Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(15):2408-2419. doi:10.7150/jca.15703 This issue Cite
Research Paper
Brown Seaweed Fucoidan Inhibits Cancer Progression by Dual Regulation of mir-29c/ADAM12 and miR-17-5p/PTEN Axes in Human Breast Cancer Cells
Szu-Yuan Wu1,2,3,4, Ming-De Yan5, Alexander T.H. Wu6, Kevin Sheng-Po Yuan7, Shing Hwa Liu1,8 ![]()
1. Institute of Toxicology, College of Medicine, National Taiwan University, Taipei, Taiwan;
2. Department of Radiation Oncology, Wan Fang Hospital, Taipei Medical University, Taipei, Taiwan;
3. Department of Internal Medicine, School of Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan;
4. Department of Biotechnology, Hungkuang University, Taichung, Taiwan;
5. Cancer Center, Wan Fang Hospital, Taipei Medical University, Taipei;
6. The Ph.D. Program for Translational Medicine, Taipei Medical University, Taipei, Taiwan;
7. Department of Otorhinolaryngology, Wan Fang Hospital, Taipei Medical University, Taipei, Taiwan;
8. Department of Medical Research, China Medical University Hospital, China Medical University, Taichung, Taiwan.
Received 2016-3-30; Accepted 2016-9-17; Published 2016-12-9
Abstract
In this study, we observed that brown seaweed fucoidan inhibited human breast cancer progression by upregulating microRNA (miR)-29c and downregulating miR-17-5p, thereby suppressing their target genes, a disintegrin and metalloproteinase 12 (ADAM12) and phosphatase and tensin homolog (PTEN), respectively. Moreover, fucoidan reduced the luciferase activity of 3'-untranslated region reporter; treatment of cells with the miR-29c mimic or miR-17-5p inhibitor also produced similar results. These effects of fucoidan inhibited the epithelial-mesenchymal transition in breast cancer cells, as evidenced by an increase in E-cadherin and a drop in N-cadherin, and inhibited breast cancer cell survival, as evidenced by the activation of the phosphoinositide 3-kinase/Akt pathway. Taken together, these findings demonstrate that fucoidan inhibits breast cancer progression by regulating the miR-29c/ADAM12 and miR-17-5p/PTEN axes. Fucoidan is a potential chemopreventive/chemotherapeutic agent for breast cancer.
Keywords: Fucoidan, miR-29c, ADAM12, miR-17-5p, PTEN, breast cancer cells.
Introduction
Breast cancer is associated with high morbidity and mortality and is the fifth leading cause of cancer-related death worldwide [1, 2]. Breast cancer has also become a critical issue in Taiwan, where it is the fourth leading cause of cancer death because of its high recurrence rate after resection and poor prognosis [3], which are primarily related to tumor invasion and distant metastasis [4, 5]. Regarding breast cancer treatment, the estrogen receptor (ER), progesterone receptor (PR), and Her2 expression levels determine the choice of therapeutic regimens in addition to predicting progression in breast cancer patients. Among the different subtypes of breast cancer, the triple-negative subtype, defined as no/low ER, PR, and Her2 expression levels, accounts for 10%-20% of all breast cancers, and this subtype is associated with a high risk of tumor recurrence because of the lack of a reliable, specific targeted chemotherapeutic agent [6]. In recent years, the trend of systemic treatment for breast cancer has shifted more toward targeted therapy and combined therapies rather than nonspecific cytotoxic chemotherapy or hormone therapy [7, 8]. Many different signaling pathways and molecular mechanisms regulate the metastatic processes of breast cancer [9]. Accumulating evidence indicates that microRNAs (miRNAs) play crucial roles in tumorigenesis and metastasis, suggesting that targeting miRNAs is a promising approach for treating human cancers [10, 11]. Moreover, miRNAs are a class of endogenous, noncoding regulatory RNAs of approximately 20-25 nucleotides in length that negatively regulate gene expression by inducing messenger RNA degradation or by inhibiting translation through base pairing with the 3'-untranslated region (UTR) of target mRNAs [12, 13].
The miR-29 family consists of miR-29a, -29b, and -29c with shared regulatory capacities. The miR-29 family exerts various effects on cellular apoptosis [14], cell cycle regulation, epigenetic modification, and metastasis inhibition [15] to prevent cancer progression and carcinogenesis. Downregulation of miR-29 expression has been reported in multiple cancers including breast cancer [16, 17]. A crucial downstream target of miR-29c is a disintegrin and metalloproteinase 12 (ADAM12) [18]. ADAM12 is a member of the metalloproteinase-disintegrin family and a breast cancer-associated gene. ADAM12 expression is strongly upregulated in preinvasive and invasive carcinomas [19-23]. In addition, miR-29b/c and miR-200b/c post-transcriptionally mediate the downregulation of ADAM12 by directly targeting the 3'-UTR of ADAM12, resulting in degradation or decreased translation of target mRNAs. A significant negative correlation was recently demonstrated between ADAM12 and miR-29c in breast invasive carcinomas [24]. Identifying agents that can modulate the miR-29c-ADAM12 axis may lead to the development of improved breast cancer treatments
The miR-17/92 cluster is a crucial oncomiR group with vital roles in cancer development. The miR-17/92 cluster is upregulated in various cancers including breast, lung, colon, pancreas, prostate, and gastric cancer [25, 26]. Caloric restriction and radiotherapy downregulate the expression of members of the miR-17/92 cluster by suppressing extracellular matrix (ECM) mRNAs that exhibit miR-17-5p binding sites, thus reducing the metastatic potential of triple-negative breast cancer (TNBC) [27, 28]. The seven members of the miR-17~92 cluster play vital roles predominantly in phosphatase and tensin homolog (PTEN) and programmed cell death 4, which promote migration and invasion of metastatic cancer cells [29]. These findings collectively demonstrate the oncogenic character of the miR-17/92 cluster. TNBC with elevated oncomiR levels is associated with low expression levels of the tumor suppressor protein PTEN [30, 31].
Previous studies have confirmed the potential of natural dietary components as cancer therapeutics. Dietary components, namely curcumin, resveratrol, genistein, epigallocatechin-3-gallate, indole-3-carbinol, and 3, 3ʹ-diindolylmethane, exerted antiproliferative and proapoptotic effects on cancer cells by regulating one or more miRNAs [32]. Therefore, natural products represent an abundant source of miRNA regulators.
