Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(4):674-682. doi:10.7150/jca.16901 This issue Cite
Review
Fibronectin: How Its Aberrant Expression in Tumors May Improve Therapeutic Targeting
Jennifer Peyling Wang, Abigail Hielscher ![]()
Department of Biomedical Sciences, Georgia-Philadelphia College of Osteopathic Medicine, Suwanee, GA 30024, USA.
Received 2016-7-19; Accepted 2016-11-23; Published 2017-2-25
Abstract
Fibronectin is a matrix glycoprotein which has not only been found to be over-expressed in several cancers, but has been shown to participate in several steps of tumorigenesis. The purpose of this review is to illustrate how aberrant fibronectin expression influences tumor growth, invasion, metastasis and therapy resistance. In particular, this review will focus on the interactions between cell receptor ligands and fibronectin and how this interaction influences downstream signaling events that aid tumor progression. This review will further discuss the possible implications of therapeutic drugs directed against fibronectin and/or cellular interactions with fibronectin and will additionally discuss novel approaches by which to limit intra- and extra-tumoral fibronectin expression and the cellular events which lead to aberrant fibronectin expression. It is anticipated that these studies will set a basis for future research that will not only aid understanding of fibronectin and its prognostic significance, but will further elucidate novel targets for therapeutics.
Keywords: Fibronectin, extracellular matrix, integrins, tumorigenesis, metastasis, therapy resistance.
Introduction
The extracellular matrix (ECM) is a protein rich entity which supports tissue structure, cell adhesion, cell-cell communication and differentiation [1]. Numerous studies have found that ECM proteins are aberrantly expressed in carcinomas and actively participate in tumor progression. In particular, fibronectin is receiving increasing interest as a result of its participation in multiple stages of tumor progression. Under homeostatic conditions, fibronectin has been shown to play a role in cell growth, differentiation, migration, and is additionally involved in processes such as wound healing and blood coagulation [2]. With regard to cancer, fibronectin is not only increased in tumors where its altered expression has been shown to promote tumor growth [3-5], migration [6, 7] and invasion [8, 9] but fibronectin has also been reported to limit tumor cell responsiveness to therapy [10-12] (Figure 1). Fibronectin has even been shown to alter mammary epithelial cell behavior. For instance, the introduction of fibronectin to differentiated mammary epithelial cell acini cultured atop the basement membrane extract Matrigel caused the cells to re-enter the cell cycle thus reversing their growth arrest and acinar morphology [13]. Work in our lab has shown that loss of caveolin-1, the main structural protein of caveolae, results in increased expression of fibronectin, tenascin-C and collagen in murine mammary glands (Thompson et al, unpublished data). The altered ECM is accompanied by changes in mammary gland architecture characterized by increased numbers of ducts that possess a larger circumference and area when compared to wild type controls, a result which may be in part related to increased fibronectin expression observed in the stroma (Thompson et al, unpublished data).
Fibronectin signaling in healthy and tumorigenic cells. In a healthy cell, fibronectin participates in the maintenance of homeostasis. Under pathological conditions, fibronectin is upregulated and associates with several integrin receptors, resulting in activation of various signaling cascades which ultimately promote tumorigenesis, metastasis and therapy resistance.
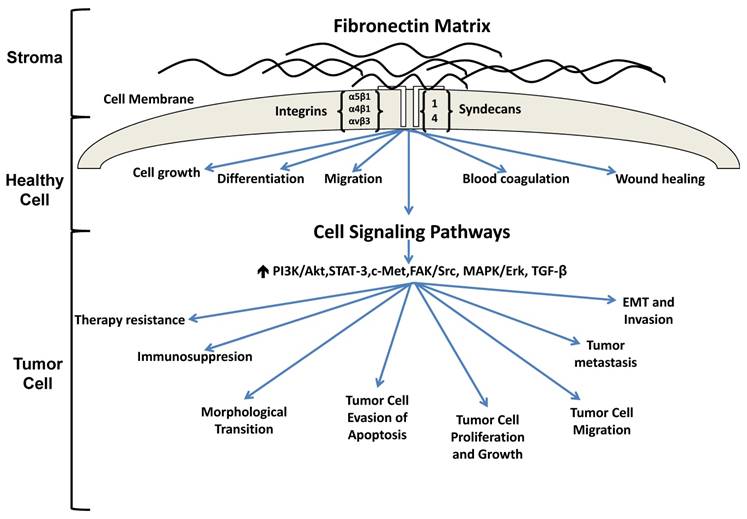
The purpose of this review is to illustrate the mechanisms in which aberrant fibronectin expression influences tumorigenesis and therapy resistance. In particular, this review will focus on the interactions between cell receptor ligands and fibronectin and how this interaction influences downstream signaling events that facilitate tumor progression. Further, this review will highlight potential strategies whereby fibronectin deposition, engagement with cell surface receptors and activation of cell signaling pathways may be exploited to halt tumor progression. In this manner, it is with anticipation that a new generation of novel therapeutics may be developed to better combat fibronectin's participation in tumorigenesis.
