid substitutions for their potential functional consequences by analysis of the evolutionary conservation across different species. Six of ten examined SNPs were predicted to lead to the loss of functionality for the different PLCε domains including Ras-GEF domain (rs17417407), catalytic domain (rs3765524) or C2 domain (rs2274223) [33].
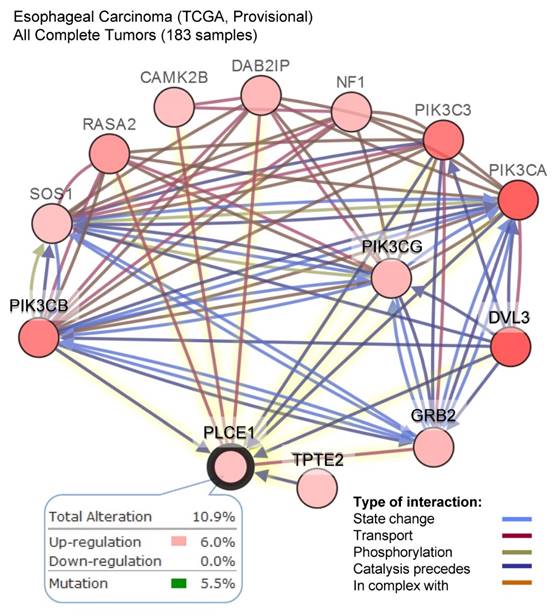
The other attempt to shed light on the role of PLCε in ESCC development has been done by the group of Chinese researchers who studied the correlation between PLCε expression and NF-kB signaling in ethnic Kazakh patients with ESCC. Authors demonstrated a strong positive correlation between the expression of PLCε and proteins from NF-kB signalling such as IKKβ and p50 [36]. Similarly to the function of PLCε in lung cancer, Li et al. showed that PLCε suppresses p53 expression in esophageal tumor cells [37]. Two ESCC cell lines, OE33 and CP-C have been analyzed in this study, and both cell lines express PLCε at a high level. Knockdown of PLCε markedly increased the expression of p53 in these cell lines. Authors suggest that PLCε can modulate p53 expression via its promoter methylation; however, there are no experimental data supporting this hypothesis. Interesting data has been obtained by Han et al. who showed that microRNA-328 (miR-328) can reduce the expression of PLCε at both mRNA and protein levels in ESCC cell lines EC109 and EC9706 [38]. Not only miR-328, but also miR-145 can have the same impact on PLCε expression in ESCC cells, as shown by Cui et al. [39]. Cui and coauthors demonstrated that PLCε expression level was elevated in tumor tissues compared to normal, and upregulation of PLCε significantly correlated with low overall survival rate in ESCC patients. The authors showed that PLCε contribute to ESCC migration and resistance to the apoptosis induced by the chemotherapeutic drugs. PLCε expression in ESCC cells is negatively regulated by tumor suppressor miR-145 [39]. Authors suggest that use of miRNAs targeting PLCε expression can be a potential therapeutic approach for esophageal cancer. These results are also supported by the analysis of the datasets from Oncomine cancer microarray database confirming a high PLCE1 gene expression in the esophageal adenocarcinoma tissues as compared to the respective normal tissues (Figure 2C). Analysis of the TCGA dataset for esophageal carcinoma suggests that frequent (10,9%) mutations of PLCE1 are associated with upregulation of the different pro-survival mechanisms such as PI3K, RAS/MAPK, WNT and calcium signaling pathways (Figure 3).
A high frequency of the alteration in the level of PLCE1 mRNA in esophageal carcinoma and its functional interaction with other genes which expression is also frequently altered in esophageal carcinoma. Data were analyzed using cBioPortal for Cancer Genomics. PIK3CB - Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Beta; SOS1 - son of sevenless homolog 1, a guanine nucleotide exchange factor for RAS proteins; RASA2 - RAS p21 protein activator 2; CAMK2B - calcium/calmodulin dependent protein kinase II beta; DAB2IP - disabled 2 (DAB2) interacting protein; NF1 - neurofibromin 1; PI3KC3 - phosphatidylinositol 3-kinase catalytic subunit type 3; PI3KCA - phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; PI3KCG - phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit gamma; DVL3 - dishevelled segment polarity protein 3; GRB2 - growth factor receptor bound protein 2; TPTE2 - transmembrane phosphoinositide 3-phosphatase and tensin homolog 2.
Gastric cancer
As it has been mentioned before, GWAS described by Wang et al. revealed that a non-synonymous SNP 2274223 A/G at 10q23 in PLCE1 gene is a shared susceptibility locus for gastric cancer and ESCC [30].
In 2011 it has been shown by Luo et al. that the presence of this SNP influences patients' survival. PLCE1 rs2274223 A/G SNP analysis in 940 gastric cancer patients from China demonstrated that patients with AA genotype survived better that those with AG and GG genotypes [40]. Later the other group of Chinese researchers showed that PLCε expression was upregulated in tumor tissues (n=74) and downregulated in non-cancerous inflammation (n=799), suggesting that PLCε can be a biomarker to distinguish between chronic gastric inflammation, normal and cancer tissue during gastric cancer development [41].
Interestingly, in 2012, Palmer et al. have found that the same genetic variants of PLCE1 gene in Caucasian population were not associated with gastric cancer [42]. For this study, authors genotyped 290 gastric cancer cases and 376 controls for the first study followed by the second study which included 306 gastric cancer, 107 esophageal adenocarcinoma, 52 esophageal squamous cancer, 376 control cases. These results might potentially suggest different association between PLCε gene polymorphism and carcinogenesis in Chinese and Caucasian populations.