Fucoidan, a heparin-like molecule with high percentages of L-fucose and sulfated ester groups and low percentages of D-xylose, D-galactose, D-mannose, and glucuronic acid, was present in the cell wall matrix of brown seaweed [33]. Brown seaweed fucoidan was reported to demonstrate various biological activities such as antioxidant, anti-inflammatory, antiproliferative, and proapoptotic activities [34, 35]. Fucoidan was also revealed to inhibit the growth of breast and lung cancer in animal models [36, 37]. Fucoidan treatment induces the degradation of transforming growth factor (TGF)-β receptor and the consequent inhibition of the epithelial-mesenchymal transition (EMT) in cancer cells. In addition to these molecular mechanisms, it is imperative to investigate the potential of fucoidan as a miRNA regulator for breast cancer treatment and thus delineate the molecular mechanisms underlying the anticancer effects of fucoidan.
In this study, we investigated the effects of fucoidan on the regulation of miR-29c and miR-17-5p in human breast cancer cells. We observed that fucoidan increased miR-29c expression and reduced miR-17-5p expression, which suppressed ADAM12 and PTEN, respectively. The effects were confirmed by the inhibition of the EMT and invasiveness of breast cancer cells. Our findings suggest a regulatory role of fucoidan in the cross-talk between miR-29c and miR-17-5p, which epigenetically control the migration and invasion of breast cancer cells.
Materials and Methods
Reagents
Fucoidan powder from Sargassum hemiphyllum was obtained from the commercial product Hi-Q Oligo-fucoidan® manufactured by Hi-Q Marine Biotech International Ltd. (New Taipei City, Taiwan). The powder was dissolved in double-distilled H2O and stirred at 25°C for 30 min. The dissolved solution was filtered through 0.22-μm sterile filters (Millipore, Billerica, MA, USA).
Cell culture
Human mammary gland epithelial adenocarcinoma (MCF-7 and MDA-MB-231) and human normal mammary gland epithelial fibrocystic (MCF-10A) cell lines were purchased from American Tissue Cell Culture (Manassas, VA, USA). The MCF-10A cells were maintained in complete MCF-10A culture medium; that is, a 1:1 mixture of Dulbecco's modified Eagle medium (DMEM) and Ham F12 supplemented with 100 ng/mL cholera enterotoxin, 10 μg/mL insulin, 0.5 μg/mL hydrocortisol, and 20 ng/mL epidermal growth factor (Life Technologies, Rockville, MD, USA). The MCF-7 and MDA-MB-231 cells were maintained in DMEM (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS). All cell lines were grown at 37°C in a humidified 5% CO2 atmosphere. These cells were also free of mycoplasma contamination.
Evaluation of cell viability using MTS assay
Cells at a density of 5 × 103 cells per well were seeded in 96-well plates in medium containing 1% FBS. The cells were allowed to adhere for 24 h and were then incubated with the indicated drug concentrations. Each condition was tested in triplicate wells. Cell viability was assessed using the Celltiter96 Aqueous1 solution reagent (Promega, Madison, WI, USA) according to the manufacturer's instructions. The amount of MTS formazan product was measured at a wavelength of 490 nm by using a PowerWave X Microplate ELISA Reader (Bio-TeK Instruments, Winoski, VT, USA).
Determination of cell cycle phases using flow cytometry
Cells were seeded in 6-well tissue culture dishes at 1 × 105 cells per well. The cells were allowed to adhere for 24 h and were then incubated with the indicated drug concentrations. Each condition was tested in triplicate wells. The cell cycle phases of the treated cells were determined by quantifying their DNA content through staining with propidium iodide staining, and the cell cycle phases were analyzed by a FACS scanner (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA) and quantified by Cell Quest software (Becton Dickinson Immunocytometry Systems).
Colony formation assay
Cells were seeded in 6-well tissue culture dishes at 4 × 103 cells per well. After 2 weeks of culture, colonies were fixed with 4% paraformaldehyde and stained with 0.05% crystal violet. The total number of colonies in each well was counted.
Invasion assay
The invasiveness of breast cancer cells was evaluated using a transwell invasion assay. Briefly, cells were plated in 200 μL of serum-free medium with or without fucoidan (200 μg/mL) in the upper chamber inserts coated with 50 μL of 1% Matrigel® (BD Biosciences, San Jose, CA, USA). Moreover, 600 μL of medium containing 10% FBS was added to the lower chamber and was used as a chemoattractant to stimulate cell migration. After 24-h incubation, cells on the upper surface of the inserts were gently removed using a cotton swab. All cells were stained with 0.1% crystal violet and counted in five fields under an inverted microscope. All experiments were repeated in three times independently.
RNA isolation and quantitative reverse-transcription polymerase chain reaction
After cells were treated with fucoidan for 48 h, total RNA was isolated using Trizol (Invitrogen, Grand Island, NY, USA) according to the manufacturer's instructions. To analyze the expression of mature miR-29c and miR-17-5p, complementary cDNA was synthesized using sequence-specific primers and the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). For cDNA amplification, quantitative reverse-transcription polymerase chain reaction (qRT-PCR) was performed on the ABI 7500 real-time system (Applied Biosystems) by using sequence-specific primers from the TaqMan MicroRNA Assays Kit. After normalization to the internal controls, the values of the vehicle-treated cells served as a basal level of miRNA expression; ΔCt values (Ctfucoidan - Ctcontrol) were used to determine their relative expression as multiples of changes. U6 was used as the internal control for evaluating miR-29c and miR-17-5p expression.
miRNA profiling
Total RNA was isolated from MDA-MB-231 cells treated with vehicle or fucoidan (200 μg/mL) for 48 h. The samples were then analyzed using the Affymetrix GeneChip miRNA 2.0 Array (Affymetrix, Santa Clara, CA, USA) containing 4560 probe sets for human small RNAs. All steps of the procedure were performed according to the Affymetrix standardized protocol for the miRNA 2.0 array.
miRNA mimic and inhibitor and construction of target gene 3'-UTR vectors
A miR-29c mimic (5'-UAGCACCAUUUGAAAUCAGUGUU-3'), miR-17-5p inhibitor (5'-GUUUCACGAAUGUCACGUCCAUC-3'), and scrambled control (5'-UUCUCCGAACGUGUCACGUTT-3') were obtained from Dharmacon (Lafayette, CO, USA). The mimic is a single-stranded RNA oligonucleotide that can bind to complimentary, mature miRNA strands. ADAM12 and PTEN 3'-UTR, which are approximately 300 bp in length and contain miR-29c and miR-17-5p binding sites, respectively, were cloned into the downstream region of the luciferase gene to create a pGL3-ADAM12 and pGL3-PTEN 3'-UTR. Site-directed mutagenesis was performed to generate the pGL3-ADAM12 and pGL3-PTEN 3'-UTR-mut, in which the miR-29c and miR-17-5p binding sites were mutated. For luciferase reporter experiments, ADAM12 and PTEN 3'-UTR segments of 978 and 823 bp, respectively, were amplified from human genomic DNA by using PCR and inserted into the pGL3-control vector with the SV40 promoter (Promega) by using the XbaI site immediately downstream from the stop codon of luciferase.