Tumor Progression
Fibronectin has been shown to play a central role in processes associated with tumor progression. In particular, α5β1 integrin and fibronectin have not only been shown to be upregulated in tumors, but have also been reported to participate in tumor cell proliferation. For example, Nam et al found that fibronectin and the extra domain (ED)-A splice variant of fibronectin were associated with higher α5β1-integrin expression in malignant as opposed to normal breast epithelial cells cultured atop Matrigel [5]. Interestingly, the authors also reported that total fibronectin, ED-A fibronectin and α5β1-integrin was markedly upregulated in the malignant breast cancer lines but not in non-malignant breast epithelial cells [5]. Similarly, Mierke et al also determined that α5β1 integrin was upregulated in MDA-MB-231 breast cancer cells and further demonstrated that fibronectin augmented the invasiveness of α5β1 expressing breast cancer cells cultured atop collagen hydrogels [3]. To investigate a mechanism responsible for α5β1-fibronectin in tumor growth, Mitra et al examined the activation of the receptor tyrosine kinase c-Met in human HeyA8 and SKOV3ip ovarian cancer cell lines following α5β1 engagement with fibronectin [4]. Results showed that binding of cellular α5β1 to fibronectin resulted in increased activation of the c-Met/FAK/Src signaling pathways in ovarian cancer cells [4]. Blocking ovarian cancer cell α5β1 interaction with fibronectin reduced c-Met mediated focal adhesion kinase (FAK) and Src phosphorylation in vitro and in vivo and additionally reduced tumor weight and proliferation in xenograft tumors [4], suggesting that α5β1-fibronectin interactions regulate cell signaling pathways important for ovarian cancer growth. In an effort to further examine the effects of integrins on ovarian tumor cell proliferation, Kenny et al examined adhesion and proliferation of human ovarian cancer cells following culture atop a de-cellularized ECM derived from human omental tumors [8]. When cultured in the presence of anti- α5β1, αvβ3 and RGD peptides, the authors observed decreased adhesion and proliferation of ovarian cancer cells cultured atop the de-cellularized ECM supporting a role for fibronectin-integrin engagement in ovarian cell proliferation [8].
A defining feature of tumors is the ability of cancer cells to evade apoptosis. In an effort to delineate a role for fibronectin-integrin interactions on tumor cell resistance to apoptosis, Han et al used Wortmannin to block PI3-K in human bronchial epithelial BEAS-2B and 16-HBE cell lines following α5β1 cellular engagement with fibronectin [14]. The authors found that Wortmannin caused an increase in DNA fragmentation indicative of apoptosis [14]. In the absence of Wortmannin, fibronectin stimulated α5β1-mediated phosphorylation of PI3-K and downstream signaling of PI3-K/Akt, which in turn, down-regulated p21 and the tumor suppressor gene p53 and upregulated cyclin D1 in the bronchial epithelial cells [14].
In addition to integrins, fibronectin binding to cell surface receptors such as syndecans has been reported to regulate tumor cell ECM attachment and growth. This is evidenced by work from Huang et al who examined the effect of tenascin-C on tumor cell adhesion to fibronectin [15]. Using MDA-MB-435 breast cancer and T98G gliobastoma cell lines, the authors found that tenascin-C bound to the FNIII13 of the HepII site on fibronectin blocked syndecan-4 mediated tumor cell binding to fibronectin [15]. Overexpressing syndecan-4 reversed the effects of tenascin-C induced inhibition of tumor cell adhesion to fibronectin, suggesting that tenascin-C bound to the FNIII13 site on fibronectin disrupts syndecan-4-fibronectin signaling [15]. Similarly, syndecan 1 and 4 engagement with fibronectin was reported to promote MDA-MB-231 breast cancer cell adhesion and migration on collagen and proliferation on fibronectin matrices, an observation which was reversed upon incubation with anti-syndecan 1 and 4 antibodies [16].
Aside from direct cellular interactions with fibronectin on tumor progression, others have reported a role for fibronectin in tumor immunosuppression. For example, Sengupta et al examined the role of siRNA against fibronectin in glioma tumor growth and immune cell activation [17]. Using GL261 glioma tumor cells, the authors observed delayed tumor cell proliferation and growth in mice injected with fibronectin silenced GL261 cells [17]. This observation was in part, attributed to a reduction in the number of T-regulatory cells, which inhibit the activity of T helper cells.
While the full length form of fibronectin plays an important role in tumorigenesis, isoforms of fibronectin, such as the ED-A and ED-B variants, have been reported to regulate tumor growth. For instance, Rybak et al demonstrated markedly higher expression of ED-A fibronectin in the vasculature of murine liver metastases from F9 teratocarcinoma cells with minimal expression observed in non-diseased organs, pointing to a role for ED-A fibronectin in angiogenesis [18]. Indeed, a later study demonstrated that ED-A overexpressing colorectal cancer cells exhibited upregulated expression of the pro-angiogenic cytokine vascular endothelial growth factor-C (VEGF-C) and promoted tumor growth and lymphangiogenesis in mice [19]. To determine a mechanism responsible for these observations, the authors report that ED-A upregulation of VEGF-C is accomplished via ED-A mediated activation of the PI3K/Akt pathway. Dose dependent reductions in VEGF-C were observed following cancer cell treatment with an inhibitor of PI3K [19]. Aside from a role in angiogenesis, ED-A fibronectin was reported to be upregulated in patient colorectal tumors, especially those that were of advanced stage [20]. Investigating CD133+/CD44+ colorectal tumor cells, a subset of cells believed to be responsible for colon cancer initiation, the authors found that these cells expressed higher levels of the ED-A receptor α9β1 and further demonstrated that ED-A interaction with α9β1 was required for tumor cell proliferation [20]. To elucidate a mechanism responsible for their observations, Ou et al found that ED-A sustains Wnt/β-catenin signaling, necessary for the progression of colorectal cancer cells, via integrin mediated activation of a FAK/ERK pathway [20]. In a more recent study, ED-A fibronectin was shown to increase the population of myeloid derived cells, known to impair immune responses to cancer cells, from osteoblasts [21]. To determine whether the absence of fibronectin impaired myeloid cell numbers and thus tumor formation, the authors subcutaneously injected melanoma cells into either a fibronectin knock out model (cKO) in which fibronectin expression in differentiating osteoblasts was conditionally deleted or a wild type animal (WT) and evaluated for tumor formation [21]. Tumor growth and the presence of myeloid cells in tumors were reduced in cKO animals but not in WT animals [21]. Although the authors don't attribute these findings to a specific reduction in the ED-A fibronectin isoform, they go on to demonstrate that culture of myeloid cells from cKO animals with ED-A fibronectin resulted in a reduction of melanoma apoptosis, suggesting that this pro-tumor effect results from ED-A activation of myeloid cells [21].
