Impact Factor ISSN: 1837-9664
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Issue 3; 2026
- Volume 17; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Special Issues
Background
Chemistry and structure-activity...
Modulators of the cell cycle
Interaction with tubulin
Multiple molecular targets
Apoptosis
Autophagy
Other anticancer properties
Application of in vivo and ex...
Experimental PBOX-containing...
Comparison with other...
Conclusion and future...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(15):2367-2377. doi:10.7150/jca.16616 This issue Cite
Review
Pre-clinical evaluation of a novel class of anti-cancer agents, the Pyrrolo-1, 5-benzoxazepines
LM Greene1 ![]() , S Butini 2, G Campiani 2, DC Williams 1, DM Zisterer 1
, S Butini 2, G Campiani 2, DC Williams 1, DM Zisterer 1
1. School of Biochemistry and Immunology, Trinity Biomedical Sciences Institute, Trinity College Dublin, Dublin 2, Ireland.
2. European Research Centre for Drug Discovery and Development, Department of Biotechnology, Chemistry and Pharmacy, and Istituto Toscano Tumori, University of Siena, via Aldo Moro 2, I-53100 Siena, Italy.
Received 2016-6-27; Accepted 2016-9-28; Published 2016-12-4
Abstract
Microtubules are currently ranked one of the most validated targets for chemotherapy; with clinical use of microtubule targeting agents (MTAs) extending beyond half a century. Recent research has focused on the development of novel MTAs to combat drug resistance and drug associated toxicities. Of particular interest are compounds structurally different to those currently used within the clinic. The pyrrolo-1, 5-benzoxazepines (PBOXs) are a structurally distinct novel group of anti-cancer agents, some of which target tubulin. Herein, we review the chemistry, mechanism of action, preclinical development of the PBOXs and comparisons with clinically relevant chemotherapeutics. The PBOXs induce a range of cellular responses including; cell cycle arrest, apoptosis, autophagy, anti-vascular and anti-angiogenic effects. The apoptotic potential of the PBOXs extends across a wide spectrum of cancer-derived cell lines, by targeting tubulin and multiple molecular pathways frequently deregulated in human cancers. Extensive experimental data suggest that combining the PBOXs with established chemotherapeutics or radiation is therapeutically advantageous. Pre-clinical highlights of the PBOXs include; cancer specificity and improved therapeutic efficacy as compared to some current first line therapeutics.
Keywords: Pyrrolo-1, 5-benzoxazepines, tubulin, apoptosis, drug resistance and G2/M arrest.
Background
Cancer is the leading cause of death in children past infancy and second only to cardiovascular disease in adults. Hence, the discovery and pre-clinical development of novel anti-cancer agents is of paramount importance. Continued development of novel drugs is necessary to improve patient survival and limit treatment-associated toxicities. Whilst the majority of anti-cancer agents are of natural origin, the significance of 'made to order' synthetic drugs in the fight against cancer cannot be disputed. Two decades have passed since the first publication of a novel series of pyrrolobenzoxazepine derivatives. The compounds were originally designed and synthesised as prospective 'peripheral-type' benzodiazepine receptor (PBR) ligands (1). However, further studies demonstrated that the pyrrolobenzoxazepine derivatives could elicit anti-proliferative effects (2) and apoptosis (3) independent of PBR status. The PBOXs were patented (WO 2001058904 A1) as potent anti-cancer agents with IC50 values in the nanomolar to micromolar range. Herein, we review for the first time the chemical synthesis and the pre-clinical evaluation of the pyrrolo-1, 5-benzoxazepines (PBOXs) using a collection of in vitro, in vivo and ex vivo cancer models spanning an extensive range of cancer types.