Transfection and luciferase reporter assay
The miR-29c, miR-17-5p, or scrambled miRNA mimic (100 nM); ADAM12 and PTEN 3'-UTR firefly luciferase report vectors (wild-type or mutated type); and the control vector containing the Renilla luciferase pRL-TK vector (Promega) were cotransfected into breast cancer cells by using Lipofectamine-2000 transfection reagent (Invitrogen), according to the manufacturer's protocol. The medium was changed after 16 h. For the luciferase reporter assay, cells were harvested after culturing for 48 h. Firefly and Renilla luciferase activities were consecutively measured using the Dual-Luciferase® Reporter Assay System (Promega). The experiments were performed in triplicate.
Western blotting
After cells were treated with fucoidan for 48 h, cell lysates were separated by electrophoresis on a sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel. After gel transference to polyvinylidene fluoride membranes (Millipore), the membranes were incubated with antibodies against E-cadherin (sc-8426, Santa Cruz, CA, USA), occludin (ab31721, Abcam, Cambridge, MA, USA), N-cadherin (sc-8424, Santa Cruz, CA, USA), vimentin (sc-6260, Santa Cruz, CA, USA), ADAM12 (ab16049, Abcam), TGF-βR2 (sc-220, Santa Cruz, CA, USA), PTEN (sc-7974, Santa Cruz, CA, USA), p-phosphoinositide 3-kinase (PI3K; ab182651, Abcam), t-PI3K (ab86714, Abcam), p-AKT (sc-16646, Santa Cruz, CA, USA), t-AKT (sc-8312, Santa Cruz, CA, USA), and glyceraldehyde-3-phosphate dehydrogenase (sc-25778, Santa Cruz, CA, USA). Subsequently, the membranes were incubated with suitable secondary antibodies. Bands for specific molecules were detected using enhanced chemiluminescence (ECL; Millipore).
Statistical analyses
All statistical analyses were performed using GraphPad Prism software (version 4.0). All values are expressed as mean ± standard error of the mean (SEM). The significance level was set at p < 0.05. Student's t test was used to compare cell viability, cell death, luciferase activity, and gene expression.
Results
Effects of fucoidan on the growth, clonogenicity, and invasiveness of human breast cancer cells
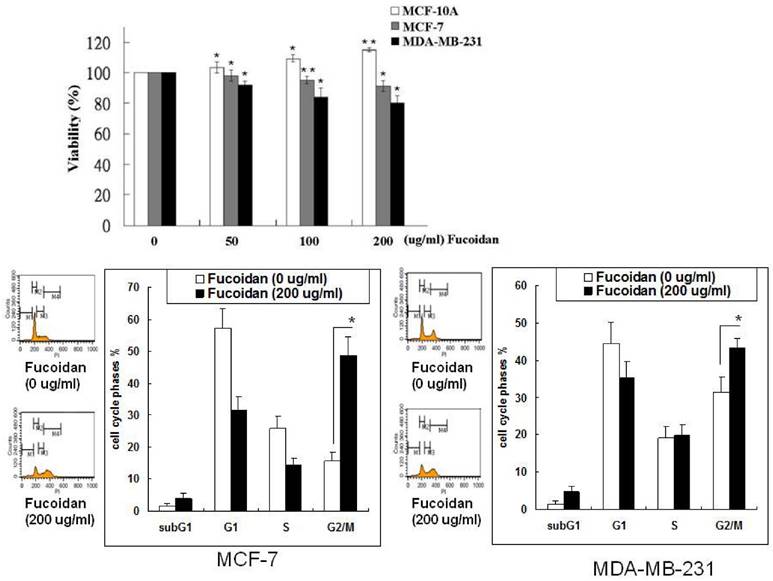
The effects of fucoidan on the growth of breast cancer cells were determined by evaluating the viability of MCF-7 and MDA-MB-231 cells in the MTS assay. Varying degrees of growth inhibition were observed after treatment with fucoidan (50-200 µg/mL) for 48 h (Figure 1). Fucoidan (200 µg/mL) inhibited 12% and 21% of cell growth in the MCF-7 and MDA-MB-231 cells, respectively. By contrast, fucoidan exerted no inhibitory effect on the growth of the normal human breast cell line MCF-10A (Figure 1), suggesting that fucoidan preferentially suppresses cancer cells. Fucoidan exerted stronger inhibitory effects on the triple-negative type MDA-MB-231 cells than on the luminal A-type MCF-7 cells. To demonstrate the effect of fucoidan on the cell cycle phases of cancer cells, we determined the changes in the cell cycle phase pattern through a flow cytometric analysis of cellular DNA content (Figure 1). The results revealed that fucoidan treatment induced G2/M arrest in both the MCF-7 and MDA-MB-231 cells.
Inhibitory effects of fucoidan on the viability of human immortalized normal breast cells and breast cancer cells. MCF-10A, MCF-7, and MDA-MB-231 cells were incubated with various concentrations of fucoidan (0, 50, 100, and 200 μg/mL) for 48 h, and the proliferation of the cells was measured using the MTS assay. Values are expressed as mean ± standard error of three independent experiments. Changes in the cell cycle phase pattern were determined by a flow cytometric analysis of cellular DNA content. Significant differences are shown (*p < 0.05 and **p < 0.01, compared with the control group).
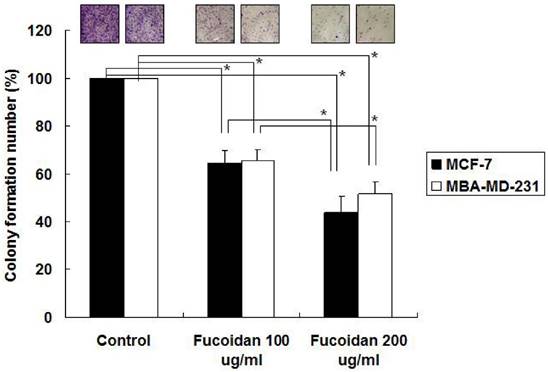
We next examined the effects of fucoidan on the clonogenicity of MCF-7 and MDA-MB-231 cells. The results revealed that fucoidan (200 µg/mL) significantly reduced colony formation in the MCF-7 and MDA-MB-231 cells to 53% and 64%, respectively, relative to the baseline values (Figure 2). To evaluate the dose-dependent effects of fucoidan, the MCF-7 and MDA-MB-231 cells were treated with 100 and 200 µg/mL fucoidan, respectively, and clonogenicity was measured. The results revealed that fucoidan affected the clonogenicity of these cells in a dose-dependent manner (Figure 2).
