Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(4):513-522. doi:10.7150/jca.17622 This issue Cite
Review
Molecular Subtyping of Pancreatic Cancer: Translating Genomics and Transcriptomics into the Clinic
Yongxing Du*, Bangbo Zhao*, Ziwen Liu, Xiaoxia Ren, Wenjing Zhao, Zongze Li, Lei You ![]() , Yupei Zhao
, Yupei Zhao ![]()
Department of General Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100730, PR China.
*Authors share co-first authorship.
Received 2016-9-18; Accepted 2016-12-23; Published 2017-2-11
Abstract
Pancreatic cancer remains one of the most lethal malignancies, and insights into both personalized diagnosis and intervention of this disease are urgently needed. The rapid development of sequencing technologies has enabled the successive completion of a series of genetic and epigenetic sequencing studies of pancreatic cancer. The mutational landscape of pancreatic cancer is generally portrayed in terms of somatic mutations, structural variations, epigenetic alterations and the core signaling pathways. In recent years, four significant molecular subtype classifications of pancreatic cancer have been proposed based on the expression of transcription factors and downstream targets or the distribution of structural rearrangements. Increasing researches focus on the identification of somatic mutations and other genetic aberrations that drive pancreatic cancer has led to a new era of precision medicine based on molecular subtyping. However, few known molecular classifications are used to guide clinical strategies. Specific scientific, regulatory and ethical challenges must be overcome before genomic and transcriptomic discoveries can be translated into the clinic.
Keywords: pancreatic cancer, molecular subtyping, precision medicine.
Introduction
Pancreatic cancer is one of the most lethal malignancies worldwide. The incidence of pancreatic cancer is gradually increasing, while the 5-year survival rate has remained stable at 7-8% [1]. According to the American Cancer Society, approximately 53,070 new cases of pancreatic cancer were diagnosed in 2016, and 41,780 affected individuals are expected to die from the disease. Pancreatic cancer is projected to be the second leading cause of death by 2030 [2]. Few improvements have been made in the diagnosis and treatment of pancreatic cancer despite extensive efforts over the past few decades. Owing to the lack of clinically validated early screening methods, over 80% of patients are diagnosed at an advanced stage, at which time the cancer is generally considered unresectable. There are a limited number of other effective treatment modalities that can significantly increase overall survival. Patients, clinicians and researchers alike are frustrated at the lack of progress, and new strategies are needed to better understand this disease [3].
Since completion of the Human Genome Project, genome sequencing technologies have advanced considerably, contributing to the emergence of a new scientific era of “omics”, which has revolutionized the study of malignant tumors. The development of next-generation sequencing (NGS) has resulted in increased efficiency and reduced costs, which have greatly facilitated both cancer genomic discoveries and their translation into the clinic [4]. In previous studies [5, 6], recurrent mutations in several genes, including KRAS, TP53, CDKN2A and SMAD4, have been identified in pancreatic cancer. These findings have improved our understanding of the initiation and progression of this type of cancer. However, unlike breast [7], prostate [8], gastric [9] and colorectal cancers [10], there is no known molecular taxonomy of pancreatic cancer that can guide therapeutic strategies. In recent years, increasing research focus on the identification of somatic mutations and other genetic alterations that drive pancreatic cancer has led to a new era of precision medicine based on molecular subtyping. However, specific scientific, regulatory and ethical challenges must be overcome before the molecular subtyping of pancreatic cancer can be applied in clinical practice. This article summarizes the recent findings of large-scale genomic analyses of pancreatic cancer, highlights the promising clinical prospects of molecular subtyping, and discusses the potential challenges of and solutions for translating genomics and transcriptomics into clinical practice. Pancreatic ducal adenocarcinoma (PDAC), more commonly called “pancreatic cancer”, is the most common solid tumor type in the pancreas; accordingly, in the following sections, “pancreatic cancer” refers to invasive ductal adenocarcinoma of the pancreas unless otherwise specified. Other pathologies of pancreatic malignancy, including variants of ductal adenocarcinoma, acinar carcinoma, endocrine tumors and malignant cystic neoplasms, are relatively less commonly observed in the clinic (Table 1) [11-13]; therefore, they are only briefly mentioned in this review.
Pathologies of major pancreatic malignancies.
| Tumor classification | Tumor type | Clinical significance |
|---|---|---|
| Pancreatic exocrine tumors (>95%) | Invasive ductal adenocarcinoma | The most common type of pancreatic exocrine neoplasm. Accounts for more than 80% of cases. Very poor prognosis. |
| Acinar carcinoma | Accounts for less than 1% of cases. Fully malignant. 15% of cases are associated with metastatic fat necrosis. Better overall prognosis than ductal adenocarcinoma. | |
| Pancreatoblastoma | Accounts for less than 1% of cases. More common in infants and children than in adults. Less aggressive and better prognosis than ductal adenocarcinoma. | |
| Variants of ductal adenocarcinoma (adenosquamous, colloid, medullary, undifferentiated, etc.) | Adenosquamous (4% of cases), colloid (2%), others rare. Most share a similarly poor long-term prognosis, except for colloid carcinoma, which has a somewhat better prognosis. | |
| Cystic neoplasm with invasive carcinoma | Intraductal papillary mucinous neoplasm (IPMN) (2-3% of cases), mucinous cystic neoplasm (MCN) (1%), and solid-pseudopapillary neoplasm (SPT) (<1%). Better prognosis than ductal adenocarcinoma. | |
| Pancreatic endocrine tumors (<5%) | Pancreatic neuroendocrine tumors (PNETs) (glucagonoma, VIPoma, gastrinoma etc.) | Commonly accompanied by a clinical syndrome due to aberrant hormone production. Fully malignant, with a 45% 10-year survival rate. |
The mutational landscape of pancreatic cancer
Researchers from the U.S., Australia, and other countries have carried out a massive number of genome sequencing studies of pancreatic cancer, with the number of recruited patients varying from 7 to 456 (Table 1) [14-21]. Using a number of advanced high-throughput techniques, several point mutations and structural variations have been detected.