Chemistry and structure-activity relationship studies of the PBOXs
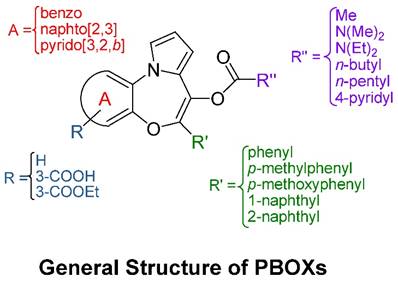
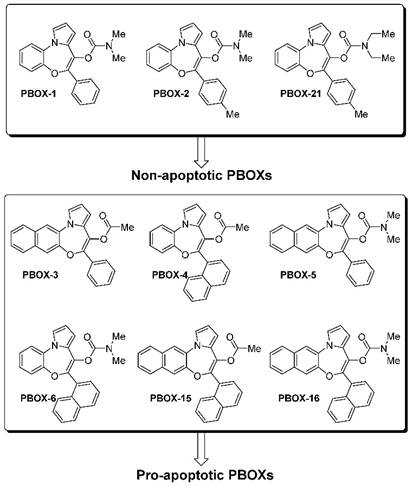
The general core structure depicted in Figure 1 is common to all PBOXs (4). The chemical structures of selected PBOXs (namely; PBOX-1, -2, -3, -4, -5, -6, -15, -16 and -21) are detailed in Figure 2. Chemical properties of the selected PBOXs are reported in Table 1.
The core structure of PBOX-based compounds and the explored modifications for structure-activity relationship studies.
Chemical structure of representative PBOXs.
Chemical properties of the PBOXS.
| Compound | Formula | mp (°C) | Recrystallization solvent | Assay | |
|---|---|---|---|---|---|
| Cytospin % cell death | Viability IC50 [µM] | ||||
| Non-apoptotic PBOXs | |||||
| PBOX-1 | C21H18N2O3 | 118-1191 | EtOH1 | 05 | 352 |
| PBOX-2 | C22H20N2O3 | 154-1551 | hexanes/Et2O1 | 05 | >1002 |
| PBOX-21 | C24H24N2O3 | 154-1551 | hexanes/Et2O1 | - | 742 |
| Apoptotic PBOXs | |||||
| PBOX-3 | C24H17NO3 | 164-1651 | EtOH1 | 475 | - |
| PBOX-4 | C24H17NO3 | 157-1581 | EtOH1 | 435 | - |
| PBOX-5 | C25H20N2O3 | 189-1911 | hexanes1 | 405 | - |
| PBOX-6 | C25H20N2O3 | 177-1794 | EtOH4 | 485 | 1.9 6 |
| PBOX-15 | C28H19NO3 | - | - | - | 0.216 |
| PBOX-16 | C29H22N2O3 | - | - | - | 0.226 |
mp, melting point.
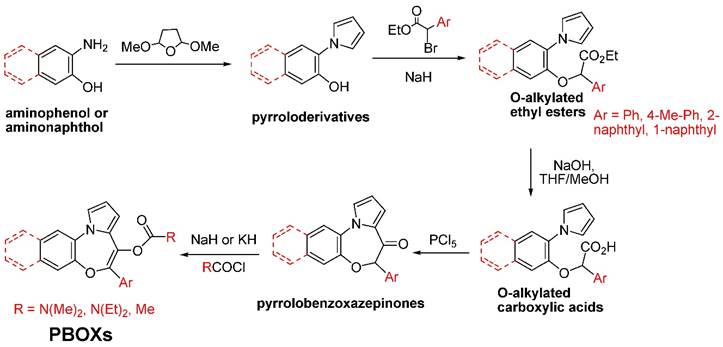
A schematic depiction of the synthetic pathway of the selected PBOXs shown in Figure 1 is outlined in Figure 3 (1). The synthesis began using the commercially available ortho-amino phenol or 3-amino-naphthalen-2-ol which then underwent a Clauson-Kaas reaction to produce pyrrole derivatives with good yields. O-alkylation of these latter compounds with the ethyl ester of the appropriate α-bromo derivative of the aryl-acetic acid was performed in the presence of NaH to give the different O-alkylated ethyl esters. After saponification of these esters, the free carboxylic function of the corresponding acids was ready for chlorination and Friedel-Crafts intramolecular acylation of the alpha-position of the pyrrole ring. This reaction was performed in the presence of phosphorus pentachloride, which works both as the chlorinating agent and as the Lewis acid for the acylation reaction. The obtained pyrrolobenzoxazepinones were then treated with sodium or potassium hydride to obtain the corresponding salts of their enolates, which act as nucleophiles for the appropriate chlorides used for the synthesis of the PBOXs bearing a carbamate or ester moiety at the C6 position.