MCF-7 and MDA-MB-231 cells were treated with 100 and fucoidan (200 μg/mL), and clonogenicity was measured. (*p < 0.05 and **p < 0.01, compared with the control group).
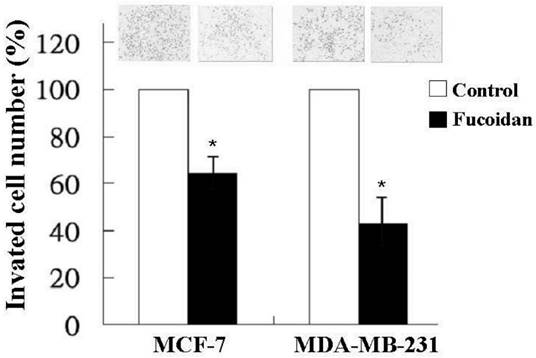
In addition to tumor growth, invasion and distant metastasis play crucial roles in the poor clinical outcomes of breast cancer. Therefore, we also investigated the effect of fucoidan on the invasiveness of MCF-7 and MDA-MB-231 cells by using a transwell invasion assay. As shown in Figure 3, fucoidan (200 µg/mL) significantly reduced the invasion of the MCF-7 and MDA-MB-231 cells to 62% and 43%, respectively, relative to the baseline values.
MCF-7 and MDA-MB-231 cells were treated with fucoidan (200 μg/mL) for 24 h, and invasiveness was measured using a two-chamber transwell assay. Significant differences are shown (*p < 0.05, compared with the control group).
Fucoidan modulates miRNA expression
Because the regulation of miRNAs has become a novel approach for cancer treatment, we analyzed the miRNA expression profile of fucoidan-treated MDA-MB-231 cells by using the Affymetrix GeneChip miRNA 2.0 Array. The results showed that fucoidan increased the expression levels of tumor-suppressive miRNAs such as miRNAs belonging to the miR-29 and miR-34 families (Table 1). The expression levels of miR-29c, miR-29b, and miR-29a were 10.6-, 8.4-, and 7.3-fold higher, respectively, in the fucoidan-treated MDA-MB-231 cells than in the control. By contrast, fucoidan markedly suppressed the expression of oncogenic miRNAs such as the miR-17~92 cluster. Moreover, the expression levels of miR-17-5p, miR-18a, and miR-92a were 9.1-, 7.9- and 6.9-fold higher, respectively, in fucoidan-treated MDA-MB-231 cells than in the control (Table 1).
Effects of fucoidan on the microRNA profile of human breast cancer cells.
| miR-ID | Increased Fold-Changes | miR-ID | Decreased Fold-Changes |
|---|---|---|---|
| miR-29c | 10.6 | miR-17-5p | 9.1 |
| miR-29b | 8.4 | miR-18a | 7.9 |
| miR-34 | 7.6 | miR-10b | 7.5 |
| miR-29a | 7.3 | miR-92a | 6.9 |
| miR-200c | 6.1 | miR-21 | 6.3 |
| miR-31 | 4.5 | miR-183 | 5.2 |
| miR-221 | 3.7 | miR-96 | 4.8 |
| miR-1258 | 3.4 | miR-210 | 3.6 |
| miR-205 | 2.8 | miR-182 | 2.7 |
| miR-200b | 2.3 | miR-154 | 2.2 |
Total RNA was isolated from MDA-MB-231 cells treated with vehicle or fucoidan (200 μg/mL) for 48 h. The samples were then analyzed using the Affymetrix GeneChip miRNA 2.0 Array containing 4560 probe sets for human small RNAs. Values represent increased or decreased multiples of change in expression.
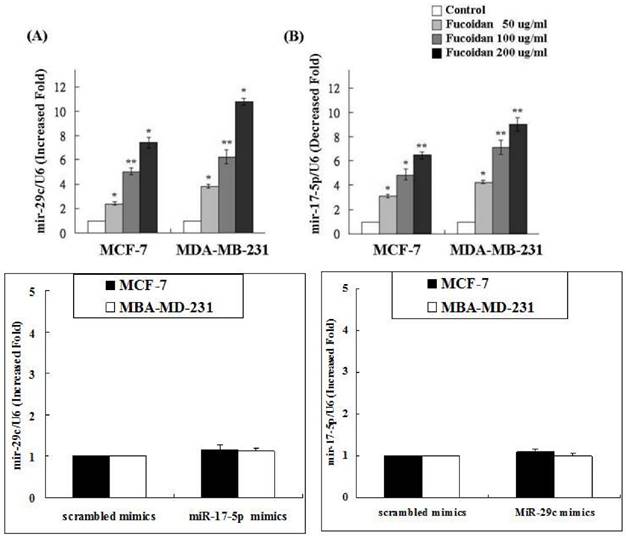
Our data (Figure 4) revealed that fucoidan upregulated miR-29c expression but downregulated miR-17-5p expression. To determine whether any cross-talk occurs between these two miRNAs, MCF-7 and MDA-MB-231 cells were treated with the miR-17 or miR-29c inhibitor followed by verifying the expression of miR-17-5p and miR-29c, respectively. The results revealed no interference between miR-29c and miR-17-5p expression (Figure 4). Thus, we conclude that the miR-29c-ADAM12 pathway and the miR-17-5p-PTEN pathway are totally independent. These results are consistent with those of the miRNA 2.0 array shown in Table 1.
Fucoidan upregulated microRNA-29c but downregulated microRNA-17-5p. To determine whether cross-talk occurs between the two micro RNAs, MCF-7 and MDA-MB-231 cells were treated with the microRNA-17 or microRNA-29c inhibitor followed by verifying the expression of microRNA-17-5p and microRNA-29c, respectively. (*p < 0.05 and **p < 0.01, compared with the control group).