ED-B fibronectin has also been reported to play a role in angiogenesis where it has been shown to promote VEGF expression, tube formation and proliferation of human vascular endothelial cells [22]. Specifically, the authors found that ED-B fibronectin was upregulated in vascular endothelial cells in response to high glucose, TGF-β1 and endothelin-1, a finding which was reversed following administration of inhibitors to TGF-β1 and endothelin-1 [22]. These findings would suggest that growth factor signaling is an important regulator of ED-B fibronectin mediated vascular morphogenesis [22]. Despite a role in angiogenesis, ED-B fibronectin expression has been documented to be more highly expressed in the stromal and epithelial compartments of colorectal and breast carcinomas, but not in endothelial cells [23], suggesting that additional work is needed in order to fully elucidate the role of ED-B fibronectin in tumorigenesis. Regardless of its role in tumorigenesis, ED-B fibronectin has more recently been exploited as a molecular marker for prostate cancer. Here, Han et al synthesized a small peptide to target upregulated ED-B expression in tumor xenografts harboring human prostate carcinomas, demonstrating that the peptide bound to ED-B fibronectin produced by the tumor cells [24]. It's conceivable that this strategy may be utilized for both imaging and diagnostic purposes as well as delivery of therapeutics.
Overall, these studies suggest that fibronectin, via engagement with several integrin receptors and interference with immune function, favors tumor cell survival and proliferation. Additionally, multiple isoforms of fibronectin also participate in tumorigenesis, highlighting the importance of understanding how the different splice variants of fibronectin not only interact with cell surface receptors, but how this interaction activates downstream signaling pathways important for tumor growth.
Tumor Invasion and Metastasis
While fibronectin appears to play a key role in multiple facets of tumor progression, it has also been shown to participate in processes associated with tumor migration, invasion and metastasis. To examine the role of fibronectin on tumor cell migration, Lou et al overexpressed SOX2 to determine its role in ovarian tumor cell metastasis [6]. Here, the overexpression of SOX2 upregulated fibronectin gene expression resulting in increased migration and invasion of the A2780 human ovarian cancer cell line in transwell chambers [6]. Downregulation of fibronectin using siRNA resulted in a reversal of cell migration and invasion despite high expression of SOX2 indicating that SOX2 signals via fibronectin during tumor cell metastasis [6]. To further explore the role of fibronectin on tumor cell migration, Wei et al sought to demonstrate whether nicotine enhanced the migration of SW480 colorectal cancer cells through nicotinic-acetylcholine receptor (7-nAChR) mediated induction of COX-2 [7], reported to regulate EMT [25]. The authors found that inhibiting 7-nAChR and COX-2 not only limited fibronectin expression, but also increased E-cadherin expression, reducing SW480 cell migration [7]. In the absence of COX-2 inhibition, nicotine enhanced colon cancer cell migration via 7-nAChR and COX-2 mediated up-regulation of fibronectin expression indicating that inhibition of COX-2 and 7-nAChR reduced migration of colorectal cancer cells as a result of a reduction in fibronectin expression [7].
Integrin engagement has been reported to play a crucial role in fibronectin mediated tumor cell invasion. A recent study reported an indispensable role for fibronectin and the α5β1 integrin on the invasive capabilities of HeyA8 and SKOV3ip1 human ovarian cancer cell lines [8]. Here, the authors first showed that 3D co-culture of ovarian cancer cells with mesothelial cells supported fibronectin expression from mesothelial cells via activation of a TGF-βR1/RAC1/SMAD3 signaling pathway, a phenomenon which was important for cancer cell adhesion, proliferation and invasion as silencing of fibronectin reduced these cellular responses [8]. In an attempt to further elucidate a role for fibronectin in tumor cell progression, the authors administered antibodies against α5 and β1 integrins to 3D co-cultures of ovarian cancer cells and mesothelial cells and discovered that tumor cell invasion and proliferation was reduced up to 40% [8]. In-vivo, the authors found that treatment of mice with antibodies against α5 and β1 reduced the number of metastases and tumor weight from orthotopically injected ovarian cancer cell lines [8]. These results suggest that ovarian cancer cells stimulate fibronectin expression from mesothelial cells via a TGF-β pathway and that tumor cells adhere, proliferate and invade in response to α5 and β1 integrin engagement with fibronectin. In a similar study, it was shown that inhibition of α5 and β1 integrins significantly reduced ovarian tumor cell adhesion to a 3D model consisting of primary human mesothelial cells and fibroblasts and additionally limited the number of metastases in ovarian cancer xenografts [26]. Downregulation of E-cadherin on ovarian cancer cells was specifically shown to augment α5 integrin expression via activation of FAK and Erk1 [26]. Further, siRNA of E-cadherin was shown to increase cancer cell adhesion to fibronectin and invasion in a Matrigel matrix [26], suggesting that ovarian cancer cell adhesion to fibronectin and invasion in a 3D matrix are regulated by decreased E-cadherin mediated α5 integrin expression. In addition to ovarian cancer, work by others have shown that MDA-MB-231 breast cancer, T24 bladder carcinoma, and 786-O renal adenocarcinoma cells expressing α5β1 integrins exhibited increased invasion in 3D collagen matrices which had been polymerized with fibronectin, further highlighting that integrin engagement with fibronectin supports tumor cell invasion in 3D [3]. Interestingly, the addition of soluble fibronectin at the start of the assay reduced tumor cell invasion in collagen matrices, suggesting that differences in the polymerization status of fibronectin may differentially regulate tumor cell behavior [3]. A role for αvβ3 interaction with fibronectin has also been reported to participate in tumor cell invasiveness. For example, 3D matrices of clotted plasma, reported to play an important role in tumor metastasis [27], or fibrin, the principal component of clots, were found to induce invadopodia, cellular protrusions which participate in the early stages of cell migration and invasion, formation from several tumor cell lines [9]. Invadopodia were associated with increased expression of αvβ3 integrin and fibronectin matrix formation [9]. Furthermore, it was found that primary metastatic kidney tumor cells exhibited an increased expression of invadopodia formation in addition to fibronectin and αvβ3 integrin which together upregulated the expression of the epithelial-mesenchymal (EMT) transcription factor Slug [9]. These results would suggest that αvβ3 and fibronectin promote tumor cell invadopodia formation via activation of the EMT transcription factor Slug.