Somatic mutations
The number of high-confidence mutations identified varies depending on the method used and number of samples assessed. The average number of mutations detected per patient is 21-857, and approximately 75% of these mutations are not silent [14, 18, 20, 21]. Similar to colorectal and brain cancer, the somatic mutation spectrum of pancreatic cancer includes a preponderance of mutations at 5'-CpG sites. Jones et al. [14] scored 924 mutations using an algorithm to evaluate missense mutations and found that 17.3% were predicted to contribute to tumorigenesis. The average number of somatic mutations in pancreatic cancer is considerably less than those in breast/colorectal cancers. One plausible explanation for this is that the cells that initiate pancreatic tumorigenesis have undergone fewer divisions than colorectal or breast cancer cells and that a greater proportion of genes are positively selected during pancreatic tumorigenesis.
Structural variations
There are fewer copy number variations (CNVs) than somatic mutations, and amplification is an even rarer occurrence in pancreatic cancer [14]. The most frequent genetic gain has been detected on chromosome 8q (96%), and the most recurrent genetic loss has been observed on chromosome 9p (78%) [22].
High-throughput sequencing studies of pancreatic cancer.
| Author | Publication Year | Case Number | Sequencing Method | Main Discovery |
|---|---|---|---|---|
| Jones S | 2008 | 24 | Exome Sequencing | Identified a core set of 12 altered cellular signaling pathways and processes. |
| Yachida S | 2010 | 7 | Exome Sequencing | Demonstrated genetic heterogeneity of metastatic cancer within primary carcinoma and a large window of opportunity for early detection. |
| Campbell PJ | 2010 | 13 | Parallel paired-end sequencing | Demonstrated genomic instability and genetic heterogeneity. |
| Collisson EA | 2011 | 2 databases | Gene expression microarray | Identified three molecular subtypes and presented evidence of differences in clinical outcomes and therapeutic responses among them. |
| Biankin AV | 2012 | 99 | Whole-genome sequencing; Copy number variation analysis | Identified 16 significantly mutated genes, as well as frequent and diverse somatic aberrations in genes involved in axon guidance, particularly SLIT/ROBO signaling. |
| Moffitt RA | 2015 | 206 | Gene expression microarray | Identified and validated the 'classical' and 'basal-like' tumor subtypes; defined the 'normal' and 'activated' stromal subtypes. |
| Waddell N | 2015 | 100 | Whole-genome sequencing; Copy number variation analysis | Classified PDAC into 4 molecular subtypes according to patterns of structural variation. |
| Bailey P | 2016 | 456 | Whole-genome sequencing; Deep exome sequencing; CNV analysis | Identified 32 recurrently mutated genes grouped into 10 pathways; defined 4 PDAC molecular subtypes by expression analysis. |
Despite the low prevalence of homozygous deletions, in most types of cancer their presence indicates the existence of a tumor suppressor gene within the deleted region, according to the allelic two-hit hypothesis. In contrast, in pancreatic cancer, approximately 10 genes are typically eradicated by homozygous deletions, thus providing appropriate targets for therapeutic strategies [14]. Two tumor suppressor genes, CDKN2A and CDKN2B, are located on chromosome 9p21.3, which is deleted at the highest frequency (63%). Other deleted genes include DCC (18q21.1, 48%), SMAD4 (18q21.1, 33%), MAP2K4 (17p12, 30%), TP53 (17p13.1, 26%) and RUNX3 (1p36.11, 22%). On the other hand, SCAP2 (SKAP2, 7p15.2) is the most frequently (63%) amplified gene. An average of 43-119 structural variations have been detected in previous studies, and they have been classified into the following 7 types: intra-chromosomal rearrangements, deletions, duplications, tandem duplications, inversions, fold-back inversions and amplified inversions [16, 20]. Intra-chromosomal rearrangements have been demonstrated to be the most frequently occurring variations, with considerable inter-individual heterogeneity in terms of the total number of events [16]. The distribution of rearrangements in pancreatic cancer differs from that in breast cancer. In particular, deletions (22% vs. 13%) and fold-back inversions (16% vs. 2%) occur more frequently in pancreatic cancer, whereas recurrent gene fusions do not occur.