Synthesis of the representative PBOXs.
The cellular location of the molecular target of the PBOXs together with a detailed structure-activity relationship (SAR) was determined by the strategic synthesis of various substituted PBOX analogues. The applied use of specific functionalities for obtaining cell penetrating (ester) or non-cell penetrating (free acid) compounds (the COOMe and COOH substituted analogues, see Figure 1) demonstrated an intra-cellular target for the PBOXs (4). A SAR study was also performed to further enhance our knowledge of the respective moieties required for apoptosis induced by the PBOXs. In particular, rational modifications were introduced in specific regions of the PBOX scaffold to explore the spatial dimensions of the lipophilic pockets of their intracellular putative target exemplified by L1, L2 and L3 in Figure 4.
Areas of interaction of the biological target explored by the development of specific PBOX-based analogues.
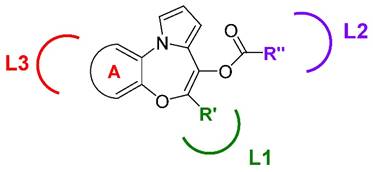
The volume of L1, L2 and L3 regions was explored by increasing the dimensions of the substituents such as ring A, R' and R'' on the molecules. It was concluded that a 2-naphthyl system within L1 was more favourable in terms of apoptotic potential. With respect to the L2 pocket the best interaction was attained with an acetyl ester group (PBOX-3, -4, and -15) or a dimethylaminocarbamoyl moiety (PBOX-5, -6 and -16). Introduction of longer chains at this position were not tolerated. With respect to ring A, interacting with the putative pocket L3, the introduction of a further aromatic system was nicely accommodated in the pocket and allowed the identification of the naphtha-[2,3]-based analogues as potent pro-apoptotic agents (e.g. PBOX-3 and -5).
To conclude, analysis of cell data obtained from PBOXs substituted at ring A and at position 7 allowed the identification of the most potent analogues of the series, namely PBOX-15 and -16. A 2-naphthyl group at position 7 of the tricyclic system appears to be better suited than a phenyl or a 1-naphthyl, for correct interactions with pocket L1. Also L3 benefits from the presence of a fused naphthyl system.
Modulators of the cell cycle
Rapid cell division is a common trait amongst cancer cells. The cell cycle progresses from the G1 phase to synthesis (S) to the G2/M phase. Each phase of the cell cycle is highly regulated by cell cycle checkpoints. Activation of such checkpoints leads to a transient or sustained cell cycle arrest. Cell cycle arrest is characterised as an increase in the percentage of cells at one or more phases of the cell cycle and can be readily quantified by flow cytometric analysis of propidium iodide-stained cells (7). The PBOXs were classified into two groups based on their ability to induce apoptosis. Group one (PBOX-1, -2, and -21) do not induce apoptosis in cancer cells (3, 5, 8, 9). Group two (PBOX-3, -4, -5, -6, -15 and -16) induce apoptosis in cancer cells (3-6, 10-28). The non-apoptotic subset of the PBOXs (PBOX-1,-2,-21) exert their anti-proliferative effects by inducing a G1 cell cycle arrest (2, 8, 9). On the other hand, the pro-apoptotic subset of PBOXs (PBOX-6, -15, -16) induce a G2/M arrest subsequent to apoptosis (6, 13-15, 19-22, 24, 29). In each case, the aforementioned cell cycle arrest induced by a representative member of each group was confirmed using several biochemical techniques. Cell cycle progression is controlled by the interaction of cyclins with their respective cyclin-dependent kinases (CDKs). CDK inhibitors such as p21 can negatively regulate CDKs. PBOX-21-induced G1 arrest was confirmed by a sequential reduction of CDK2, CDK4, CDK6 and cyclins D3 and D2; a reduction in complex formation between the kinases and cyclins; increased P21 and a decrease in Rb hyper-phosphorylation (8).