Fucoidan increases miR-29c expression to suppress ADAM12
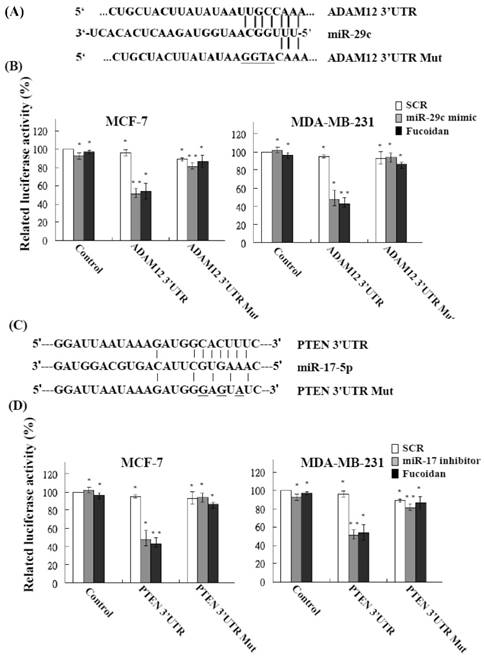
The functional targets of miR-29c were predicted using the PicTar (http://pictar.mdc-berlin.de/) and TargetScan (http://targetscan.org/) databases. The results showed that ADAM12 had a higher score and appeared to be a potential miR-29c target gene. The 3'-UTR of the ADAM12 gene matches the seed sequence of miR-29c and represents the putative binding site. To validate the repression of ADAM12 by miR-29c in breast cancer cells, we created luciferase reporter constructs containing the 300-bp 3'-UTR of ADAM12 (nt 1182-1209 of ADAM12 mRNA) with a wild-type or mutated miR-29c binding site, as described previously (Figure 5A). In accordance with the marked increase in miR-29c expression by fucoidan (Figure 4), the luciferase activity of the wild-type, but not the mutated, ADAM12 3'-UTR reporter was repressed to approximately 50%-60% in MCF-7 and MDA-MB-231 cells after treatment with fucoidan for 48 h (Figure 5B, black bars). As expected, similar and compatible results were observed for MCF-7 and MDA-MB-231 cells transfected with the miR-29c mimic (Figure 5B, gray bars). These findings indicate that fucoidan increases miR-29c expression to negatively regulate ADAM12 in breast cancer cells.
Fucoidan modulates microRNAs to target the 3'-UTR region of ADAM12 and PTEN and represses their expression in human breast cancer cells. (A) (C) Schematic representation of 3'-UTRs of ADAM12 and PTEN showing the putative miR-29c and miR-17-5p target sites. (B) (D) After transfection of cells, the ADAM12- and PTEN-dependent luciferase activity of breast cancer cells was measured using the Dual-Luciferase® Reporter Assay System 48 h after treatment, as indicated. The experiments were performed in triplicate. Values were normalized to Renilla luciferase activity (transfection efficiency control) and are plotted as relative luciferase activities. SCR, scrambled control. Significant differences are shown (*p < 0.05 and **p < 0.01, compared with the control group).
Fucoidan reduces miR-17-5p expression to restore PTEN
The functional targets of miR-17-5p were predicted using the PicTar (http://pictar.mdc-berlin.de/) and TargetScan (http://targetscan.org/) databases. The results showed that PTEN had a higher score and appeared to be a potential miR-17-5p target gene. The 3'-UTR of the PTEN gene matches the seed sequence of miR-17-5p and represents the putative binding site. To validate the repression of PTEN by miR-17-5p in breast cancer cells, we created luciferase reporter constructs containing the 300-bp 3'-UTR of PTEN (nt 915-942 of PTEN mRNA) with a wild-type or mutated miR-17-5p binding site, as described previously (Figure 5C). In accordance with the marked increase in miR-17-5p expression by fucoidan (Figure 4), the luciferase activity of the wild-type, but not the mutated, PTEN 3'-UTR reporter was repressed to approximately 50%-60% in MCF-7 and MDA-MB-231 cells after treatment with fucoidan for 48 h (Figure 5D, black bars). As expected, similar and compatible results were observed for MCF-7 and MDA-MB-231 cells transfected with the miR-17-5p inhibitor (Figure 5D, gray bars). These findings indicate that fucoidan reduces miR-17-5p expression to negatively regulate PTEN in breast cancer cells.
Fucoidan modulates protein expression of ADAM12, PTEN, EMT markers, and survival markers
miRNAs can reduce protein expression by inducing mRNA degradation. Western blotting was performed to confirm the repression of ADAM12 by fucoidan, as demonstrated by the luciferase reporter assay results shown in Figure 5. A previous study reported ADAM12 overexpression in clinical breast cancer specimens [20]. In this study, the protein levels of ADAM12 and TGF-βRII were dose-dependently repressed in fucoidan-treated MCF-7 and MDA-MB-231 cells (Figure 6A). MCF-7 is a weakly metastatic and E-cadherin-expressing breast cancer cell line that expresses little or no N-cadherin and ADAM12; MDA-MB-231 is a highly invasive breast cancer cell line that lacks E-cadherin expression but expresses high levels of N-cadherin and ADAM12 [38, 39]. Gruel et al. reported that the loss of TGF-βRII in cancers may reduce junction stability, which ultimately promotes the EMT and metastasis [19]. Accordingly, in this study, the increase in E-cadherin and occludin and the reciprocal drop in N-cadherin and vimentin (Figure 6A) indicated that fucoidan inhibited the EMT in breast cancer cells.
Fucoidan regulates the miR-29c/ADAM12 and miR-17-5p/PTEN axes to inhibit the epithelial-mesenchymal transition (EMT) and cancer cell survival in MCF-7 and MDA-MB-231 cells. (A) After treatment with fucoidan (0, 50, 100, 200 μg/mL) for 48 h, protein levels of E-cadherin, occludin, N-cadherin, vimentin, ADAM12, TGF-βRII, and were analyzed by Western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B) Protein levels of PTEN, phosphor- and total-PI3K, and phosphor- and total-AKT were analyzed by Western blotting. GAPDH was used as a loading control.
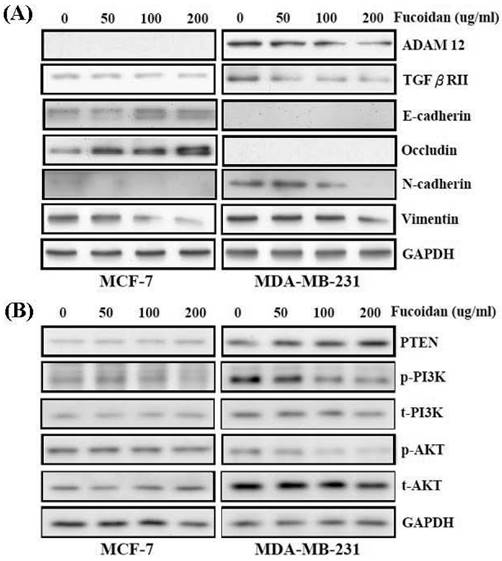
As shown in Figure 6B, fucoidan dose-dependently modulated the protein expression levels of PTEN, PI3K, and AKT in MCF-7 and MDA-MB-231 cells. Fucoidan dose-dependently increased PTEN protein expression in these breast cancer cells (Figure 6B). Moreover, the diminished phosphorylation of PI3K and AKT proteins (Figure 6B) indicated that fucoidan inhibited the survival of breast cancer cells.