The findings of Luo et al. have been confirmed by Wang and coauthors who demonstrated that 2 SNPs, rs2274223 and rs11187870, are significantly associated with a higher risk of gastric cancer in Han Chinese patients (cancer: n=1059; control n=1240) [43]. Later, Zhang et al. performed the meta-analysis for the eligible case-control studies which included 8281 cases and 10532 controls and showed that Asian patients, but not Europeans carrying PLCE1 rs2274223 A>G polymorphism are under the higher risk of digestive tract cancer development (particularly gastric and esophageal cancer) [44]. In 2014 the same SNPs were proved to be associated with gastric cancer development in Korean population (cancer: n=3245; control n=1700) [32, 45]. Finally, as it was above discussed, Cui et al and then Xue and coauthors conducted a large meta-analysis and summarized that PLCε could be a biomarker for ESCC and gastric adenocarcinoma [27, 35].
Intestine cancer
Not so many experiments have been done to reveal the role of PLCε in development of intestine cancer. In 2009 the group of Japanese researchers used intestine cancer mouse model (Min-/- mice) and showed that Min-/- PLCε-/- mice developed significantly smaller number of intestine tumors during their lifetime compared to Min-/- PLCε+/+ mice [24]. Also authors showed that PLCε deletion contributed to the lack of transition from low-grade adenoma to high-grade sarcoma. Blood vessel formation was decreased in low-grade PLCε-/- adenomas compared to PLCε+/+ ones; however, no such difference was observed in high-grade adenomas, indicating that PLCε expression can augment blood vessel formation in tumors of intestine. Authors propose the following model of PLCε-mediated intestine cancer development: stem cells first undergo neoplastic transformation and begin to express more PLCε, and then this leads to the expression of angiogenic factors, which drives further tumor development.
Colorectal cancer
Study of Danielsen and coauthors was one of the first findings demonstrating the role of PLCE1 as tumor suppressor. PLCE1 gene expression was significantly downregulated in colorectal cancer samples (n=137) as compared to normal colonic mucosa specimens (n=10), and low levels of PLCε expression were strongly associated with mutations in KRAS gene [46]. Due to the decreasing of PLCε levels during cancer progression, it has been postulated that PLCε downregulation is important for this process. Interestingly that PLCε level rose when tumors reached the metastatic stage, but PLCε promotor showed no signs of methylation suggesting that the other mechanisms underlying this phenomenon could exist. According to these findings authors suggest that PLCε can play a tumor suppressor role for colon cancer.
In 2012 Wang et al. analyzed 100 colon cancer samples and found the downregulation of PLCε expression in 46% of them, which also correlated with patients' age and tumor stage [47]. The overexpression of full-length PLCε in colon cancer cell lines resulted in higher apoptosis rate, slower growth rate and decreased migration ability; cells overexpressing PLCε formed smaller tumors in xenograft mice.
A tag SNP (tSNP) analysis of 203 colorectal cancer samples and 296 controls showed that PLCE1 gene had one of the SNPs believed to be responsible for cancer development [48]. Further analysis [49] conducted in European population (controls: n=382, colorectal tumor: n=192) showed the common genetic variants of PLCE1 identified earlier in GWAS study [30] were not associated with colorectal cancer development. However, the study conducted by Ezgi et al. (controls: n=210, colorectal tumor: n=200) clearly showed that rs2274223 SNP was associated with higher risk of colorectal cancer development in Turkish and Caucasian people [50]. Interestingly, the study of Wang and coauthors demonstrated that rs2274223 A>G change might decrease level of PLCE1 expression and the variant G phenotype is associated with a high susceptibility to colorectal cancer in Chinese population (controls: n=416, colorectal tumor: n=417) [51]. Later, Zhang et al. performed a study of multiple PLCE1 SNPs for their potential correlation with a high risk of colorectal cancer development in Han Chinese population (controls: n=385, colorectal tumor: n=276). This study demonstrated that rs753724 and rs11187842 polymorphisms significantly differ between cancer patients and healthy individuals [52].
Therefore, a number of studies have shown evidence to support the tumor suppressor role of PLCε in colorectal cancer development. This is also consistent with a higher PLCE1 gene expression in the normal colorectal tissues as compared to the colorectal adenocarcinoma in the Oncomine cancer microarray dataset (Figure 2D). However, the molecular mechanisms behind this tumor suppressor role of PLCε warrant further investigation.
Head and neck cancer
It has been shown that PLCε may contribute to the development of head and neck cancer. Ma et al. have analyzed three potentially functional SNPs of PLCE1 in 1,098 patients with head and neck squamous cell carcinoma (HNSCC) and 1,090 controls matched by age and sex in non-hispanic whites [53]. It has been shown that PLCE1 variants may have an effect on risk of HNSCC associated with tobacco and alcohol exposure (particularly for those tumors aroused at non-oropharyngeal sites). Bourguignon et al. showed that in head and neck cancer cell line HSC-3 PLCε activation through RhoA-GTP can be blocked by overexpression of PZD domain of leukemia Rho-GEF (LARG) protein, suggesting that PZD domain of LARG can be a potential inhibitor of RhoA/PLCε-mediated production of inositol-3-phosphates, release of Ca2+ from internal storages and thus starting the cascade of signaling events involved in development of head and neck cancer [54].
Bladder cancer
Recent works devoting to the involvement of PLCε in bladder cancer development have been focused mainly on the experiments on bladder cancer cell lines. In 2010 Ou et al. studied the effect of PLCε gene silencing with small hairpin sh RNA on the invasive properties of T24 cells and showed the significant decrease of the invasive cell potential and downregulation of BCL2, MMP2 and MMP9 gene expression suggesting that PLCε may act as an oncogene for bladder cancer [55]. Using the same approach for BIU-87 cells, Ling et al. demonstrated that knockdown of PLCε expression led to the inhibition of cell proliferation and accumulation of the cells in G0/G1 phase of cell cycle [56]. In addition, Cheng and coauthors showed the cyclin D downregulation in xenograft tumors derived from cells with knocked down PLCε [57]. Recent data obtained by Yang et al. links PLCε expression to inflammatory-associated pathways, particularly to the phosphorylation of STAT3 transcription factor in transitional cell carcinoma of bladder (TCCB) [58]. Taken together these data suggest that PLCε may have an impact on bladder cancer development, although these experimental results need to be explained by investigation of the molecular mechanisms of PLCε signaling.