The absolute levels of cyclin B1 are a critical determinant of cell cycle progression through M phase. Increased levels of cyclin B1 will in turn lead to the activation of the cyclin B1/cyclin-dependent kinase 1 (CDK1) complex and cell cycle progression from G2 to M phase. Decreased cyclin B1 inactivates CDK1 and allows mitotic exit. PBOX-6-induced G2/M arrest was verified by an increase in cyclin B1 and activation of CDK1 complex (13). Whilst CDK1 remains the most prominent mitotic kinase controlling entry and exit from mitosis, other signalling cascades such as the spindle assembly checkpoint (SAC) also influence mitotic exit.
The main components of the SAC are BUBR1, BUB1, BUB3, MAD1, MAD2 and MAD3 (30). The SAC signal is initiated at an unattached kinetochore. Given that the tubulin-targeting pro-apoptotic PBOXs destabilise the microtubules, which in turn compromises kinetochore attachment, the SAC is critical in delaying progression to anaphase. Activation of the spindle assembly checkpoint by means of phosphorylation and activation of BUBR1 was also observed in cancer cells following PBOX-6 and -15 exposure (15, 18). PBOX-15- induced mitotic block was compromised following BUBR1 siRNA, conclusively demonstrating the critical importance of the SAC initiated by BUBR1 in maintaining PBOX-15-induced mitotic arrest (15). Reduction of BUBR1 protein levels associated with a decline in PBOX-15-induced mitotic block and the onset of apoptosis (15, 18, 21). Furthermore, increased polyploidy was observed in BUBR1-depleted cells exposed to PBOX-15 thus confirming the importance of SAC in preventing genomic instability. More importantly, the absolute levels of BUBR1 inversely correlated with PBOX-induced apoptosis (15, 21) a trend observed by independent researchers with other MTAs (31). In the future, predictive biomarker screening may be available in the clinic. BUBR1 may ultimately serve as a biomarker to predict response to tubulin targeting PBOXs and other MTAs. It is worth noting that PBOX-15 also induced mitosis-independent apoptosis in primary non-cycling B-chronic lymphocytic leukaemia (CLL) cells (32). Disruption of tubulin networks in interphase CLL was observed. Whilst the metaphase microtubules are highly dynamic and very susceptible to PBOX-induced destabilisation of the mitotic spindle; in the absence of mitosis the PBOXs can also effectively destabilise interphase tubulin and induce apoptosis. Hereafter, unless otherwise stated the PBOXs refer to the pro-apoptotic subgroup.
Interaction with tubulin
Microtubules play a central role in a number of cellular events required for tumour formation and progression and remain one of the most effective targets for cancer therapeutics (33, 34). Agents that interact with tubulin can disrupt microtubule-dependent processes by interfering with the natural equilibrium between free tubulin dimers and assembled polymers. Microtubule targeting agents (MTAs) are classified into two groups according to their effect on tubulin mass at high concentrations (35). Agents that inhibit the assembly of tubulin dimers are classified as tubulin depolymerisers whereas agents promoting the assembly of tubulin are defined as tubulin polymerisers. The vinca alkaloids and the taxanes been the most significant example of the former and the latter. MTAs can also influence microtubule dynamics without altering microtubule mass (33). The pro-apoptotic subset of the PBOXs (-6, -15 and -16) are tubulin depolymerisers (13, 15, 19, 32, 36). It is worth noting that PBOX-21 a representative G1 arresting or non-apoptotic PBOX did not interact with tubulin (13). We hypothesise that the presence of an extra naphthyl ring at position 7 of the tricyclic system may be a key determinant for improving tubulin interactions and subsequent effects such as G2/M arrest and apoptosis.