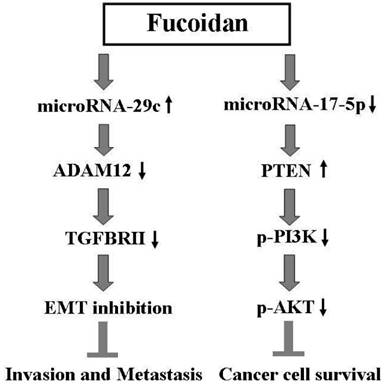
The results of this study indicate that fucoidan inhibits the invasion, metastasis, and survival of human breast cancer cells by regulating the miR-29c/ADAM12 and miR-17-5p/PTEN axes. This proposed molecular mechanism of action of fucoidan is illustrated in Figure 7.
Scheme for the proposed molecular mechanism of action of fucoidan in inhibiting breast cancer progression through the regulation of the miR-29c/ADAM12 and miR-17-5p/PTEN axes.
Discussion
Evidence accumulated over the past two decades indicates that fucoidan is a potent anticancer agent with chemopreventive and chemotherapeutic potential. Hsu et al. demonstrated that fucoidan inhibited the EMT and invasion of lung and breast cancer cells through TGF-β receptor degradation [36, 37]. TGF-β can also suppress miR-29c expression [40]. The induction of miR-29c by fucoidan in breast cancer cells might contribute to TGF-β receptor degradation. Additional studies should investigate the association of fucoidan-induced TGF-β receptor degradation with miR-29c expression in cancer cells. However, the precise mode of action of fucoidan is not completely understood, hindering its incorporation into mainstream clinical treatment regimens.
Recent experimental evidence reveals that dietary nutraceuticals such as curcumin and resveratrol have great potential as epigenetic agents. In contrast to genetic changes, epigenetic changes can be modified by environmental, dietary, or pharmacological interventions. To investigate the epigenetic modification mechanism of fucoidan, we used MCF-7 and MDA-MB-231 cells for in vitro studies. Our results demonstrate that fucoidan treatment caused a moderate reduction in tumorigenesis in MDA-MB-231 cells (Figure 1), because of alterations in the miRnome expression profile. Specifically, the fucoidan-treated MDA-MB-231 cells exhibited significant increases in the expression of miRNAs involved in the EMT (miR-29c, miR-29b, miR-29a, miR-200c, and miR-200b) and significant increases in the expression of tumor suppressors (miR-34a and miR-205). Significant reductions were observed in the expression of oncomiRNAs promoting tumorigenesis (the miR-17/92 cluster) [41] and oncomiRNAs promoting metastasis (the miR-96-182-183 family) (Table 1). The MCF10A human breast epithelial cell line is the most commonly used normal breast cell model. Western blotting was performed to determine whether fucoidan targets the protein expression of PTEN in MCF10A cells. High PTEN expression was observed in MCF10A cells; thus, PTEN expression was not inhibited by fucoidan treatment (Figure S1). Moreover, p-Akt expression was not observed in MCF10A cells. We conclude that the regulation of cell growth in the MCF10A cell line is different from that in the MCF-7 and MDA-MB-231 cells. Taken together, those studies demonstrated for the first time that fucoidan can inhibit the growth of MDA-MB-231 cells. Moreover, fucoidan exerts its anticancer effect through the dysregulation of miRNA expression, which is a hallmark of numerous carcinomas and consistent with the less-aggressive phenotype.
Fucoidan exhibits unique activities that regulate miRNA expression during multiple disease states, including cancer. miRNAs are small, noncoding RNA molecules that can inhibit target gene expression by binding to the 3'-UTR of target mRNAs, resulting in RNA degradation or translation inhibition. Thus, aberrant miRNA expression has been correlated with the development of tumors, progression of cancer, formation of cancer stem cells, and acquisition of the EMT phenotype. This study identified, for the first time, fucoidan as a novel agent that can alter miRNAs regulating the EMT and metastasis in breast cancer cells. Previous studies have also revealed that resveratrol and curcumin can alter miRNAs involved in breast cancer prevention and apoptosis, respectively [32]. These results indicate the existence of a mechanism by which dietary agents can modulate the activity of miRNAs crucial for cancer progression. Moreover, miRNAs are characterized as biomarkers of diagnosis and prognosis; thus, they are becoming attractive targets for cancer therapy. We found that in addition to the induction of the tumor-suppressive miR-29 family, fucoidan repressed the oncogenic miR-17/92 cluster. The finding demonstrates that fucoidan has multiple functions in miR regulation. The members of the miR-17/92 cluster have been suggested to play crucial roles in normal development; the dysregulation of their expression may lead to diseases including cancers and diseases related to the immune, nervous, and cardiovascular systems [41]. Such miR-17/92 cluster repression might contribute to fucoidan-induced inhibition of growth and invasion in breast cancer cells. Additional studies should clarify this hypothesis. Accumulating evidence suggests that natural agents are useful for targeting miRNAs and can subsequently enhance the efficacy of conventional cancer therapies. Therefore, the use of natural agents such as fucoidan, curcumin, and resveratrol to target miRNAs is a potential novel strategy toward designing combination approaches with conventional therapies for preventing tumor recurrence and achieving successful treatment outcomes for cancer patients.
Loss of expression of epithelial adherens junction proteins (e.g., E-cadherin and occludin) with a concomitant gain of mesenchymal marker expression (e.g., N-cadherin and vimentin) is a biochemical hallmark of EMT [42]. Loss of E-cadherin expression is associated with increased invasion of breast cancer cells [43]. In the current study, as shown in Figure 6A, the expression levels of E-cadherin and occludin were extremely low in the MDA-MB-231 cell line, which is a highly invasive “triple negative” type that lacks the three molecular markers (the expression of ER and PR and amplification of HER-2/Neu). By contrast, the MCF7 cell line is “luminal A” type and is ER and PR positive [44, 45]. Therefore, Western blotting could not detect N-cadherin expression in this study. Moreover, miRNAs frequently mediate post-transcriptional gene regulation. The 3'-UTR of the mouse ADAM12 gene contains a well-conserved potential target site for the miR-29 family, comprising miR-29a, miR-29b, and miR-29c. According to a recent study, miR-29b regulates the expression of ADAM12 in human trabecular meshwork and NIH-3T3 fibroblast cells [18, 46]. In the current study, the protein expression of ADAM12 was not observed in MCF-7 cells (Figure 6A). Therefore, we examined whether ADAM12 expression in MCF-7 cells is modulated by miR-29c (Figure 5). Fucoidan may downregulate E-cadherin expression by mediating the binding of Snail, Zeb, and Twist to the E-cadherin promoter directly. We plan to clarify the mechanism by which fucoidan downregulates E-cadherin expression in an additional study. ADAM12 expression has been reported to be increased in human breast cancer and breast cancer cell lines [47]. ADAM12 confers tumor cells with enhanced resistance against apoptosis and contributes to tumor progression [48]. ADAM12 is associated with the early onset and progression of breast cancer [49]. Loss of expression of PTEN is associated with poor outcomes in breast cancer [50]. PTEN may also suppress breast cancer cell growth by downregulating PI3K signaling, leading to the blockage of cell cycle progression and the induction of cell death sequentially [51]. Loss of expression of PTEN is a predictor of early-onset and familial breast cancer in women [52].