Gallbladder cancer
Interesting research has been carried out by the group of Indian researchers who studied the association of some genetic variants of PLCε with susceptibility to gallbladder cancer in North Indian population. Gallbladder cancer is a relatively rare disease which is mostly abundant in populations from South America, Central and Eastern Europe and Northern India. Authors genotyped 641 patients (416 with gallbladder cancer and 225 controls) and proved that PLCε polymorphisms previously found in GWAS study can be associated with gallbladder cancer [30]; moreover, authors suggest the involvement of inflammation process in PLCε-mediated gallbladder cancer development [59].
Prostate cancer
The role of PLCε in prostate cancer has not been studied till recently, when the first article on this topic by Wang et al. has been published [60]. Authors investigated the expression of PLCε in 37 prostate cancer samples derived from cancer patients compared to 10 benign prostatic hyperplasia (BPH) specimens and also studied the relations among PLCε, androgen receptor AR and Notch1. This study showed that PLCε expression was elevated in prostate cancer samples, and targeted silencing of PLCε inhibited cell growth and proliferation of LNCaP and PC3 cells, decreased the expression levels of androgen receptor, Hes-1 and Notch, and blocked AR translocation to the nucleus. The authors concluded that PLCε may contribute to the development of prostate cancer through the independent regulation of both Notch-1 and AR pathways.
PLCε: a critical but controversial player in carcinogenesis
There is considerable evidence that metabolome changes induced in response to microenvironmental stimuli e.g. inflammation, hypoxia, nutrient deficiency, cancer therapy such as chemo- or radiotherapy may lead to the development of various diseases including malignant tumors [61, 62]. Metabolic reprogramming is one of the common cancer hallmarks [63]. A high biosynthetic and energetic demand of cancer cells drives the broad disregulation of the metabolic pathways that enable tumor cells to survive and grow in the harsh microenvironmetal conditions. Fast growing tumors have considerable alterations in the lipid metabolism including de novo lipogenesis. This not only serves as additional energetic resource, but also generates a number of biologically active molecules such as diacylglycerol, cholesterol, ceramide, sphingosine, PIP2, IP3 which are involved in the activation of a variety of signaling pathways associated with cancer progression, metastases and therapy resistance e.g. GPCR, AKT/PI3K, PKC, PLC [1, 63, 64].
Being the largest member of phospholipase C enzyme family, PLCε has unique features enabling it to be involved in the different signaling pathways and thus to play a role in different tumor entities. Up to now the functions of PLCε within the cell is not absolutely clear. It is known that PLCε can be activated by EGF, through GPCR receptors and via Rho pathway. Within the cell PLCε has not only classical phospholipase C function, but is also able to interact with Ras family GTPases and activate different signaling pathways leading to the changes in cell proliferation, survival etc.
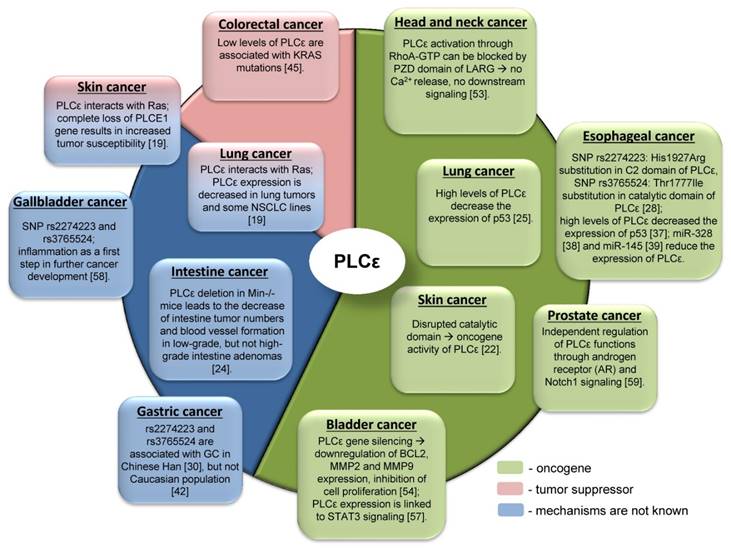
Due to the complex domain structure and ability to interact with multiple signaling molecules PLCε can play role in cancer development. Interestingly, that PLCε can act either as an oncogene or as a tumor suppressor for different tumor entities (Table 1, Figure 4). One of the possible reasons for such differences in functions might be hidden behind the complex structure of PLCε and thus its involvement in the different signaling pathways. For some types of cancer distinct pathways can be more important than others, that could be in part attributed to the oncogenic addiction e.g. in the KRAS driven lung tumors or to the tumor microenvironment. Thus, depending on the tissue context, PLCε can be critical molecule that enhances or suppresses cancer development [65]. The study of Song and coauthors which demonstrated that changes in the subcellular localization of PLCε in response to EGFR activation depend on the activated upstream regulators, H-Ras or Rap1A, in a nice example of the contextual physiological role of this protein in cancer cells [6]. An interesting example of another metabolic protein which serves as contextual oncogene of tumor suppressor is the adenosine monophosphate (AMP) activated protein kinase (AMPK), which maintains energy homeostasis and promotes cell survival under bioenergetics stress conditions. However, when it is highly activated, it might lead to the inhibition of tumor growth [66]. The reported conflicting role of AMPK in regulating carcinogenesis might at least partially depend on other oncogenes driving tumor growth such as Myc and H-Ras [67, 68].