In order to metastasize to distant sites, tumor cells make use of altered cell signaling pathways and matrix metalloproteinases (MMPs), which degrade and remodel the ECM allowing tumor cell migration. With regard to a role for fibronectin in altered cell signaling, Balanis et al showed that adhesion of MDA-MB-231 breast cancer cells to fibronectin elicited robust STAT3 activation, a result which was dependent on cellular expression of FAK and the tyrosine kinase PYK2 [28]. The authors further demonstrated that administration of fibronectin to MDA-MB-231 cells cultured atop 3D hydrogels resulted in cellular outgrowth and a phenotype previously linked with enhanced metastatic properties [28]. Inhibition of β1 integrin or the use of a small inhibitor against STAT3 reversed these findings, suggesting that in 3D, MDA-MB-231 breast cancer cells utilize fibronectin-β1 integrin mediated STAT3 signaling during invasion [28]. To illustrate a cooperative role of fibronetin and MMPs in tumor invasion, Meng et al sought to determine whether A549 lung cancer cell invasion occurred as a result of fibronectin-induced FAK activation of MMP9 [29]. The authors found that fibronectin stimulated FAK phosphorylation and Src recruitment, resulting in activation of downstream targets, ERK1/2 and PI3K/Akt in A549 cells, promoting invasion and migration of the cancer cells in transwell chambers [29]. Inhibition of ERK1/2 and PI3K resulted in a reduction of migrating and invasive A549 cells as a result of downregulation of MMP9, implicating a role for fibronectin-FAK-MMP9 in lung cancer invasion [29]. Together, these findings not only suggest an important role for integrin engagement with fibronectin, but cooperation between fibronectin and altered cell signaling and MMP expression in processes associated with tumor invasion and metastasis.
Therapy Resistance
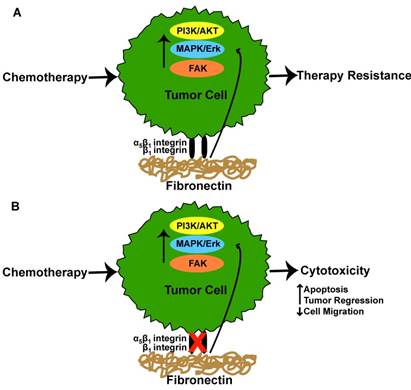
Chemotherapeutic agents have undoubtedly contributed to improved survival rates for individuals with cancer. Despite these successes, tumor cell resistance to therapies and relapse are still a common feature of the disease. While a number of factors have been implicated in chemotherapy resistance, fibronectin has received interest. In a study by Pontiggia et al the efficacy of tamoxifen, an estrogen receptor antagonist, was used to examine how the microenvironment limits tumor cell sensitivity to tamoxifen [12]. Here, the authors co-incubated human and mouse breast tumor cells with fibronectin and measured tumor cell sensitivity to tamoxifen [12]. The addition of fibronectin promoted tamoxifen resistance via fibronectin-mediated activation of β1-integrin which in turn, stimulated the PI3K/Akt and MAPK/Erk 1/2 signaling pathways in both tumor cell lines [12]. Disrupting the interaction between the β1-integrin and fibronectin reversed the conferred resistance, leading to drug-induced apoptosis of the tumor cells [12]. Similarly, Yuan et al found that β1 integrin and fibronectin were markedly increased in tamoxifen resistant as opposed to tamoxifen sensitive MCF7 breast cancer cells [30]. The overexpression of β1 was associated with increased expression of the epidermal growth factor receptor (EGFR)/ERK signaling pathway. Inhibition of β1 improved tamoxifen sensitivity of resistant MCF7 cells and further reduced cellular migration following culture in the presence of conditioned media from carcinoma associated fibroblasts (CAFs) [30]. Interestingly, culture of tamoxifen resistant MCF7 cells in CAF conditioned media resulted in enhanced cell migration due to CAF produced fibronectin, a result which was diminished following cellular treatment with the β1 integrin inhibitor [30]. In addition to tamoxifen, cetuximab, an antibody that inhibits cell proliferation when bound to the EGFR, has been reported to have limited efficacy as a result of tumor cell resistance [10]. For example, A549 human lung adenocarcinoma cells and H1299 human non-small cell lung carcinoma cells were reported to have diminished cell cytotoxicity to cetuximab and radiation following culture atop fibronectin coated surfaces [10]. The observed reduction of cytotoxicity was a result of tumor cell α5β1 integrin engagement with fibronectin as silencing these integrins restored tumor cell sensitivity to cetuximab [10]. Interestingly, cetuximab was also shown to promote increased fibronectin expression from both tumor cell lines, a finding which was reported to result from activation of the p38-MAPK-ATF2 signaling pathway [10]. siRNA silencing of fibronectin improved cytotoxicity of cetuximab in H1299 and A549 tumor cell lines, demonstrating that excess fibronectin in combination with α5β1 integrin engagement with fibronectin facilitates cetuximab resistance [10]. In a similar study, changes in tumor cell apoptosis were analyzed following etoposide treatment of H69 small cell lung cancer cells which had been cultured in fibronectin coated dishes [31]. The results showed that tumor