The colchicine, taxol, vinca alkaloid and the nucleotide binding sites on tubulin are all well characterized (37-40). Other recently identified tubulin binding sites include the rhizoxin binding site on β-tubulin (41) and the peloruside and laulimalide binding sites on βI-tubulin (42, 43). Tubulin binding sites are primarily identified using inhibition based studies. A general principle for inhibition-based experiments is that competitive inhibition can result from isosteric effects at a single common binding site or allosteric effects between two separate binding sites, whereas non-competitive inhibition between two ligands must result from an allosteric binding at two separate binding sites. However, owing to the complex molecular structure and dynamic nature of tubulin, the exact binding site of some MTAs remain unidentified (44). The challenge of identifying tubulin binding sites is further complicated by the fact that some sites are large and overlap resulting in competitive inhibition of tubulin binding ligands. For example, it was originally assumed that rhizoxin bound to the vinca alkaloid site based on results obtained from a vinblastine binding assay. However, recent x-ray crystallography results clearly reveal a novel binding site for rhizoxin on β-tubulin (41). It was hypothesised that rhizoxin and β-tubulin interactions interfere with the formation of the vinca alkaloid binding domain. These findings suggest that x-ray crystallography is the gold standard to accurately define tubulin binding sites. To date, the exact location of PBOXs' binding site within tubulin remains to be clarified. Future x-ray crystallography experiments will ultimately resolve this issue.
Multiple molecular targets
The causes of cancer are multifactorial with current advances in cancer research often failing to keep up with evolving cancer cells. The last decade has seen the introduction of novel anti-cancer agents with very specific single targets. Imatinib mesylate, been one such example, specifically targets the tyrosine kinases Bcr-Abl, c-kit (CD117) and platelet-derived growth factor receptor and is thus confined to treating carcinomas overexpressing such kinases (45). Whilst initially hailed with great triumph, Imatinib and its successors soon succumbed to resistance. Like other MTAs, the PBOXs primarily target tubulin and as a consequence downregulate several core signalling pathways involved in cancer development and progression. Table 2 summarises the extensive molecular analysis of PBOX signalling pathways across a diverse range of established cancer cell lines, primary cells and cells derived directly from patient samples (see section on application of in vivo and ex vivo models for further details). Modification of the signalling pathways detailed in Table 2 occurred following exposure to concentrations of PBOXs required to depolymerise the microtubules. Molecular profiling of pro-apoptotic PBOX-induced signalling identified early phosphorylation and activation of c-Jun n-terminal kinase (JNK); followed by late downregulation of signalling pathways which are frequently associated with a wide variety of human cancers including; Bcr-Abl, c-kit, B-cell lymphoma 2 (Bcl-2), B-cell lymphoma-extra large (Bcl-xL), Myeloid cell leukaemia 1 (Mcl-1), phosphoinositide 3-kinase P13K/(protein kinase B (AKT)/mechanistic target of rapamycin (mTOR) and inhibitors apoptosis proteins (IAPs) (Table 2). In more detail, genes in the P13K/AKT pathway are the most frequently deregulated genes in cancer, as such agents targeting this pathway often reach clinical trials (47). Similarly, overexpression of IAPs in cancer cells can mediate drug resistance by inhibiting caspase activation and promoting the expression of various anti-apoptotic proteins; hence IAPs represent a valid target for cancer therapy, with novel agents targeting IAPs in constant demand (48). On the other hand, JNK displays both oncogenic and tumour suppressive roles (49). Activation of JNK is commonly associated with various types of cytotoxic insults. JNK is a critical mediator of PBOX-induced cell death (12, 32, 46) and responsible for the phosphorylation and inactivation of the anti-apoptotic protein Bcl-2 (12). Data obtained to date suggests that JNK acts a pro-apoptotic effector during PBOX-induced cell death. The PBOXs are suitable to treat a diverse range of cancer types by targeting tubulin and consequently downregulating several oncogenes responsible for tumour growth and development.
Apoptotic hallmarks and molecular targets of the pro-apoptotic PBOXs in HUVECs and cancer cells.