The role of PLCε in the different tumor entities.
| Oncogene | Tumor suppressor | ||||
|---|---|---|---|---|---|
| Tumor entity | Model | Ref | Tumor entity | Model | Ref |
| Gastric cancer | Gastric cancer tissue samples from Chinese patients (N=2766) and healthy controls (N=11013) | [30] | Colorectal cancer | Transcriptome datasets from colorectal cancer samples (N=137) and normal mucosa (N=10) | [46] |
| Gastric cancer tissue samples from Chinese patients (N=1059) and healthy controls (N=1240) | [43] | ||||
| Meta-analysis where gastric and esophageal cancer cases (N=8281) and healthy controls (N=10532) were compared | [44] | ||||
| Gastric cancer cell lines AGS, SGC7901, MGC803; tissue samples from patients with gastric cancer (N=74), tissue samples from patients with chronic atrophic gastritis (N=799) | [41] | Colorectal cancer tissue samples obtained from patients and their pair-matched normal tissues (N=50) | [47] | ||
| Meta-analysis where gastric and esophageal cancer cases (N=761) and healthy controls (N=457) were compared | [27] | ||||
| Tissue samples from gastric cancer patients (N=940) | [40] | ||||
| Tissue samples from patients with gastric cancer (N=108) and healthy controls (N=195) from Kashmir Valley | [32] | ||||
| Tissue samples from Korean patients with gastric cancer (N=3245) | [45] | ||||
| Esophageal cancer | ESCC tissue samples (N=222) and controls (N=326); Eca109, TE-1, KYSE-150, KYSE-450 human ESCC cell lines | [27, 39] | Skin cancer | Transgenic PLCε-/- mice developed by authors | [19] |
| ESCC cell lines EC109 and EC9706, | [38] | ||||
| Tissue samples from ESCC patients and pair-matched controls (N=132) | [37] | ||||
| Tissue samples from patients with ESCC (N=135) and age and gender matched controls (N=195) | [32] | ||||
| Tissue samples obtained from patients with ESCC and their age and gender-matched controls (N=550) | [34] | ||||
| GWAS performed on ESCC patients (N=1077) and healthy controls (N=1733), and then repetition of 18 promising SNP on additional number of ESCC patients (N=7673) and healthy controls (N=11013) | [30] | ||||
| Colorectal cancer (rs2274223A >G transition) | Colorectal cancer samples obtained from patients (N=203) and normal tissue samples (N=296); | [48] | Lung cancer | Tissue samples obtained from patients with lung adenocarcinoma - microarray data from Oncomine database | [26] |
| tSNPs in PLCε gene analyzed in colorectal cancer samples from European patients (N=192) and non-cancerous tissues (N=382) | [49] | ||||
| Head and neck cancer | Human oral squamous cell carcinoma HSC-3 cell line | [54] | |||
| Tissue samples from patients with HNSCC (N=1098) and normal tissue (N=1090) | [53] | ||||
| Lung (NSCLC cells) | NSCLC cells obtained from patients with lung cancer (N=36) | [25] | |||
| Transgenic PLCε-/- mice developed by authors | [19] | ||||
| Bladder cancer | Xenograft tumors obtained from cells with knockdown of PLCε; human bladder cancer cell lines BIU-87 | [57] | |||
| Human bladder cancer cell line BIU-87 | [56] | ||||
| Human bladder cancer cell line T24 | [55] | ||||
| Bladder cancer cell lines BIU-87, T24; bladder carcinoma tissue samples (N=48) and adjacent normal tissue (N=21) | [58] | ||||
| Gallbladder cancer | Gallbladder tissue samples from patients (N=416) and controls (N=225) | [59] | |||
| Prostate cancer | Prostate cancer tissue samples (N=37) and benign prostatic hyperplasia (N=10) | [60] | |||
| Skin cancer | Transgenic PLCε-/- mice developed by authors | [22] | |||
| [23] | |||||
| Intestine cancer | Transgenic mouse model of intestine cancer (Min-/-PLCε-/-, Min-/- PLCε+/+) | [24] | |||
The controversial role of PLCε in carcinogenesis and associated signaling mechanisms.
Currently the most studied tumor entities where the role of PLCε has been clearly shown include esophageal squamous cell carcinoma and gastric cancer. The large GWAS analysis discovered 3 SNPs in PLCE1 gene that are significantly correlating with development of ESCC in Chinese Han population and some other populations. It has been shown that patients with ESCC bearing those SNPs have significantly lower survival rate compared to the patients lacking them. Interestingly, this correlation has not been shown for the South African populations. There are some potential reasons for these controversy including small sample size, unavailability of some patient-related data including smoking and drinking status, age and sex, and finally, a lower linkage disequilibrum (LD) in African population compared to Chinese population [35].
On the other hand, SNP polymorphism in one gene might have a little impact on cancer development and therefore be loosely related to the tumorigenesis. For example, instead of one-by-one SNP analysis, Tan and coworkers employed analysis of GWAS data for 54 genes involved in the inositol phosphate metabolic pathway in eight different types of tumors that could potentially pave the way for the GWAS-based analysis of the metabolism-related biomarkers [69]. Interestingly, this study confirmed the highly significant association of PLCE1 as an individual gene with ESCC and GC development. The pathway-based analysis demonstrated that inositol phosphate metabolism is significantly associated with lung cancer, ESCC, GC and renal cell carcinoma. This study illustrates distinct metabolic demands for the different tumor types and might at least in part explain reported discrepancy in the role of PLCε for the development of different tumor types.
Despite ESCC and GC can be considered as the most studied tumor entities where PLCε plays oncogenic role, the biological function of PLCE1 SNPs which correlate with cancer risk is not yet clear. On one hand, the high level PLCE1 expression correlates with ESCC and GC progression. However, on the other hand most of the examined SNPs might lead to the loss of functionality of the different PLCε domains including the core domain and therefore potentially disrupt downstream signaling pathways [33]. But this hypothesis has to be supported by further experimental data.