cell α2, α3, α6, and β1 integrin interaction with fibronectin inhibited chemotherapy induced apoptosis, evidenced by decreased caspase 3-activity [31]. Using breast cancer as a model, Spangenberg et al reported that inducible expression of the ERBB2 proto-oncogene in MCF7 breast cancer cells resulted in upregulated gene expression of α5 and β1 integrins [32]. The authors found that MCF7 cells exhibited increased resistance to cisplatin and 5-fluorouracil as a result of ERBB2 induced α5β1 integrin expression and exhibited a 2 fold reduction in responsiveness to these agents when the cells were cultured on fibronectin. To overcome chemotherapy resistance from integrin-fibronectin interactions, Nam et al showed that disrupting the peptide bond between fibronectin and α5β1-integrin in combination with radiation therapy promoted apoptosis and reduced the expression of α5β1 in cultures of malignant human breast cancer cells cultured atop a 3D Matrigel [5]. The authors found that the pro-survival effect of malignant tumor cell α5β1 interaction with fibronectin in a 3D environment was a result of α5β1 mediated activation of Akt signaling as Akt kinase and Akt phosphorylation were downregulated following α5β1 inhibition [5]. Similarly, it was found that primary glioblastomas resistant to the anti-angiogenic agent bevacizumab exhibited a higher expression of β1 integrins and FAK, a phenomenon the authors attributed to high intra-tumoral hypoxia following anti-angiogenic therapy [33]. To determine whether inhibition of β1 integrin improved therapeutic responses, the authors treated subcutaneously grown U87MG glioblastomas with alternating doses of a β1 inhibitor and low dose (1mg/kg) bevacizumab [33]. The results showed a significant improvement in tumor regression in the dual treated animals [33]. These results were comparable to a high dose regimen (10mg/kg) of bevacizumab alone, suggesting that the addition of a β1 integrin inhibitor improves the therapeutic efficiency of bevacizumab [33]. Figure 2 provides a graphical summary of the findings addressed above. Given these and other similar studies, it's apparent that targeting the cellular interactions with fibronectin and/or fibronectin itself may be a desirable strategy to improve patient outcomes.
Integrin-fibronectin interaction in tumor therapy responses. (A) Tumor cell α5β1 or β1 integrin interaction with fibronectin results in activation of cell signaling pathways which ultimately culminate in tumor cell resistance to chemotherapeutic agents. (B) Inhibition of α5β1 or β1 integrin interaction with fibronectin restores tumor cell sensitivity to chemotherapeutic agents resulting in increased cytotoxicity.
Future Outlook: Potential Strategies for Targeting Fibronectin
While the aforementioned studies have provided encouraging evidence to address therapeutic targets against integrin engagement with fibronectin, further research is necessary in order to find novel strategies for inhibiting tumor growth via targeting fibronectin biosynthesis and/or fibronectin regulated cell signaling.
Matrix rigidity is a common feature of tumors and has been shown to support tumor growth, invasion and metastasis [34]. In response to matrix rigidity, focal adhesions, complexes of integrin receptors which facilitate cellular attachments with the ECM, are formed. FAK, a structural and signaling component of focal adhesions, has not only been reported to be upregulated in response to matrix rigidity [35], but has additionally been shown to regulate fibronectin-directed matrix fibrillar organization [36]. For instance, Ilic et al reported that fibronectin fibrils were more sparse and thinner in FAK null embryos compared to wildtype littermates and fibroblasts, a result which was demonstrated to be independent of fibronectin gene or protein expression and synthesis, suggesting that FAK activity is necessary for fibronectin matrix organization [36]. Interestingly, in a separate study, fibronectin expression was shown to drive FAK signaling [29], suggesting that both FAK and fibronectin may interact in a positive feedback loop. Given the role of FAK in fibronectin expression, it's conceivable that targeting FAK expression may be a mechanism to reduce fibronectin expression and thus tumor progression. Meng et al showed that fibronectin-induced FAK tumor cell migration and invasion was decreased following treatment of A549 lung tumor cells with the Src inhibitor PP2 [29]. Specifically, the authors reported that inhibition of Src resulted in decreased FAK phosphorylation suggesting that a reduction of FAK activity may be a useful strategy to reduce fibronectin mediated effects on tumor cell migration and invasion [29]. In addition to indirect targets of FAK, it may be possible to directly reduce FAK activity via use of a novel ATP competitive kinase inhibitor, PF-04554878, which was recently shown to promote apoptosis and reduce proliferation of pancreatic neuroendocrine tumor cells [37].