Patients with hormone receptor-positive breast cancer should receive hormone therapy to reduce the risk of breast cancer recurrence and breast cancer-related mortality [53-55]. Moreover, patients with HER2-positive breast cancer should receive trastuzumab after undergoing surgery with or without chemotherapy [56]. However, no endocrine therapy or target therapy exists for TNBC. In this study, the promising results of the anticancer effect of fucoidan for TNBC are obvious. Thus, fucoidan might be a therapeutic option for TNBC patients for reducing the relatively high risk of tumor recurrence. In the future, a large-scale randomized trial should administer a suitable fucoidan regimen to carefully selected breast cancer patients and should compare standard approaches to obtain crucial information on population specificity and disease occurrence.
In this study, we investigated the functional significance of fucoidan for miR-29c and miR-17-5p in human breast cancer cells and identified novel targets for these miRNAs. Fucoidan inhibits breast cancer progression by regulating the miR-29c/ADAM12 and miR-17-5p/PTEN axes (Figure 7). To the best of our knowledge, this study is the first to define the epigenetic effects of fucoidan on miRNAs and report the direct biological consequences of fucoidan in breast cancer cells. Considering our results and the relative nontoxicity of fucoidan, we suggest that fucoidan offers a valuable chemopreventive or therapeutic advantage in the clinical management of refractory breast cancer over other standard treatments.
Supplementary Material
Supplementary figure S1.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Baselga J, Norton L. Focus on breast cancer. Cancer cell. 2002;1:319-22
2. DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA: a cancer journal for clinicians. 2014;64:52-62
3. Chen SM, Chou WC, Hu LY, Hsiung CN, Chu HW, Huang YL. et al. The Effect of MicroRNA-124 Overexpression on Anti-Tumor Drug Sensitivity. PloS one. 2015;10:e0128472
4. Khotskaya YB, Goverdhan A, Shen J, Ponz-Sarvise M, Chang SS, Hsu MC. et al. S6K1 promotes invasiveness of breast cancer cells in a model of metastasis of triple-negative breast cancer. American journal of translational research. 2014;6:361-76
5. Su CM, Wang MY, Hong CC, Chen HA, Su YH, Wu CH. et al. miR-520h is crucial for DAPK2 regulation and breast cancer progression. Oncogene. 2015
6. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. The New England journal of medicine. 2010;363:1938-48
7. Gluz O, Liedtke C, Gottschalk N, Pusztai L, Nitz U, Harbeck N. Triple-negative breast cancer-current status and future directions. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2009;20:1913-27
8. Finn RS, Press MF, Dering J, Arbushites M, Koehler M, Oliva C. et al. Estrogen receptor, progesterone receptor, human epidermal growth factor receptor 2 (HER2), and epidermal growth factor receptor expression and benefit from lapatinib in a randomized trial of paclitaxel with lapatinib or placebo as first-line treatment in HER2-negative or unknown metastatic breast cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:3908-15
9. Mulrane L, McGee SF, Gallagher WM, O'Connor DP. miRNA dysregulation in breast cancer. Cancer research. 2013;73:6554-62
10. Samantarrai D, Dash S, Chhetri B, Mallick B. Genomic and epigenomic cross-talks in the regulatory landscape of miRNAs in breast cancer. Molecular cancer research: MCR. 2013;11:315-28
11. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97
12. Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nature reviews Drug discovery. 2010;9:775-89
13. Iorio MV, Croce CM. microRNA involvement in human cancer. Carcinogenesis. 2012;33:1126-33
14. Xiong Y, Fang JH, Yun JP, Yang J, Zhang Y, Jia WH. et al. Effects of microRNA-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology. 2010;51:836-45
15. Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S. et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53:209-18
16. Sengupta S, den Boon JA, Chen IH, Newton MA, Stanhope SA, Cheng YJ. et al. MicroRNA 29c is down-regulated in nasopharyngeal carcinomas, up-regulating mRNAs encoding extracellular matrix proteins. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:5874-8
17. Cittelly DM, Finlay-Schultz J, Howe EN, Spoelstra NS, Axlund SD, Hendricks P. et al. Progestin suppression of miR-29 potentiates dedifferentiation of breast cancer cells via KLF4. Oncogene. 2013;32:2555-64
18. Li H, Solomon E, Duhachek Muggy S, Sun D, Zolkiewska A. Metalloprotease-disintegrin ADAM12 expression is regulated by Notch signaling via microRNA-29. The Journal of biological chemistry. 2011;286:21500-10
19. Gruel J, Leborgne M, LeMeur N, Theret N. In silico investigation of ADAM12 effect on TGF-beta receptors trafficking. BMC research notes. 2009;2:193
20. Frohlich C, Nehammer C, Albrechtsen R, Kronqvist P, Kveiborg M, Sehara-Fujisawa A. et al. ADAM12 produced by tumor cells rather than stromal cells accelerates breast tumor progression. Molecular cancer research: MCR. 2011;9:1449-61
21. Duhachek-Muggy S, Li H, Qi Y, Zolkiewska A. Alternative mRNA splicing generates two distinct ADAM12 prodomain variants. PloS one. 2013;8:e75730
22. Ieguchi K, Tomita T, Omori T, Komatsu A, Deguchi A, Masuda J. et al. ADAM12-cleaved ephrin-A1 contributes to lung metastasis. Oncogene. 2014;33:2179-90
23. Aghababaei M, Hogg K, Perdu S, Robinson WP, Beristain AG. ADAM12-directed ectodomain shedding of E-cadherin potentiates trophoblast fusion. Cell death and differentiation. 2015
24. Duhachek-Muggy S, Zolkiewska A. ADAM12-L is a direct target of the miR-29 and miR-200 families in breast cancer. BMC cancer. 2015;15:93
25. Petrocca F, Visone R, Onelli MR, Shah MH, Nicoloso MS, de Martino I. et al. E2F1-regulated microRNAs impair TGFbeta-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer cell. 2008;13:272-86
26. Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F. et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2257-61