Interesting questions have been raised during the investigation of PLCε role in development of skin cancer. A major part of work in this field has been done by Kataoka's group showing that PLCε acts as oncogene for skin cancer [6]. However, the findings obtained by Martins et al. were completely opposite, raising the discussion about the real role of PLCε in skin cancer [19]. Both groups agree that inflammation mechanisms can be important triggers of skin cancer. The idea of inflammation as an intermediate mechanism that can lead to further tumor development has been given by authors who studied PLCε involvement in intestine cancer [24] and prostate cancer [60]. The other studies support this idea, showing that PLCε is significantly involved in neuroinflammation (through the activation of NF-kB signaling) [70] and inflammatory response of epithelial cells during bronchial asthma (through the upregulation of inflammatory cytokines) [71]. The hypothesis of the link between PLCε, inflammation and cancer has been also confirmed by Yang et al., who showed that knockdown of PLCε by shRNA decreased not only PLCε expression itself, but also the expression of inflammatory cytokines IL-6, TNF-α, IL-2β and inflammation-associated genes TLR-4, MyD88 and phosphorylated STAT-3 [58]. This can be the other way of how PLCε can be involved in development of different cancers. In addition to its potential contribution to the development of a number of solid tumors, our previous protein-protein interaction studies demonstrated that PLCε can bind to the PH domain of Bcr-Abl oncogene that potentially indicates its involvement in the Bcr-Abl mediated leukemogenesis [72].
Taken together, the current experimental and clinical data suggest that PLCε might play a pivotal role in regulation of cancer development and progression However, there is a long way ahead before PLCε could be potentially employed as diagnostic marker and therapeutic target. Additional functional studies and more clinical investigations are needed with larger sample size and improved study design to verify PLCε association with cancer risk.
Abbreviations
Abl: Abelson murine leukemia viral oncogene homolog 1
AMP: adenosine monophosphate
AMPK: AMP activated protein kinase
AR: androgen receptor
Bcr: breakpoint cluster region protein
BPH: benign prostatic hyperplasia
DAG: diacylglycerol
EC: esophageal cancer
EGF: epidermal growth factor
ESCC: esophageal squamous cell carcinoma
GAP: guanidine nucleotide exchange factor
GA: gastric adenocarcinoma
GC: gastric cancer
GCA: gastric cardia adenocarcinoma
GEF: guanine nucleotide exchange factor
GPCR: G-protein coupled receptors
GTP: guanosine-5'-triphosphate
GWAS: genome-wide association study
HNSCC: head and neck squamous cell carcinoma
IL: interleukin
IP3: inositol-1,4,5-phosphate
LD: linkage disequilibrium
LPA: lysophosphatidic acid
MAPK: mitogen-activated protein kinase
MEF: mouse embryonic fibroblasts
MMP: matrix metalloproteinase
NF-κB: nuclear factor kappa light chain enhancer of activated B cells
NSCLC: non-small cell lung carcinoma
PDGF: platelet-derived growth factor receptor
PH domain: pleckstrin-homology domain
PI3K: phosphoinositide 3-kinase
PIP2: phosphoinositol 4,5-bisphosphates
PKC: protein kinase C
PLC: phospholipase C
RA domain: Ras-associating domain
RTK: receptor tyrosine kinases
shRNA: small hairpin ribonucleic acid
S1P: sphingosine-1-phosphate
SNP: single nucleotide polymorphisms
STAT3: signal transducer and activator of transcription 3
TCCB: transitional cell carcinoma of bladder
TLR-4: Toll-like receptor 4
TNF-α: tumor necrosis factor α
TPA: 12-O-tetradecanoyl-phorbor-13-acetate
Acknowledgements
This works was supported by BMBF grant (ZIK OncoRay; A. Dubrovska) and by Sächsischen Landesstipendium (A. Tyutyunnykova).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Park JB, Lee CS, Jang JH, Ghim J, Kim YJ, You S, Hwang D, Suh PG, Ryu SH. Phospholipase signalling networks in cancer. Nature reviews Cancer. 2012;12(11):782-792
2. Yang YR, Follo MY, Cocco L, Suh PG. The physiological roles of primary phospholipase C. Advances in biological regulation. 2013;53(3):232-241
3. Shibatohge M, Kariya K, Liao Y, Hu CD, Watari Y, Goshima M, Shima F, Kataoka T. Identification of PLC210, a Caenorhabditis elegans phospholipase C, as a putative effector of Ras. The Journal of biological chemistry. 1998;273(11):6218-6222
4. Kelley GG, Reks SE, Ondrako JM, Smrcka AV. Phospholipase C(epsilon): a novel Ras effector. The EMBO journal. 2001;20(4):743-754
5. Lopez I, Mak EC, Ding J, Hamm HE, Lomasney JW. A novel bifunctional phospholipase c that is regulated by Galpha 12 and stimulates the Ras/mitogen-activated protein kinase pathway. The Journal of biological chemistry. 2001;276(4):2758-2765
6. Song C, Hu CD, Masago M, Kariyai K, Yamawaki-Kataoka Y, Shibatohge M, Wu D, Satoh T, Kataoka T. Regulation of a novel human phospholipase C, PLCepsilon, through membrane targeting by Ras. The Journal of biological chemistry. 2001;276(4):2752-2757
7. Sorli SC, Bunney TD, Sugden PH, Paterson HF, Katan M. Signaling properties and expression in normal and tumor tissues of two phospholipase C epsilon splice variants. Oncogene. 2005;24(1):90-100