Aside from targeting FAKs, it may be possible to directly reduce matrix stiffness in tumors, thereby preventing FAK-induced fibronectin expression. Levental et al demonstrated that expression of lysyl oxidase (LOX), an enzyme which cross-links collagen fibers [38], not only increased FAK expression, but augmented matrix rigidity and breast tumor progression, a phenomenon which was reversed upon treatment with an inhibitor of LOX [39]. In support of a role for LOX in fibronectin expression, Adam et al showed that atrial samples from patients with atrial fibrillation exhibited increased expression of LOX and fibronectin [40]. Administration of a small molecule inhibitor of LOX to neonatal cardiac fibroblasts attenuated LOX-induced fibronectin expression via downregulation of angiotensin II and connective tissue growth factor [40]. Although this work was conducted in cardiac fibroblasts, it's foreseeable that inhibiting LOX may be a means to reduce tumoral fibronectin expression. Overall, it is likely that targeting the molecular machinery which is responsible for supporting matrix rigidity will reduce both fibrosis and fibronectin expression and in effect decrease tumor progression.
While indirect targets against fibronectin should undoubtedly yield more favorable outcomes for cancer patients, it would be ideal to directly target the biosynthesis of fibronectin. To this end, Tomasini-Johansson et al developed a small peptide inhibitor of fibronectin, pUR4B, which was shown to limit fibronectin polymerization in the matrix [41]. Work by Chiang et al has shown that administration of pUR4B in animals which had received a partial ligation of the external or internal carotid arteries resulted in reduction of intima, media and adventitial thickness of the vessels and further decreased the accumulation of fibronectin and collagen [42]. Furthermore, Hielscher et al demonstrated that treatment of co-cultures of fibroblasts and breast cancer cells with pUR4B not only reduced fibronectin deposition and organization in the ECM, but also inhibited the deposition of other matrix proteins [43]. Additionally, inhibition of fibronectin matrix assembly also attenuated vascular morphogenesis of endothelial cells cultured atop the fibronectin-deficient matrix [43]. These studies illustrate that fibronectin not only serves as a substrate regulating the assembly of other ECM proteins but is additionally important for supporting angiogenesis. As such, direct inhibition of fibronectin polymerization via a small peptide may impair the assembly of other matrix proteins and capillaries, thus depriving tumors of a supportive scaffold and the oxygen and nutrients necessary for continued growth. Indeed, the use of such anti-cancer peptides have not only been used to directly treat cancer but have also been used as cytotoxic drug carriers [44]. As such, it's possible that delivery of a peptide inhibitor of fibronectin polymerization may be utilized in conjunction with a chemotherapy regimen to improve outcomes for cancer patients. A benefit from use of an inhibitor of fibronectin polymerization is that the off target effects of the peptide should be minimal as fibronectin isn't actively produced in tissues with the exception of wounds and during development. One drawback may be concentrating the peptide to achieve maximal responses. As such, one may need to complex the inhibitor in a delivery vector to improve both tumor targeting and sustained delivery of the peptide at the tumor site.
RNA interference is rapidly becoming a powerful tool to silence the expression of genes and has been extensively investigated for its utility as a form of cancer therapy. Previous studies have shown that silencing fibronectin reduced tumor cell proliferation and growth [17] and tumor cell migration [6]. As such, silencing fibronectin gene expression via RNA silencing may be an effective strategy to enhance therapeutic responses. Despite its obvious advantages, the major drawback associated with use of gene based therapies is the delivery of these to the tumor. Traditionally, siRNA have been delivered locally where they've had much success with regard to treating ocular diseases [45]. For delivery of fibronectin siRNA to tumors, it will be important to utilize vectors which not only target the tumor, but penetrate the tumor cell membrane to deliver the packaged siRNA particles. Cell-penetrating peptides (CPP), which consist of short peptide sequences of 10-30 amino acids, have been shown to cross the cellular plasma membrane [46]. Derived from viral proteins or mammalian proteins with translocation capabilities, CPPs have been used to deliver siRNA to in-vitro cell cultures [47] and in transvascular delivery of siRNA to mice [48]. Although additional work will be necessary, the use of these vectors may be an avenue by which to deliver fibronectin siRNA to tumors.
Another means by which to target fibronectin expression could come from use of vaccinia viruses, double stranded DNA viruses used to treat cancer. Use of vaccinia viruses has received increased interest as strategies to engineer more potent oncolytic forms of vaccinia viruses has not only resulted in greater tumor specificity but have been shown to be an efficient means by which to lyse tumor cells [49]. Recently, a genetically engineered vaccinia virus was reported to target both malignant ovarian cancer cells in addition to the fibroblasts in the tumor stroma [50]. Given that the vaccinia virus was able to infect fibroblasts, it's possible that utilizing a similar strategy may halt the production of fibronectin from cancer associated fibroblasts as fibroblasts are a principle cell type responsible for ECM production.
Together, the aforementioned studies have provided a clearer perspective on the mechanisms whereby fibronectin exerts its tumorigenic effects in cancer. These studies suggest that fibronectin is not only aberrantly expressed in tumors, but also promotes tumor progression, metastasis and therapy resistance via activation of a number of downstream signaling pathways. With advances in this knowledge, it's foreseeable that employing strategies to inhibit tumor cell association with fibronectin, fibronectin-mediated cell signaling and/or fibronectin biosynthesis in the ECM will not only result in unprecedented discoveries about the mechanisms fibronectin exerts on tumor cell behavior, but will additionally result in development of novel therapeutic approaches to treat cancer.