Epigenetic alterations
The importance of epigenetic modifications, such as DNA methylation, in tumorigenesis is increasingly being acknowledged. Hypermethylation of CpG islands and promoter regions is associated with the transcriptional silencing of tumor suppressor genes, and conversely, hypomethylation is associated with the over-expression of oncogenes and genomic instability [23]. Omura et al. observed that MDF1, miR-9-1, ZNF415, CNTNAP2 and EVOLV-4 were the most frequently methylated loci by assessing 88,000 probes in high-throughput analysis comparing the whole methylomes of PDAC and healthy tissues [24]. In addition, analysis of the methylation profiles of 167 untreated resected PDACs using high-density arrays revealed that a total of 11,634 CpG sites associated with 3,522 genes were significantly differentially methylated (DM) [25]. As expected, PDAC hypermethylation was the most prevalent in the 5' region of genes, which includes the proximal promoter, 5' untranslated region (5'UTR) and CpG islands. Approximately 33% of the DM genes were significantly inversely correlated with the mRNA expression levels. Moreover, alteration of miRNA expression by aberrant DNA methylation is an early event that occurs during PDAC carcinogenesis, but the consequences of these modifications remain unclear [26].
Genes and pathways affected by mutations and structural variations
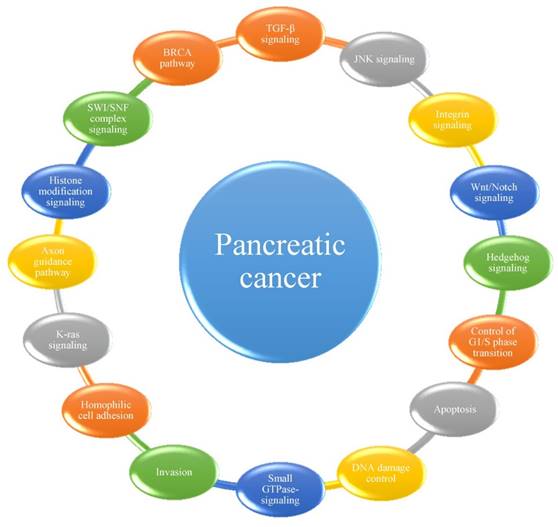
Among the mutations and structural variations described above, a number of significantly mutated genes have been confirmed or newly detected. Classically, KRAS has been demonstrated to be activated in 92-100% of pancreatic cancer patients, whereas TP53, SMAD4 and CDKN2A have been shown to be inactivated in 74-83%, 31-33% and 35-75% of these patients, respectively [14, 18, 20, 21]. Analyses of passenger mutation rates have resulted in the detection of numerous other genes of potential biological interest, including the transcriptional activator MLL315, the DNA damage response gene ATM15, and the SWI/SNF interacting partner KDM6A12. Many of these genes had not been previously identified to play an important role in this type of cancer. Considering that most cellular pathways and processes involve multiple proteins that function in a concerted manner, Jones et al. [14] first grouped significantly genetically altered gene sets in pancreatic cancer into 12 core signaling pathways or processes. In subsequent studies [18, 20, 21], several other pathways were determined to play important roles in the progression of pancreatic cancer (Figure 1). It has been suggested that the best hope for therapeutic advances may lie in the discovery of significantly altered pathways and processes rather than in their individual genetic components, highlighting the importance of identifying the core pathways and processes in pancreatic cancer [14].
The 16 pathways whose component genes are genetically altered in most pancreatic cancers.
Molecular subtypes of pancreatic cancer
Genome- and gene expression-based subtypes have been widely accepted as methods of disease stratification. However, the translation of this subtyping into the clinic has been hindered by inconsistent results, which are likely due to differences in the data processing methods, algorithms applied to diverse patient cohorts, sample preparation methods, and sequencing technologies used, as well as the criteria used for subtyping. Pancreatic cancer has been divided into four main molecular subtypes based on the expression of transcription factors and downstream targets or the distribution of structural rearrangements (Table 3).
Molecular subtyping studies of pancreatic cancer
| First author | Subtyping method | Molecular subtypes | Clinical significance |
|---|---|---|---|
| Collisson EA | Transcriptional profiles | 3 subtypes: classical, quasi-mesenchymal, exocrine-like | Classical subtype - GATA6 - erlotinib; QM subtype - KRAS - gemcitabine |
| Moffitt RA | Transcriptional profiles | 4 subtypes: classical and basal-like tumor subtypes and normal and activated stromal subtypes | / |
| Waddell N | Structural variations | 4 subtypes: stable, locally rearranged, scattered, unstable | Unstable subtype - BRCA pathway - platinum-based therapy |
| Bailey P | Transcriptional profiles | 4 subtypes: squamous, pancreatic progenitor, immunogenic, aberrantly differentiated endocrine exocrine | / |
Collisson classification
The first impactful PDAC molecular subtyping study was published by Collisson in 2011 [17]. This group performed combined analysis of the transcriptional profiles of primary PDAC samples from several studies, as well as those of human and mouse PDAC cell lines, to overcome the limitation of an insufficient number of tumor specimens. They attempted to identify the PDAC subtypes by first detecting intrinsically variable genes in two gene expression microarray datasets for resected PDAC and then performing nonnegative matrix factorization (NMF) analysis with consensus clustering. They identified three subtypes, which were designated as classical, quasi-mesenchymal (QM-PDA) and exocrine-like based on differences in subtype-specific gene expression. The classical subtype was characterized by high expression of adhesion-associated and epithelial genes, such as transmembrane protein 45B (TMEM45B), trefoil factor 1 (TFF1) and mucin 13 (MUC13); the QM-PDA subtype had high expression of mesenchyme-associated genes, such as absent in melanoma 2 (AIM2), glycoprotein m6b (GPM6B) and 5'-nucleotidase, ecto (NT5E); and the exocrine-like subtype had relatively high expression of tumor cell-derived digestive enzyme genes, such as regenerating islet-derived 1 beta (REG1B), pancreatic lipase-related protein 2 (PNLIPRP2) and cystic fibrosis transmembrane conductance regulator (CFTR). Analysis of the expression of PDAC signature genes in other published expression datasets of unique origin, platform or processing supported the identities of these three subtypes, demonstrating the robust nature of subtype classification in the early stages of PDAC [27, 28].