| Cancer type | PBOX | Markers of apoptosis | Molecular targets | Refs |
|---|---|---|---|---|
| AML, T-ALL Lymphoma | PBOX-3,-4,-5, -6* | DNA fragmentation, morphological features, caspase activation*, caspase-dependent*, cytosolic cytochrome c* | 3 | |
| CML | PBOX-3,-4,-5,-6*,-7 | Morphological features, DNA fragmentation*, caspase-3 activation*, PARP cleavage*. | Bcr-Abl ↓ | 5 |
| CML | PBOX-6 | JNK ↑, C-Jun -P ↑, ATF-2 -P ↑ | 46 | |
| CML*, ALL | PBOX-6 | Caspase-independent* | Bcl-2 -P↑, Bcl-xL P ↑ | 12 |
| Breast | PBOX-6 | PARP cleavage | 25 | |
| Breast | PBOX-6 | CDK1 ↑ , Cyclin B1 ↑, Tubulin depolymerisation | 13 | |
| CML | PBOX-6 | DNA fragmentation, Caspase-independent, Serine protease dependent,, MMP↓ | 47 | |
| CML, AML, breast, cervical* | PBOX-6 PBOX-15 | PARP cleavage | Tubulin depolymerisation *, BubR1 -P | 14 |
| Lymphoma | PBOX-15 | Tubulin | 36 | |
| CML | PBOX-6 | Translocation of cyclophilin A and Pin A | 29 | |
| Ex vivo CLL | PBOX-15 | Annexin V+, partial caspase dependence, caspase-8 cleavage | Tubulin depolymerisation, JNK-P↑ | 32 |
| CML, ovarian, HUVECs | PBOX-6 PBOX-16 | Caspase-3/7 activation | Tubulin depolymerisation | 19 |
| Ex vivo CML | PBOX-6 PBOX-15 | Annexin V + | 17 | |
| AML | PBOX-6 PBOX-15 PBOX-16 | Cleaved PARP, caspase-3 activation | Cleaved Bcl-2↑ | 6 |
| Prostate | PBOX-6, PBOX-15 | Cleaved PARP | Tubulin depolymerisation, BubR1 -P Bcl-2-P, Bcl-xL-P, BubR↓, Bcl-2↓, Bcl-xL↓ | 18 |
| Multilple myeloma Ex vivo Multilple myeloma* | PBOX-15 | Caspase-dependent, Cleaved caspase-8, Annexin V +* , MMP↓, Cytosolic cytochrome c | Tubulin depolymerisation, BubR↓, DR5↑, Bid↓, BimEL↓ | 21 |
| Prostate | PBOX-15 | Caspase-3/7 activation | HIF-α↑ | 20 |
| T-ALL B-ALL | PBOX-15 | Caspase-dependent Annexin V + | Tubulin depolymerisation | 22 |
| Colon | PBOX-6 | PARP cleavage, cleaved caspase-3 | LC3B-II↑, Bcl-2↓, Mcl-1↓, Bcl-XL-P | 23 |
| Neuroblastoma | PBOX-6 | PARP cleavage, cleaved caspase-3,-8,-9, MMP↓, Cytosolic cytochrome c | Cleaved Bcl-2↑, Bak↑, Mcl-1↓ | 24 |
| OSCC | PBOX-6 PBOX-15 | Tubulin depolymerisation, full-length PARP↓, | 26 | |
| T-ALL | PBOX-15 | PARP cleavage↑, active caspase-3,-8,-9↑, MMP↓ caspase-dependent | DR5↑, Bcl-XL↓, Mcl-1↓,Bim↓,CIAP2↓ livin↓, AKT↓,AKT-P↓,c-RAF-P↓,PTEN-P↓ | 28 |
| GIST | PBOX-15 | PARP cleavage↑ | CDC37↓, CDC37-P↓, Mcl-1↓,c-kit-P↓, AKT↓, AKT-P↓, HSP90↓, CKIIα↓ | 27 |
PARP, poly (ADP-ribose) polymerase; JNK, c-Jun n-terminal kinase; -P, phosphorylated; ATF-2-P, activating transcription factor 2 phosphorylated), HUVECs, human umbilical vein endothelial cells; MMP, mitochondrial membrane permeabilisation; DR5, death receptor 5; HIF-α, hypoxia-inducible factor; LC3B-II, microtubule-associated protein light chain 3 (marker of autophagy); OSCC, oral squamous cell carcinoma; GIST, gastrointestinal stromal tumour; Bcl-XL, , B-cell lymphoma-extra large; Mcl-1, Myeloid cell leukaemia 1; AKT, protein kinase B; PTEN, phosphatase and tensin homolog; HSP90, heat shock protein 90, CKIIα, casein kinase 2. *, links markers of apoptosis/molecular targets to PBOX or cell line.