Acknowledgements
Funding for this work was supported by the Philadelphia College of Osteopathic Medicine Centers for Chronic Disorders of Aging.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jarvelainen H, Sainio A, Koulu M, Wight TN, Penttinen R. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacological reviews. 2009;61:198-223
2. Pankov R, Yamada KM. Fibronectin at a glance. Journal of cell science. 2002;115:3861-3
3. Mierke CT, Frey B, Fellner M, Herrmann M, Fabry B. Integrin alpha5beta1 facilitates cancer cell invasion through enhanced contractile forces. Journal of cell science. 2011;124:369-83
4. Mitra AK, Sawada K, Tiwari P, Mui K, Gwin K, Lengyel E. Ligand-independent activation of c-Met by fibronectin and alpha(5)beta(1)-integrin regulates ovarian cancer invasion and metastasis. Oncogene. 2011;30:1566-76
5. Nam JM, Onodera Y, Bissell MJ, Park CC. Breast cancer cells in three-dimensional culture display an enhanced radioresponse after coordinate targeting of integrin alpha5beta1 and fibronectin. Cancer research. 2010;70:5238-48
6. Lou X, Han X, Jin C, Tian W, Yu W, Ding D. et al. SOX2 targets fibronectin 1 to promote cell migration and invasion in ovarian cancer: new molecular leads for therapeutic intervention. Omics: a journal of integrative biology. 2013;17:510-8
7. Wei PL, Kuo LJ, Huang MT, Ting WC, Ho YS, Wang W. et al. Nicotine enhances colon cancer cell migration by induction of fibronectin. Annals of surgical oncology. 2011;18:1782-90
8. Kenny HA, Chiang CY, White EA, Schryver EM, Habis M, Romero IL. et al. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. The Journal of clinical investigation. 2014;124:4614-28
9. Knowles LM, Gurski LA, Engel C, Gnarra JR, Maranchie JK, Pilch J. Integrin alphavbeta3 and fibronectin upregulate Slug in cancer cells to promote clot invasion and metastasis. Cancer research. 2013;73:6175-84
10. Eke I, Storch K, Krause M, Cordes N. Cetuximab attenuates its cytotoxic and radiosensitizing potential by inducing fibronectin biosynthesis. Cancer research. 2013;73:5869-79
11. Murphy LC, Seekallu SV, Watson PH. Clinical significance of estrogen receptor phosphorylation. Endocrine-related cancer. 2011;18:R1-14
12. Pontiggia O, Sampayo R, Raffo D, Motter A, Xu R, Bissell MJ. et al. The tumor microenvironment modulates tamoxifen resistance in breast cancer: a role for soluble stromal factors and fibronectin through beta1 integrin. Breast cancer research and treatment. 2012;133:459-71
13. Williams CM, Engler AJ, Slone RD, Galante LL, Schwarzbauer JE. Fibronectin expression modulates mammary epithelial cell proliferation during acinar differentiation. Cancer research. 2008;68:3185-92
14. Han SW, Roman J. Fibronectin induces cell proliferation and inhibits apoptosis in human bronchial epithelial cells: pro-oncogenic effects mediated by PI3-kinase and NF-kappa B. Oncogene. 2006;25:4341-9
15. Huang W, Chiquet-Ehrismann R, Moyano JV, Garcia-Pardo A, Orend G. Interference of tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumor cell proliferation. Cancer research. 2001;61:8586-94
16. Teng Y, Qiu J, Zheng Y. et al. Effects of type I collagen and fibronectin on regulation of breast cancer cell biological and biochemical characteristics. Journal of Medical and Biological Engineering. 2014;34:62-8
17. Sengupta S, Nandi S, Hindi ES, Wainwright DA, Han Y, Lesniak MS. Short hairpin RNA-mediated fibronectin knockdown delays tumor growth in a mouse glioma model. Neoplasia. 2010;12:837-47
18. Rybak JN, Roesli C, Kaspar M, Villa A, Neri D. The extra-domain A of fibronectin is a vascular marker of solid tumors and metastases. Cancer research. 2007;67:10948-57
19. Xiang L, Xie G, Ou J, Wei X, Pan F, Liang H. The extra domain A of fibronectin increases VEGF-C expression in colorectal carcinoma involving the PI3K/AKT signaling pathway. PloS one. 2012;7:e35378
20. Ou J, Deng J, Wei X, Xie G, Zhou R, Yu L. et al. Fibronectin extra domain A (EDA) sustains CD133(+)/CD44(+) subpopulation of colorectal cancer cells. Stem cell research. 2013;11:820-33
21. Rossnagl S, Altrock E, Sens C, Kraft S, Rau K, Milsom MD. et al. EDA-Fibronectin Originating from Osteoblasts Inhibits the Immune Response against Cancer. PLoS biology. 2016;14:e1002562
22. Khan ZA, Chan BM, Uniyal S, Barbin YP, Farhangkhoee H, Chen S. et al. EDB fibronectin and angiogenesis - a novel mechanistic pathway. Angiogenesis. 2005;8:183-96
23. Midulla M, Verma R, Pignatelli M, Ritter MA, Courtenay-Luck NS, George AJ. Source of oncofetal ED-B-containing fibronectin: implications of production by both tumor and endothelial cells. Cancer research. 2000;60:164-9
24. Han Z, Zhou Z, Shi X, Wang J, Wu X, Sun D. et al. EDB Fibronectin Specific Peptide for Prostate Cancer Targeting. Bioconjugate chemistry. 2015;26:830-8
25. Neil JR, Johnson KM, Nemenoff RA, Schiemann WP. Cox-2 inactivates Smad signaling and enhances EMT stimulated by TGF-beta through a PGE2-dependent mechanisms. Carcinogenesis. 2008;29:2227-35