Moffitt classification
A key hallmark of PDAC is extensive stromal involvement, which makes it difficult to capture precise tumor-specific molecular information. Moffitt et al. [19] overcame this obstacle by applying blind source separation to a diverse collection of PDAC gene expression microarray data. They identified two factors that described gene expression in the stroma. Consensus clustering of exemplar genes according to these two stromal factors resulted in division of the tumor samples into two stromal subtypes: 'normal' and 'activated'. 'Normal' stroma was characterized by relatively high expression of known markers of pancreatic stellate cells, such as ACTA2, VIM and DES. In contrast, 'activated' stroma was characterized by high expression of a more diverse set of genes associated with macrophages, such as the integrin ITGAM and the chemokine ligands CCL13 and CCL18. The expression of other genes in 'activated' stroma, including the secreted protein SPARC, the Wnt family members WNT2 and WNT5A, MMP9 and MMP11, demonstrated its role in tumor promotion. Moffitt also proposed that another two tumor-specific factors defined the 'classical' and 'basal-like' subtypes of PDAC. Gene expression in the basal-like subtype is consistent with that in basal subtypes previously identified in bladder and breast cancers, with the upregulation of genes such as VGLL1, UCA1 and S100A2. In contrast, the 'classical' subtype exhibits low expression of the abovementioned genes and overexpression of genes such as BTNL8, FAM3D and ATAD4. Basal-like and classical tumors have been observed in both the normal and activated stromal subtypes, and considering this information, 4 molecular subtypes of PDAC were ultimately proposed: 'basal-like tumor and normal stroma', 'basal-like tumor and activated stroma', 'classical tumor and normal stroma' and 'classical tumor and activated stroma'.
Bailey classification
Recently, Bailey et al. [21] also defined a form of molecular subtyping of PDAC based on integrated genomic analysis of 456 PDAC tumors. Initial unsupervised clustering of RNA-Seq data for 96 tumors with a high epithelial content (≥40%) to balance stromal gene expression resulted in the identification four stable classes of pancreatic cancer, namely squamous, pancreatic progenitor, immunogenic and aberrantly differentiated endocrine exocrine (ADEX), on the basis of differences in the expression of transcription factors and downstream targets. Squamous tumors contained mutated TP53 and KDM6A genes, an upregulated TP63∆N transcriptional network and hypermethylated pancreatic endodermal cell fate-determining genes. Pancreatic progenitor tumors preferentially expressed genes involved in early pancreatic development (FOXA2/3, PDX1 and MNX1). Immunogenic tumors exhibited upregulation of immune networks, including pathways involved in acquired immune suppression. ADEX tumors contained highly expressed genes involved in KRAS activation, exocrine differentiation (NR5A2 and RBPJL), and endocrine differentiation (NEUROD1 and NKX2-2). These four subtypes were also found to be represented among an extended set of 232 PDAC tumors by analysis of array-based mRNA expression profiles covering the full range of tumor cellularity (from 12-100%).
Waddell classification
In addition to the classification of PDAC according to gene expression profiles, the prevalence of aberrations in key driver genes and pathways in this disease suggests that structural variations represent an important mutational mechanism in pancreatic carcinogenesis. Inspired by this observation, Waddell et al. [20] performed CNV analysis of 100 PDACs and observed a high prevalence of chromosomal rearrangements involving genes known to be important in pancreatic cancer (TP53, SMAD4, CDKN2A, ARID1A and ROBO2), as well as new candidate drivers of pancreatic carcinogenesis (KDM6A and PREX2). PDAC was classified into four subtypes based on the patterns of structural variation. Subtype 1 was classified as 'stable' (20% of all samples); the genomes of these tumors contained ≤50 structural variation events and often exhibited widespread aneuploidy, suggesting defects in the cell cycle/mitosis. The point mutation rates for KRAS and SMAD4 were similar to those in the other subtypes, and the prevalence of TP53 mutations was only slightly lower. Subtype 2 was classified as 'locally rearranged' (30% of all samples). The genomes of these tumors exhibited significant focal events on one or two chromosomes. Approximately one-third of the tumors of this subtype exhibited copy number gains in regions harboring known oncogenes, including common focal amplifications in KRAS, SOC9 and GATA6 and in therapeutic targets, such as ERBB2, MET, CDK6, PIK3CA and PIK3R3; however, they each exhibited a low individual prevalence. Subtype 3 was classified as 'scattered' (36% of all samples), and these tumors displayed a moderate range of non-random chromosomal damage and less than 200 structural variation events. Subtype 4 was classified as 'unstable' (14% of all samples), and these tumors exhibited a large number of structural variation events (>200). This scale of genomic instability suggests defects in DNA maintenance, which potentially indicates sensitivity to DNA-damaging agents.