8. Bunney TD, Katan M. Phospholipase C epsilon: linking second messengers and small GTPases. Trends in cell biology. 2006;16(12):640-648
9. Bunney TD, Harris R, Gandarillas NL, Josephs MB, Roe SM, Sorli SC, Paterson HF, Rodrigues-Lima F, Esposito D, Ponting CP. et al. Structural and mechanistic insights into ras association domains of phospholipase C epsilon. Molecular cell. 2006;21(4):495-507
10. Song C, Satoh T, Edamatsu H, Wu D, Tadano M, Gao X, Kataoka T. Differential roles of Ras and Rap1 in growth factor-dependent activation of phospholipase C epsilon. Oncogene. 2002;21(53):8105-8113
11. Kelley GG, Reks SE, Smrcka AV. Hormonal regulation of phospholipase Cepsilon through distinct and overlapping pathways involving G12 and Ras family G-proteins. The Biochemical journal. 2004;378(Pt 1):129-139
12. Cojoc M, Peitzsch C, Trautmann F, Polishchuk L, Telegeev GD, Dubrovska A. Emerging targets in cancer management: role of the CXCL12/CXCR4 axis. OncoTargets and therapy. 2013;6:1347-1361
13. Kelley GG, Kaproth-Joslin KA, Reks SE, Smrcka AV, Wojcikiewicz RJ. G-protein-coupled receptor agonists activate endogenous phospholipase Cepsilon and phospholipase Cbeta3 in a temporally distinct manner. The Journal of biological chemistry. 2006;281(5):2639-2648
14. Hains MD, Wing MR, Maddileti S, Siderovski DP, Harden TK. Galpha12/13- and rho-dependent activation of phospholipase C-epsilon by lysophosphatidic acid and thrombin receptors. Molecular pharmacology. 2006;69(6):2068-2075
15. Wing MR, Snyder JT, Sondek J, Harden TK. Direct activation of phospholipase C-epsilon by Rho. The Journal of biological chemistry. 2003;278(42):41253-41258
16. Yun S, Moller A, Chae SK, Hong WP, Bae YJ, Bowtell DD, Ryu SH, Suh PG. Siah proteins induce the epidermal growth factor-dependent degradation of phospholipase Cepsilon. The Journal of biological chemistry. 2008;283(2):1034-1042
17. Tadano M, Edamatsu H, Minamisawa S, Yokoyama U, Ishikawa Y, Suzuki N, Saito H, Wu D, Masago-Toda M, Yamawaki-Kataoka Y. et al. Congenital semilunar valvulogenesis defect in mice deficient in phospholipase C epsilon. Molecular and cellular biology. 2005;25(6):2191-2199
18. Wang H, Oestreich EA, Maekawa N, Bullard TA, Vikstrom KL, Dirksen RT, Kelley GG, Blaxall BC, Smrcka AV. Phospholipase C epsilon modulates beta-adrenergic receptor-dependent cardiac contraction and inhibits cardiac hypertrophy. Circulation research. 2005;97(12):1305-1313
19. Martins M, McCarthy A, Baxendale R, Guichard S, Magno L, Kessaris N, El-Bahrawy M, Yu P, Katan M. Tumor suppressor role of phospholipase C epsilon in Ras-triggered cancers. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(11):4239-4244
20. Lowik MM, Groenen PJ, Levtchenko EN, Monnens LA, van den Heuvel LP. Molecular genetic analysis of podocyte genes in focal segmental glomerulosclerosis-a review. European journal of pediatrics. 2009;168(11):1291-1304
21. Hinkes B, Wiggins RC, Gbadegesin R, Vlangos CN, Seelow D, Nurnberg G, Garg P, Verma R, Chaib H, Hoskins BE. et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nature genetics. 2006;38(12):1397-1405
22. Bai Y, Edamatsu H, Maeda S, Saito H, Suzuki N, Satoh T, Kataoka T. Crucial role of phospholipase Cepsilon in chemical carcinogen-induced skin tumor development. Cancer research. 2004;64(24):8808-8810
23. Ikuta S, Edamatsu H, Li M, Hu L, Kataoka T. Crucial role of phospholipase C epsilon in skin inflammation induced by tumor-promoting phorbol ester. Cancer research. 2008;68(1):64-72
24. Li M, Edamatsu H, Kitazawa R, Kitazawa S, Kataoka T. Phospholipase Cepsilon promotes intestinal tumorigenesis of Apc(Min/+) mice through augmentation of inflammation and angiogenesis. Carcinogenesis. 2009;30(8):1424-1432
25. Luo XP. Phospholipase C epsilon-1 inhibits p53 expression in lung cancer. Cell biochemistry and function. 2014;32(3):294-298
26. Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6(1):1-6
27. Cui XB, Peng H, Li S, Li TT, Liu CX, Zhang SM, Jin TT, Hu JM, Jiang JF, Liang WH. et al. Prognostic value of PLCE1 expression in upper gastrointestinal cancer: a systematic review and meta-analysis. Asian Pacific journal of cancer prevention: APJCP. 2014;15(22):9661-9666
28. Smrcka AV, Brown JH, Holz GG. Role of phospholipase Cepsilon in physiological phosphoinositide signaling networks. Cellular signalling. 2012;24(6):1333-1343
29. Abnet CC, Freedman ND, Hu N, Wang Z, Yu K, Shu XO, Yuan JM, Zheng W, Dawsey SM, Dong LM. et al. A shared susceptibility locus in PLCE1 at 10q23 for gastric adenocarcinoma and esophageal squamous cell carcinoma. Nature genetics. 2010;42(9):764-767
30. Wang LD, Zhou FY, Li XM, Sun LD, Song X, Jin Y, Li JM, Kong GQ, Qi H, Cui J. et al. Genome-wide association study of esophageal squamous cell carcinoma in Chinese subjects identifies susceptibility loci at PLCE1 and C20orf54. Nature genetics. 2010;42(9):759-763
31. Wu C, Hu Z, He Z, Jia W, Wang F, Zhou Y, Liu Z, Zhan Q, Liu Y, Yu D. et al. Genome-wide association study identifies three new susceptibility loci for esophageal squamous-cell carcinoma in Chinese populations. Nature genetics. 2011;43(7):679-684