26. Sawada K, Mitra AK, Radjabi AR, Bhaskar V, Kistner EO, Tretiakova M. et al. Loss of E-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer research. 2008;68:2329-39
27. Malik G, Knowles LM, Dhir R, Xu S, Yang S, Ruoslahti E. et al. Plasma fibronectin promotes lung metastasis by contributions to fibrin clots and tumor cell invasion. Cancer research. 2010;70:4327-34
28. Balanis N, Wendt MK, Schiemann BJ, Wang Z, Schiemann WP, Carlin CR. Epithelial to mesenchymal transition promotes breast cancer progression via a fibronectin-dependent STAT3 signaling pathway. The Journal of biological chemistry. 2013;288:17954-67
29. Meng XN, Jin Y, Yu Y, Bai J, Liu GY, Zhu J. et al. Characterisation of fibronectin-mediated FAK signalling pathways in lung cancer cell migration and invasion. British journal of cancer. 2009;101:327-34
30. Yuan J, Liu M, Yang L, Tu G, Zhu Q, Chen M. et al. Acquisition of epithelial-mesenchymal transition phenotype in the tamoxifen-resistant breast cancer cell: a new role for G protein-coupled estrogen receptor in mediating tamoxifen resistance through cancer-associated fibroblast-derived fibronectin and beta1-integrin signaling pathway in tumor cells. Breast cancer research: BCR. 2015;17:69
31. Rintoul RC, Sethi T. Extracellular matrix regulation of drug resistance in small-cell lung cancer. Clinical science. 2002;102:417-24
32. Spangenberg C, Lausch EU, Trost TM, Prawitt D, May A, Keppler R. et al. ERBB2-mediated transcriptional up-regulation of the alpha5beta1 integrin fibronectin receptor promotes tumor cell survival under adverse conditions. Cancer research. 2006;66:3715-25
33. Carbonell WS, DeLay M, Jahangiri A, Park CC, Aghi MK. beta1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer research. 2013;73:3145-54
34. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. The Journal of cell biology. 2012;196:395-406
35. Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28:4326-43
36. Ilic D, Kovacic B, Johkura K, Schlaepfer DD, Tomasevic N, Han Q. et al. FAK promotes organization of fibronectin matrix and fibrillar adhesions. Journal of cell science. 2004;117:177-87
37. Francois RA, Maeng K, Nawab A, Kaye FJ, Hochwald SN, Zajac-Kaye M. Targeting Focal Adhesion Kinase and Resistance to mTOR Inhibition in Pancreatic Neuroendocrine Tumors. Journal of the National Cancer Institute. 2015:107
38. Csiszar K. Lysyl oxidases: a novel multifunctional amine oxidase family. Progress in nucleic acid research and molecular biology. 2001;70:1-32
39. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891-906
40. Adam O, Theobald K, Lavall D, Grube M, Kroemer HK, Ameling S. et al. Increased lysyl oxidase expression and collagen cross-linking during atrial fibrillation. Journal of molecular and cellular cardiology. 2011;50:678-85
41. Tomasini-Johansson BR, Kaufman NR, Ensenberger MG, Ozeri V, Hanski E, Mosher DF. A 49-residue peptide from adhesin F1 of Streptococcus pyogenes inhibits fibronectin matrix assembly. The Journal of biological chemistry. 2001;276:23430-9
42. Chiang HY, Korshunov VA, Serour A, Shi F, Sottile J. Fibronectin is an important regulator of flow-induced vascular remodeling. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:1074-9
43. Hielscher A, Ellis K, Qiu C, Porterfield J, Gerecht S. Fibronectin Deposition Participates in Extracellular Matrix Assembly and Vascular Morphogenesis. PloS one. 2016;11:e0147600
44. Thundimadathil J. Cancer treatment using peptides: current therapies and future prospects. Journal of amino acids. 2012;2012:967347
45. de Fougerolles AR. Delivery vehicles for small interfering RNA in vivo. Human gene therapy. 2008;19:125-32
46. Foerg C, Ziegler U, Fernandez-Carneado J, Giralt E, Merkle HP. Differentiation restricted endocytosis of cell penetrating peptides in MDCK cells corresponds with activities of Rho-GTPases. Pharmaceutical research. 2007;24:628-42
47. Endoh T, Ohtsuki T. Cellular siRNA delivery using cell-penetrating peptides modified for endosomal escape. Advanced drug delivery reviews. 2009;61:704-9
48. Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL. et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39-43
49. Jefferson A, Cadet VE, Hielscher A. The mechanisms of genetically modified vaccinia viruses for the treatment of cancer. Critical reviews in oncology/hematology. 2015;95:407-16
50. Lopez MV, Rivera AA, Viale DL, Benedetti L, Cuneo N, Kimball KJ. et al. A tumor-stroma targeted oncolytic adenovirus replicated in human ovary cancer samples and inhibited growth of disseminated solid tumors in mice. Molecular therapy: the journal of the American Society of Gene Therapy. 2012;20:2222-33
Author contact
![]() Corresponding author: A. Hielscher, Department of Biomedical Sciences, Georgia Philadelphia College of Osteopathic Medicine, Suwanee, GA 30024 Telephone: (678) 225-7463. Fax: (678) 225-7479. E-mail: abigailhiedu
Corresponding author: A. Hielscher, Department of Biomedical Sciences, Georgia Philadelphia College of Osteopathic Medicine, Suwanee, GA 30024 Telephone: (678) 225-7463. Fax: (678) 225-7479. E-mail: abigailhiedu