Relevant studies of clinical applications of PDAC molecular subtyping
Prognostic indicator
Genomic and transcriptomic analyses can provide a comprehensive overview of an individual patient's cancer, and this information is beginning to impact clinical decision making [29].
Survival after PDAC resection has been associated with many factors, including stage (tumor size and nodal involvement) and grade (degree of differentiation), but no single factor has been consistently associated with prognosis. These inconsistencies support the use of subtypes as independent prognostic indicators in resected PDAC. In Collisson's study [17] supported by one PDAC dataset (UCSF), patients with classical-subtype tumors fared better than those with QM-PDA-subtype tumors after resection. Models including stage and subtype have revealed that subtype is an independent predictor of overall survival. Similarly, according to Moffitt's classification [19], patients with the activated stromal subtype or basal-like tumor subtype have a worse median survival time and 1-year survival rate than those with the normal stromal subtype or classical tumor subtype. Another transcriptomic analysis conducted by Bailey et al. [21] resulted in identification of the following 4 subtypes associated with specific histological characteristics: squamous with adenosquamous carcinoma; pancreatic progenitor and immunogenic with mucinous non-cystic (colloid) adenocarcinoma; carcinoma arising from IPMN, which is mucinous; and ADEX with rare acinar cell carcinoma. The squamous subtype was determined to be an independent prognostic factor for poor survival, with the shortest median survival time of 13.3 months compared with survival times of 30.3, 25.6 and 23.7 months for the three other subtypes, respectively.
Predictive biomarker
Clinicians prefer to use predictive biomarkers for selection of the optimal therapies for individual patients, and these therapeutic decisions are based on associations between the biomarker results and response rates to certain therapies. In practice, predictive biomarkers often identify the molecular targets of relevance to targeted anticancer drugs, and accordingly, molecular subtypes could represent promising predictive biomarkers. It has been shown that classical PDA cell lines are relatively more dependent on KRAS than QM-PDA cell lines, suggesting that KRAS-directed therapy might have the greatest efficacy against classical PDA [17]. Additionally, QM-PDA cell lines have been shown to be more sensitive to gemcitabine than classical subtype cell lines, which are more sensitive to erlotinib [17]. These results further establish phenotypic differences between the classical and QM-PDA subtypes and indicate that gemcitabine and erlotinib are preferentially active in different PDA subtypes. Therefore, the current practice of combining these drugs may increase toxicity without increasing efficacy for many patients. Alternatively, combining agents with similar subtype specificities may be preferable. As mentioned above, the basal-like subtype of Moffitt's classification is associated with a worse prognosis, independent of race and stroma; however, patients with basal-like tumors show a strong tendency toward a better response to adjuvant therapy [19]. Drug resistance is likely an insurmountable obstacle that prevents the successful treatment of PDAC. Studies have confirmed that cytochrome P450 3A5 (CYP3A5) contributes to acquired drug resistance in QM-PDA and classical PDAC by promoting drug metabolism [30]. These findings indicate that CYP3A5 is a predictor of the therapeutic response in patients with QM-PDA and classical PDAC.
Waddell et al. [20] examined the relationships between the unstable subtype and mutations in BRCA pathway genes and found that the majority of unstable-subtype tumors fell within the top quintile of the BRCA mutational signature. To determine the putative genotypes associated with platinum responsiveness, 8 patients were administered platinum-based therapy, and 7 patients derived xenografts (PDXs) were treated with gemcitabine and cisplatin. Among 5 patients with unstable genomes and/or a high BRCA mutational burden, 2 had exceptional responses, and 2 had robust partial responses. Three patients who did not have any of these characteristics ('off genotype') showed no response. Defining biomarkers of platinum responsiveness would significantly alter the current treatment approaches to PDAC and improve overall outcomes. Furthermore, diagnostic whole-genome sequencing (WGS) for the detection of surrogate measures of defects in DNA maintenance may ultimately be a better method for identifying potential responders to platinum and PARP inhibitor therapies.
Opportunities and challenges in translating molecular subtyping of PDAC into clinic practice
Bringing NGS-based cancer genomic testing up to clinical-grade standards to support clinical decision making necessitates knowledge of and adherence to standards for molecular diagnostics and management. Research discoveries derived from cancer genomic and transcriptomic studies have potential clinical impacts as diagnostic, prognostic, and predictive biomarkers [10].
Major technical considerations in molecular subtyping of PDAC
Whole genome or exome?
WGS and whole-exome sequencing are two fundamental technologies that enable the in-depth sequencing of cancer genomes. Because of financial constraints, many cancer genome projects involve the sequencing of exomes rather than whole genomes; however, it remains debatable whether this is the best approach [31-33]. WGS of a lung cancer cell line resulted in the identification of 22,910 point mutations, only 134 (0.6%) of which were in exomes, and the consequences of the other 22,776 mutations were not known [34]. The identification of mutations in noncoding regions may greatly improve our understanding of cancer and uncover new mechanisms of cancer pathogenesis [35]. Moreover, the time and cost invested in characterizing mutations in noncoding regions impedes progress in understanding the consequences of mutations within coding regions [31-33]. Several influential WGS analyses of PDAC [14, 18, 20, 21] have contributed to our understanding of tumorigenesis. Nevertheless, in the clinical setting, treatment decisions must be based on a variety of factors.
Subtyping based on transcriptional or mutational profiles?