32. Malik MA, Umar M, Gupta U, Zargar SA, Mittal B. Phospholipase C epsilon 1 (PLCE1 rs2274223A>G, rs3765524C>T and rs7922612C>T) polymorphisms and esophageal cancer risk in the Kashmir Valley. Asian Pacific journal of cancer prevention: APJCP. 2014;15(10):4319-4323
33. Bye H, Prescott NJ, Lewis CM, Matejcic M, Moodley L, Robertson B, Rensburg C, Parker MI, Mathew CG. Distinct genetic association at the PLCE1 locus with oesophageal squamous cell carcinoma in the South African population. Carcinogenesis. 2012;33(11):2155-2161
34. Qu Y, Zhang S, Cui L, Wang K, Song C, Wang P, Zhang J, Dai L. Two novel polymorphisms in PLCE1 are associated with the susceptibility to esophageal squamous cell carcinoma in Chinese population. Diseases of the esophagus: official journal of the International Society for Diseases of the Esophagus / ISDE. 2016
35. Xue W, Zhu M, Wang Y, He J, Zheng L. Association between PLCE1 rs2274223 A > G polymorphism and cancer risk: proof from a meta-analysis. Scientific reports. 2015;5:7986
36. Cui XB, Pang XL, Li S, Jin J, Hu JM, Yang L, Liu CX, Li L, Wen SJ, Liang WH. et al. Elevated expression patterns and tight correlation of the PLCE1 and NF-kappaB signaling in Kazakh patients with esophageal carcinoma. Medical oncology. 2014;31(1):791
37. Li Y, An J, Huang S, Liao H, Weng Y, Cai S, Zhang J. PLCE1 suppresses p53 expression in esophageal cancer cells. Cancer investigation. 2014;32(6):236-240
38. Han N, Zhao W, Zhang Z, Zheng P. MiR-328 suppresses the survival of esophageal cancer cells by targeting PLCE1. Biochemical and biophysical research communications. 2016;470(1):175-180
39. Cui XB, Li S, Li TT, Peng H, Jin TT, Zhang SM, Liu CX, Yang L, Shen YY, Li SG. et al. Targeting oncogenic PLCE1 by miR-145 impairs tumor proliferation and metastasis of esophageal squamous cell carcinoma. Oncotarget. 2016;7(2):1777-1795
40. Luo D, Gao Y, Wang S, Wang M, Wu D, Wang W, Xu M, Zhou J, Gong W, Tan Y. et al. Genetic variation in PLCE1 is associated with gastric cancer survival in a Chinese population. Journal of gastroenterology. 2011;46(11):1260-1266
41. Chen J, Wang W, Zhang T, Ji J, Qian Q, Lu L, Fu H, Jin W, Cui D. Differential expression of phospholipase C epsilon 1 is associated with chronic atrophic gastritis and gastric cancer. PloS one. 2012;7(10):e47563
42. Palmer AJ, Lochhead P, Hold GL, Rabkin CS, Chow WH, Lissowska J, Vaughan TL, Berry S, Gammon M, Risch H. et al. Genetic variation in C20orf54, PLCE1 and MUC1 and the risk of upper gastrointestinal cancers in Caucasian populations. European journal of cancer prevention: the official journal of the European Cancer Prevention Organisation. 2012;21(6):541-544
43. Wang M, Zhang R, He J, Qiu L, Li J, Wang Y, Sun M, Yang Y, Wang J, Yang J. et al. Potentially functional variants of PLCE1 identified by GWASs contribute to gastric adenocarcinoma susceptibility in an eastern Chinese population. PloS one. 2012;7(3):e31932
44. Zhang X, Zhang Y, Gu D, Cao C, Zhang Q, Xu Z, Gong Y, Chen J, Tang C. Increased risk of developing digestive tract cancer in subjects carrying the PLCE1 rs2274223 A>G polymorphism: evidence from a meta-analysis. PloS one. 2013;8(10):e76425
45. Song HR, Kim HN, Kweon SS, Choi JS, Shim HJ, Cho SH, Chung IJ, Park YK, Kim SH, Choi YD. et al. Common genetic variants at 1q22 and 10q23 and gastric cancer susceptibility in a Korean population. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35(4):3133-3137
46. Danielsen SA, Cekaite L, Agesen TH, Sveen A, Nesbakken A, Thiis-Evensen E, Skotheim RI, Lind GE, Lothe RA. Phospholipase C isozymes are deregulated in colorectal cancer-insights gained from gene set enrichment analysis of the transcriptome. PloS one. 2011;6(9):e24419
47. Wang X, Zhou C, Qiu G, Yang Y, Yan D, Xing T, Fan J, Tang H, Peng Z. Phospholipase C epsilon plays a suppressive role in incidence of colorectal cancer. Medical oncology. 2012;29(2):1051-1058
48. Duan X, Li X, Lou H, Geng T, Jin T, Liang P, Li S, Long Y, Chen C. Genetic association of PLCE1, C11orf92-C11orf93, and NOC3L with colorectal cancer risk in the Han population. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35(3):1813-1817
49. Kupcinskas J, Gyvyte U, Bruzaite I, Leja M, Kupcinskaite-Noreikiene R, Pauzas H, Tamelis A, Jonaitis L, Skieceviciene J, Kiudelis G. Common Genetic Variants of PSCA, MUC1 and PLCE1 Genes are not Associated with Colorectal Cancer. Asian Pacific journal of cancer prevention: APJCP. 2015;16(14):6027-6032
50. Ezgi O, Merve A, Hakan YT, Gul O. Genetic Variations in Phospholipase C-epsilon 1 (PLCE1) and Susceptibility to Colorectal Cancer Risk. Biochemical genetics. 2016
51. Wang Q, Chen P, Chen D, Liu F, Pan W. Association between phospholipase C epsilon gene (PLCE1) polymorphism and colorectal cancer risk in a Chinese population. The Journal of international medical research. 2014;42(2):270-281
52. Zhang Y, Gong Y, Du S, Yan M, Geng T, Feng T, Wang J, Jin T. The association between phospholipase C epsilon gene (PLCE1) polymorphisms and colorectal cancer risk in a Chinese Han population: a case-control study. International journal of clinical and experimental medicine. 2015;8(10):19360-19366