27. Jin L, Lim M, Zhao S, Sano Y, Simone BA, Savage JE. et al. The metastatic potential of triple-negative breast cancer is decreased via caloric restriction-mediated reduction of the miR-17~92 cluster. Breast cancer research and treatment. 2014;146:41-50
28. Wu SY, Lin KC, Chiou JF, Jeng SC, Cheng WH, Chang CI. et al. MicroRNA-17-5p post-transcriptionally regulates p21 expression in irradiated betel quid chewing-related oral squamous cell carcinoma cells. Strahlentherapie und Onkologie: Organ der Deutschen Rontgengesellschaft [et al]. 2013;189:675-83
29. Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J. et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nature immunology. 2008;9:405-14
30. Iqbal J, Thike AA, Cheok PY, Tse GM, Tan PH. Insulin growth factor receptor-1 expression and loss of PTEN protein predict early recurrence in triple-negative breast cancer. Histopathology. 2012;61:652-9
31. Jin YY, Andrade J, Wickstrom E. Non-Specific Blocking of miR-17-5p Guide Strand in Triple Negative Breast Cancer Cells by Amplifying Passenger Strand Activity. PloS one. 2015;10:e0142574
32. Phuah NH, Nagoor NH. Regulation of microRNAs by natural agents: new strategies in cancer therapies. BioMed research international. 2014;2014:804510
33. Senthilkumar K, Manivasagan P, Venkatesan J, Kim SK. Brown seaweed fucoidan: biological activity and apoptosis, growth signaling mechanism in cancer. International journal of biological macromolecules. 2013;60:366-74
34. Atashrazm F, Lowenthal RM, Woods GM, Holloway AF, Dickinson JL. Fucoidan and cancer: a multifunctional molecule with anti-tumor potential. Marine drugs. 2015;13:2327-46
35. Kwak JY. Fucoidan as a marine anticancer agent in preclinical development. Marine drugs. 2014;12:851-70
36. Hsu HY, Lin TY, Hwang PA, Tseng LM, Chen RH, Tsao SM. et al. Fucoidan induces changes in the epithelial to mesenchymal transition and decreases metastasis by enhancing ubiquitin-dependent TGFbeta receptor degradation in breast cancer. Carcinogenesis. 2013;34:874-84
37. Hsu HY, Lin TY, Wu YC, Tsao SM, Hwang PA, Shih YW. et al. Fucoidan inhibition of lung cancer in vivo and in vitro: role of the Smurf2-dependent ubiquitin proteasome pathway in TGFbeta receptor degradation. Oncotarget. 2014;5:7870-85
38. Albrechtsen R, Kveiborg M, Stautz D, Vikesa J, Noer JB, Kotzsh A. et al. ADAM12 redistributes and activates MMP-14, resulting in gelatin degradation, reduced apoptosis and increased tumor growth. Journal of cell science. 2013;126:4707-20
39. Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. The Journal of cell biology. 2000;148:779-90
40. Yan B, Guo Q, Fu FJ, Wang Z, Yin Z, Wei YB. et al. The role of miR-29b in cancer: regulation, function, and signaling. OncoTargets and therapy. 2015;8:539-48
41. Mogilyansky E, Rigoutsos I. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell death and differentiation. 2013;20:1603-14
42. Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED. et al. Epithelial-mesenchymal and mesenchymal-epithelial transitions in carcinoma progression. Journal of cellular physiology. 2007;213:374-83
43. Tomaskovic-Crook E, Thompson EW, Thiery JP. Epithelial to mesenchymal transition and breast cancer. Breast cancer research: BCR. 2009;11:213
44. Baranwal S, Alahari SK. Molecular mechanisms controlling E-cadherin expression in breast cancer. Biochemical and biophysical research communications. 2009;384:6-11
45. Blick T, Widodo E, Hugo H, Waltham M, Lenburg ME, Neve RM. et al. Epithelial mesenchymal transition traits in human breast cancer cell lines. Clinical & experimental metastasis. 2008;25:629-42
46. Luna C, Li G, Qiu J, Epstein DL, Gonzalez P. Role of miR-29b on the regulation of the extracellular matrix in human trabecular meshwork cells under chronic oxidative stress. Molecular vision. 2009;15:2488-97
47. Lendeckel U, Kohl J, Arndt M, Carl-McGrath S, Donat H, Rocken C. Increased expression of ADAM family members in human breast cancer and breast cancer cell lines. Journal of cancer research and clinical oncology. 2005;131:41-8
48. Kveiborg M, Frohlich C, Albrechtsen R, Tischler V, Dietrich N, Holck P. et al. A role for ADAM12 in breast tumor progression and stromal cell apoptosis. Cancer research. 2005;65:4754-61
49. Nyren-Erickson EK, Bouton M, Raval M, Totzauer J, Mallik S, Alberto N. Urinary concentrations of ADAM 12 from breast cancer patients pre- and post-surgery vs. cancer-free controls: a clinical study for biomarker validation. Journal of negative results in biomedicine. 2014;13:5
50. Depowski PL, Rosenthal SI, Ross JS. Loss of expression of the PTEN gene protein product is associated with poor outcome in breast cancer. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2001;14:672-6
51. Weng LP, Smith WM, Dahia PL, Ziebold U, Gil E, Lees JA. et al. PTEN suppresses breast cancer cell growth by phosphatase activity-dependent G1 arrest followed by cell death. Cancer research. 1999;59:5808-14
52. Phuah SY, Looi LM, Hassan N, Rhodes A, Dean S, Taib NA. et al. Triple-negative breast cancer and PTEN (phosphatase and tensin homologue) loss are predictors of BRCA1 germline mutations in women with early-onset and familial breast cancer, but not in women with isolated late-onset breast cancer. Breast cancer research: BCR. 2012;14:R142
53. Dowsett M, Forbes JF, Bradley R, Ingle J, Aihara T, Bliss J. et al. Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet. 2015;386:1341-52
54. Smith IE, Dowsett M. Aromatase inhibitors in breast cancer. The New England journal of medicine. 2003;348:2431-42
55. Davies C, Godwin J, Gray R, Clarke M, Cutter D, Darby S. et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011;378:771-84
56. Moja L, Tagliabue L, Balduzzi S, Parmelli E, Pistotti V, Guarneri V. et al. Trastuzumab containing regimens for early breast cancer. Cochrane database of systematic reviews. 2012;4:CD006243
Author contact
![]() Corresponding author: Institute of Toxicology, College of Medicine, National Taiwan University, Taipei, 10051, Taiwan. E-mail: shinghwaliuedu.tw.
Corresponding author: Institute of Toxicology, College of Medicine, National Taiwan University, Taipei, 10051, Taiwan. E-mail: shinghwaliuedu.tw.