Gene expression profiling studies have enabled the molecular classification of pancreatic cancer into clinically relevant subtypes and have provided new tools for predicting disease recurrence and patient responses to different treatments, as well as new insights into various oncogenic pathways and the process of metastatic progression [31]. A major challenge for gene expression profiling studies, especially those with clear clinical implications, is independent validation [36]. Most of these studies are retrospective, with analysis of data obtained many years prior. Therefore, they do not provide the required level of evidence that can be achieved in a single, high-powered, prospective, randomized controlled trial or meta-analysis or overview of several well-designed studies [37]. Molecular subtyping based on so-called somatic mutational profiles has not been previously published. Previous studies have revealed that somatic mutational profiles are extremely sparse, with typically fewer than 100 mutated bases in an entire exome [7, 38]. These profiles are also remarkably heterogeneous, and it is very common for clinically identical patients to share no more than a single mutation. Notably, researchers have recently developed a new promising method for integrating somatic tumor genomes with gene networks, referred to as network-based stratification (NBS), which allows for the stratification of cancer into informative subtypes by the clustering together of patients with mutations in similar network regions [39, 40]. NBS has been successfully used in studies of ovarian, uterine and lung cancers, and for each tissue type, it identifies subtypes that are predictive of clinical outcomes. As the whole genomic landscape of PDAC has been elucidated, NBS offers an intriguing opportunity for the subtyping of this disease.
Integration of genomic and transcriptomic data
The advent of NGS and the resulting rapid increase in the generation of genome-scale data have created many challenges for data integration. Genomic data integration among multiple samples and techniques is essential for making reliable inferences [20] because in most cases, data obtained using only a single technique tells only part of the story. Genomic sequencing can identify structural variations, but only the addition of a technique that assesses RNA levels, such as RNA-Seq, can reveal whether the structural variations affect gene transcription [20]. As genomic sequencing and RNA-Seq are the two main methods of tumor subtyping, the use of both may establish a more clinically meaningful taxonomy of cancer. The main hurdles to their integration are the heterogeneity of experimental and analytic protocols, differing levels of data quality, and differences in data representation [41, 42].
Challenges in defining diagnostic, prognostic and predictive biomarkers
The characteristics of each molecular subtype of PDAC can be regarded as diagnostic biomarkers, but these biomarkers are far from being ready to be applied in the clinical setting. All of the methods based on whole-genome or exome analyses pose psychological, technical and economic obstacles to clinicians. Gray et al. surveyed 160 physicians at an academic cancer center and found that 22% reported “low confidence in their genomic knowledge,” suggesting a need for guidelines and education of physicians to support increased understanding of genomic testing [43]. In addition, patients have reported being concerned with incidental findings, discrimination, and obtaining additional information or genetic counseling [44]. Technically, WGS using small diagnostic samples that are preserved in a fixative, such as formalin, analytical demands and the reporting of results within a clinically relevant timeframe are significant hurdles that urgently need to be overcome [20]. Despite the declining costs of genomic sequencing with the advent of NGS technologies, the subsequent costs of molecular-targeted drugs remain a considerable challenge for both patients and the medical insurance system.
In the course of evaluating the prognosis of patients with malignant tumors by molecular subtyping, prognostic gene signatures are extremely useful, in addition to traditional prognostic markers, such as tumor size, histological grade, patient age, and the statuses of the resection margin and local lymph nodes [45]. In early breast cancer, several independently validated gene expression signatures related to prognosis, such as the Amsterdam and Rotterdam signatures, have been identified [46]. Each of these signatures contains tens to hundreds of genes, contributing to increases in both the efficacy and accuracy of judgment. However, in PDAC, specific individual genes or a whole molecular subtype have been demonstrated to be associated with prognosis. Future studies should focus on the identification of more instructive variables, such as such prognostic gene signatures.
Another valuable translational approach is to develop therapeutic agents that target genetic aberrations. The identification of markers that can be used to predict responses to particular drugs remains challenging, as commonly used therapeutic agents are ineffective in many unselected patients, and side effects often develop [45]. As mentioned above, the excellent response to platinum-based therapy of patients classified with Waddell's unstable subtype is informative for the identification of biomarkers of platinum responsiveness [20]. Mutations in BRCA pathway component genes and surrogate measures of defects in DNA maintenance (genomic instability and the BRCA mutational signature) have potential implications for therapeutic selection for PDAC, defining a putative biomarker hypothesis that requires testing in a clinical trial. Indeed, these gene predictors have been established and validated in studies with small sample sizes, leading to questions regarding their robustness. Moreover, these studies have been highly heterogeneous with respect to the endpoints, treatment regimens used, and patient populations included, and many have been complicated by statistical issues [45]. Thus, the identification of reliable and effective predictive biomarkers for each molecular subtype is still a long way off.