53. Ma H, Wang LE, Liu Z, Sturgis EM, Wei Q. Association between novel PLCE1 variants identified in published esophageal cancer genome-wide association studies and risk of squamous cell carcinoma of the head and neck. BMC cancer. 2011;11:258
54. Bourguignon LY, Gilad E, Brightman A, Diedrich F, Singleton P. Hyaluronan-CD44 interaction with leukemia-associated RhoGEF and epidermal growth factor receptor promotes Rho/Ras co-activation, phospholipase C epsilon-Ca2+ signaling, and cytoskeleton modification in head and neck squamous cell carcinoma cells. The Journal of biological chemistry. 2006;281(20):14026-14040
55. Ou L, Guo Y, Luo C, Wu X, Zhao Y, Cai X. RNA interference suppressing PLCE1 gene expression decreases invasive power of human bladder cancer T24 cell line. Cancer genetics and cytogenetics. 2010;200(2):110-119
56. Ling Y, Chunli L, Xiaohou W, Qiaoling Z. Involvement of the PLCepsilon/PKCalpha pathway in human BIU-87 bladder cancer cell proliferation. Cell biology international. 2011;35(10):1031-1036
57. Cheng H, Luo C, Wu X, Zhang Y, He Y, Wu Q, Xia Y, Zhang J. shRNA targeting PLCepsilon inhibits bladder cancer cell growth in vitro and in vivo. Urology. 2011;78(2):474 e477-411
58. Yang X, Ou L, Tang M, Wang Y, Wang X, Chen E, Diao J, Wu X, Luo C. Knockdown of PLCepsilon inhibits inflammatory cytokine release via STAT3 phosphorylation in human bladder cancer cells. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015;36(12):9723-9732
59. Sharma KL, Umar M, Pandey M, Misra S, Kumar A, Kumar V, Mittal B. Association of potentially functional genetic variants of PLCE1 with gallbladder cancer susceptibility in north Indian population. Journal of gastrointestinal cancer. 2013;44(4):436-443
60. Wang Y, Wu X, Ou L, Yang X, Wang X, Tang M, Chen E, Luo C. PLCepsilon knockdown inhibits prostate cancer cell proliferation via suppression of Notch signalling and nuclear translocation of the androgen receptor. Cancer letters. 2015;362(1):61-69
61. Peitzsch C, Perrin R, Hill RP, Dubrovska A, Kurth I. Hypoxia as a biomarker for radioresistant cancer stem cells. International journal of radiation biology. 2014;90(8):636-652
62. Peitzsch C, Cojoc M, Hein L, Kurth I, Mabert K, Trautmann F, Klink B, Schrock E, Wirth MP, Krause M. et al. An Epigenetic Reprogramming Strategy to Resensitize Radioresistant Prostate Cancer Cells. Cancer research. 2016;76(9):2637-2651
63. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674
64. Huang C, Freter C. Lipid metabolism, apoptosis and cancer therapy. International journal of molecular sciences. 2015;16(1):924-949
65. Torti D, Trusolino L. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: promises and perils. EMBO molecular medicine. 2011;3(11):623-636
66. Liang J, Mills GB. AMPK: a contextual oncogene or tumor suppressor? Cancer research. 2013;73(10):2929-2935
67. Rios M, Foretz M, Viollet B, Prieto A, Fraga M, Costoya JA, Senaris R. AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer research. 2013;73(8):2628-2638
68. Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B. et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell metabolism. 2013;17(1):113-124
69. Tan J, Yu CY, Wang ZH, Chen HY, Guan J, Chen YX, Fang JY. Genetic variants in the inositol phosphate metabolism pathway and risk of different types of cancer. Scientific reports. 2015;5:8473
70. Dusaban SS, Purcell NH, Rockenstein E, Masliah E, Cho MK, Smrcka AV, Brown JH. Phospholipase C epsilon links G protein-coupled receptor activation to inflammatory astrocytic responses. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(9):3609-3614
71. Nagano T, Edamatsu H, Kobayashi K, Takenaka N, Yamamoto M, Sasaki N, Nishimura Y, Kataoka T. Phospholipase cepsilon, an effector of ras and rap small GTPases, is required for airway inflammatory response in a mouse model of bronchial asthma. PloS one. 2014;9(9):e108373
72. Miroshnychenko D, Dubrovska A, Maliuta S, Telegeev G, Aspenstrom P. Novel role of pleckstrin homology domain of the Bcr-Abl protein: analysis of protein-protein and protein-lipid interactions. Experimental cell research. 2010;316(4):530-542
73. Skrzypczak M, Goryca K, Rubel T, Paziewska A, Mikula M, Jarosz D, Pachlewski J, Oledzki J, Ostrowski J. Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability. PloS one. 2010:5 (10)
74. Kim SM, Park YY, Park ES, Cho JY, Izzo JG, Zhang D, Kim SB, Lee JH, Bhutani MS, Swisher SG. et al. Prognostic biomarkers for esophageal adenocarcinoma identified by analysis of tumor transcriptome. PloS one. 2010;5(11):e15074
75. Hou J, Aerts J, den Hamer B, van Ijcken W, den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens JA, Hoogsteden HC. et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PloS one. 2010;5(4):e10312
Author contact
![]() Corresponding author: Anna Dubrovska, Technische Universität Dresden, Fetscherstr. 74, Dresden 01307, Germany. Phone: 4935-1458-7150; Fax: 493-5145-87311; E-mail: Anna.Dubrovskade
Corresponding author: Anna Dubrovska, Technische Universität Dresden, Fetscherstr. 74, Dresden 01307, Germany. Phone: 4935-1458-7150; Fax: 493-5145-87311; E-mail: Anna.Dubrovskade