Issues involved in personalized medicine clinical trials of PDAC
The emerging molecular taxonomy of PDAC supported by NGS analyses is providing unprecedented opportunities to accelerate the development of personalized medicine strategies [38]. Clinical trials are being increasingly designed based on molecular characteristics identified using genomic technologies; however, the translation of molecularly guided oncologic care into practice presents complex challenges. A clinical trial based on Waddell's classification, called the Individualized Molecular Pancreatic Cancer Therapy (IMPaCT) trial, was initially designed to exploit results obtained from the genomic sequencing of pancreatic cancer patients in Australia [38]. However, among the 93 recruited patients, 17 could not be screened because their tissue samples were deemed unsuitable for molecular testing or were inaccessible. Among the 76 patients who underwent molecular testing, 22 eligible candidates were identified after a median waiting time of 21.5 days, none of whom had been successfully treated in the IMPaCT study. Given that the median survival time of PDAC patients is only a few months, there is also a significant ethical issue regarding technological obstacles, as waiting the median length of time of 20-26 days from consent to reporting of the final genomic analysis results [47, 48] may be catastrophic for recruited patients who have not been treated with the first-line chemotherapy regimen. Likewise, in a Phase I Clinical Trial Program conducted at the University of Texas MD Anderson Cancer Center, 34 pancreatic cancer patients were screened (2.9% of 1,144 patients with adequate tissue samples for molecular analysis), with only 1 proceeding to receive matched targeted therapy [49]. Analogous clinical trials have experienced similar difficulties due to the need to exclude patients because of an inability to obtain biopsy, insufficient or no tumor content in the available specimen, deteriorating performance status, or patient withdrawal of consent or selection of an alternative treatment [50-53].
A new organizational structure should be established to advance molecular-targeted therapy trials. This would involve clinicians in specific clinical disciplines, such as interventional radiology and molecular pathology, who are responsible for the skilled procurement of these specimens, at centers of excellence that can provide rapid turnaround times. With regard to the abovementioned ethical issue, studies could be designed that appeal to patients, such as nonrandomized studies of novel agents, and that allow for standard-of-care treatment to begin while molecular analysis is underway [38].
Conclusion
Pancreatic cancer remains one of the most lethal malignancies, and it poses a major health burden. The advent of NGS technologies has provided the opportunity to more conveniently and affordably portray the whole-genome landscape of PDAC. The use of these technologies has resulted in the identification of numerous novel somatic mutations, CNVs, structural variations and epigenetic variations. Based on the gene expression profiles and patterns of structural variation observed, four types of molecular classifications have been established that will contribute to the translation of novel discoveries of diagnostic, prognostic and predictive biomarkers into the clinic. Hurdles such as the technical challenges of WGS, the clinical interpretation of tumor sequencing results and economic factors should be addressed. Additional resources and funding are also necessary to support the ongoing profiling efforts in basic genomics research, tumor sequencing in the clinic, and data-sharing networks to enable precision cancer medicine.
Abbreviations
NGS: next-generation sequencing; CNVs: copy number variations; PDAC: pancreatic ductal adenocarcinoma; IPMN: intraductal papillary mucinous neoplasm; MCN: mucinous cystic neoplasm; SPT: solid pseudopapillary neoplasm; PNETs: pancreatic neuroendocrine tumors; DM: differentially methylated; NMF: nonnegative matrix factorization; QM-PDA: quasi-mesenchymal pancreatic ductal adenocarcinoma; TMEM45B: transmembrane protein 45B; TFF1: trefoil factor 1; MUC13: mucin 13; AIM2: absent in melanoma 2; GPM6B: glycoprotein m6b; NT5E: 5'-nucleotidase, ecto; REG1B: regenerating islet-derived 1 beta; PNLIPRP2: pancreatic lipase-related protein 2; CFTR: cystic fibrosis transmembrane conductance regulator; ADEX: aberrantly differentiated endocrine exocrine; PDXs: patient-derived xenografts; WGS: whole-genome sequencing; NBS: network-based stratification; IMPaCT: Individualized Molecular Pancreatic Cancer Therapy.
Acknowledgements
This research was supported by the National Nature Science Foundation of China (81572459, 81672960, and 81672443) and Special Funds for Health Public Welfare Projects (201402001).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30
2. Rahib L, Smith BD, Aizenberg R. et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913-21
3. Makohon-Moore A, Iacobuzio-Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer. 2016;16:553-65
4. Mardis ER. Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif). 2013;6:287-303
5. Mimeault M, Brand RE, Sasson AA. et al. Recent advances on the molecular mechanisms involved in pancreatic cancer progression and therapies. Pancreas. 2005;31:301-16
6. Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157-88
7. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61-70
8. Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011-25
9. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202-9
10. Guinney J, Dienstmann R, Wang X. et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350-6
11. Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK. et al. Recent progress in pancreatic cancer. CA: a cancer journal for clinicians. 2013;63:318-48
12. Hruban RH, Pitman MB, Klimstra DS. Tumors of the pancreas. In: Atlas of tumor pathology, Fascicle 6, Armed Forces Institute of Pathology, Washington, DC. 2007 Vol 4th series
13. Chaper 12 - Tumours of the Pancreas. In: WHO Classification of Tumours of the Digestive System, Bosman FT, Carneiro F, Hruban RH, Theise ND (Eds), IARC Press, Lyon 2010.
14. Jones S, Zhang X, Parsons DW. et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801-6
15. Yachida S, Jones S, Bozic I. et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114-7
16. Campbell PJ, Yachida S, Mudie LJ. et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109-13
17. Collisson EA, Sadanandam A, Olson P. et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500-3
18. Biankin AV, Waddell N, Kassahn KS. et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399-405
19. Moffitt RA, Marayati R, Flate EL. et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015;47:1168-78
20. Waddell N, Pajic M, Patch AM. et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495-501
21. Bailey P, Chang DK, Nones K. et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47-52
22. Harada T, Chelala C, Bhakta V. et al. Genome-wide DNA copy number analysis in pancreatic cancer using high-density single nucleotide polymorphism arrays. Oncogene. 2008;27:1951-60
23. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415-28
24. Omura N, Li CP, Li A. et al. Genome-wide profiling of methylated promoters in pancreatic adenocarcinoma. Cancer Biol Ther. 2008;7:1146-56
25. Nones K, Waddell N, Song S. et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int J Cancer. 2014;135:1110-8
26. Delpu Y, Hanoun N, Lulka H. et al. Genetic and epigenetic alterations in pancreatic carcinogenesis. Curr Genomics. 2011;12:15-24
27. Balagurunathan Y, Morse DL, Hostetter G. et al. Gene expression profiling-based identification of cell-surface targets for developing multimeric ligands in pancreatic cancer. Mol Cancer Ther. 2008;7:3071-80
28. Grützmann R, Pilarsky C, Ammerpohl O. et al. Gene expression profiling of microdissected pancreatic ductal carcinomas using high-density DNA microarrays. Neoplasia. 2004;6:611-22
29. Roychowdhury S, Chinnaiyan AM. Translating cancer genomes and transcriptomes for precision oncology. CA Cancer J Clin. 2016;66:75-88
30. Noll EM, Eisen C, Stenzinger A. et al. CYP3A5 mediates basal and acquired therapy resistance in different subtypes of pancreatic ductal adenocarcinoma. Nat Med. 2016;22:278-87
31. Teer JK, Mullikin JC. Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet. 2010;19:R145-51
32. Ng SB, Turner EH, Robertson PD. et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272-6
33. Maher B. Exome sequencing takes centre stage in cancer profiling. Nature. 2009;459:146-7
34. Pleasance ED, Stephens PJ, O'Meara S. et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184-90
35. Walter MJ, Graubert TA, Dipersio JF. et al. Next-generation sequencing of cancer genomes: back to the future. Personalized Med. 2009;6:653-62
36. Swanton C, Govindan R. Clinical implications of genomic discoveries in Lung cancer. N Engl J Med. 2016;374:1864-73
37. Shi L, Reid LH, Jones WD. et al. The MicroArray Quality control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol. 2006;24:1151-61
38. Chantrill LA, Nagrial AM, Watson C. et al. Precision medicine for advanced pancreas cancer: the individualized Molecular Pancreatic Cancer Therapy (IMPaCT) Trial. Clin Cancer Res. 2015;21:2029-37
39. Lawrence MS, Stojanov P, Polak P. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214-8
40. Hofree M, Shen JP, Carter H. et al. Network-based stratification of tumor mutations. Nat Methods. 2013;10:1108-15
41. Saeys Y, Inza I, Larrañaga P. A review of feature selection techniques in bioinformatics. BioInformatics. 2007;23:2507-17
42. Stein LD. Integrating biological databases. Nat Rev Genet. 2003;4:337-45
43. Gray SW, Hicks-Courant K, Cronin A. et al. Physicians' attitudes about multiplex tumor genomic testing. J Clin Oncol. 2014;32:1317-23
44. Gray SW, Hicks-Courant K, Lathan CS. et al. Attitudes of patients with cancer about personalized medicine and somatic genetic testing. J Oncol Pract. 2012;8:329-35
45. Sotiriou C, Piccart MJ. Taking gene-expression profiling to the clinic: when will molecular signatures become relevant to patient care? Nat Rev Cancer. 2007;7:545-53
46. Wang Y, Klijn JG, Zhang Y. et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671-9
47. Tran B, Dancey JE, Kamel-Reid S. et al. Cancer genomics: technology, discovery, and translation. J Clin Oncol. 2012;30:647-60
48. Roychowdhury S. Cancer genomics meets clinical trials: the challenge ahead. Personalized Med. 2012;9:459-61
49. Tsimberidou AM, Ringborg U, Schilsky RL. Strategies to overcome clinical, regulatory, and financial challenges in the implementation of personalized medicine. Am Soc Clin Oncol Educ Book. 2013:118-25
50. Le Tourneau C, Kamal M, Trédan O. et al. Designs and challenges for personalized medicine studies in oncology: focus on the SHIVA trial. Targeted Oncol. 2012;7:253-65
51. Von Hoff DD, Stephenson JJ Jr, Rosen P. et al. Pilot study using molecular profiling of patients' tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol. 2010;28:4877-83
52. Thompson AM, Jordan LB, Quinlan P. et al. Prospective comparison of switches in biomarker status between primary and recurrent breast cancer: the breast recurrence in Tissues Study (BRITS). Breast Cancer Res. 2010;12:R92
53. Kim ES, Herbst RS, Wistuba II. et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discovery. 2011;1:44-53
Author contact
![]() Corresponding authors: Yupei Zhao, Department of General Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100730, PR China. Phone: 86-10-69156007, Fax: 86-10-65124875, E-mail: zhao8028net. Lei You, Department of General Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100730, PR China. Phone: 86-10-69156016, Fax: 86-10-65124875, E-mail: florayocom.
Corresponding authors: Yupei Zhao, Department of General Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100730, PR China. Phone: 86-10-69156007, Fax: 86-10-65124875, E-mail: zhao8028net. Lei You, Department of General Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100730, PR China. Phone: 86-10-69156016, Fax: 86-10-65124875, E-mail: florayocom.