Apoptosis
Most anti-cancer agents destroy malignant cells by inducing apoptosis (50). Other types of cell death include; autophagy, necrosis, necroptosis and pyroptosis (51). It is worth noting that the true existence of cell death induced by autophagy remains questioned by many. It is however, more widely accepted that cell death can associate with features of autophagy as opposed to been directly induced by autophagy (52). Like most other anti-cancer agents, the PBOXs destroy cancer cells by initiating apoptosis. The morphological features of apoptosis include cell shrinkage with chromatin condensation or fragmentation (50). This mode of cell death is complex, highly organised, energy-dependent and conserved amongst species.
The caspase cascade is central to apoptosis and once activated cannot be reversed. Some instances of caspase-independent apoptosis have been reported. Use of caspase inhibitors and siRNA identified a variable dependence on caspases for PBOX-induced apoptosis which was cell-dependent and influenced by the addition of other anti-cancer agents (Table 2 and 3). Apoptosis can be manifested by two main pathways, the intrinsic or extrinsic. However, both pathways are not always mutually exclusive with some overlap detected at the caspase-3 level. Accordingly, PBOXs can activate both the intrinsic and extrinsic apoptotic pathways with some overlap at caspase-3 detected. Data presented in Table 2 depicts that PBOX-induced apoptosis is predominately executed by the intrinsic pathway.
Summary of ex vivo data from normal donors and patients samples exposed to selected pro-apoptotic PBOXs.
| PBOX | Patient number | Sample type | Main outcome | Reference | |
|---|---|---|---|---|---|
| PBOX-15 | n=55 | CLL including: Unmutated IgV (H) (n=13), CD38 +ve (n=13), ZAP-70 +ve (n=8), 17p deletions (n=3) | IC50 0.55 µM, induced apoptosis in low risk and hign risk patients, more effective that fludarabine (n=11) | 32 | |
| PBOX-15 | n=12 | Normal bone marrow (n=8), normal PBMC (n=4) | No significant increase in cell death | 32 | |
| PBOX-6 PBOX-15 | n=6 | CML including: T315I +ve (n=2), F317I (n=1) | Significant increase in apoptosis | 17 | |
| PBOX-15 | n=5 | Multiple myeloma | Induced apoptosis | 21 | |
ZAP-70, Zeta-associated protein 70; PBMC, peripheral blood mononuclear cells.
The intrinsic or mitochondrial apoptotic pathway is controlled by the interaction of Bcl-2 family members with the mitochondria, which in turn increases mitochondrial permeability and allows the release of cytochrome c (50). The PBOXs also extensively modulate several members of the Bcl-2 family facilitating mitochondrial-mediated apoptosis. The Bcl-2 pathway is central to whether or not a cell commits to apoptosis as it proceeds to the point of irreversible commitment to cell death (53). The PBOXs (-6, -15) target the anti-apoptotic subset of the Bcl-2 family in multiple ways, specifically by phosphorylation/inactivation and also by downregulation and/or cleavage; the former associated with G2/M arrest and the latter with the onset of apoptosis. It is noteworthy that PBOX-induced apoptosis can also occur independent of change to Bcl-2 (32, 21). To date, PBOX-6- and PBOX-15-induced apoptosis associates with a decrease in Mcl-1 expression (21, 23, 24, 27, 28). SiRNA-mediated downregulation of Mcl-1 augmented PBOX-6-induced apoptosis, suggesting that downregulation of Mcl-1 enhances or facilitates PBOX-6-induced apoptosis, rather than being the effective target (16).
The pro-apoptotic members of the Bcl-2-associated X protein (BAX) and Bcl-2-antagonist/killer 1 (BAK) are the ultimate executioners of the intrinsic pathway. PBOX-6-induced apoptosis was associated with increased BAK but not BAX in neuroblastoma cells (24). BAK or BAX dimers interact with the mitochondria leading to permeabilisation (54). Increased mitochondrial outer-membrane permeability (MOMP) leads to a loss in mitochondrial membrane potential. We have conclusively established that PBOX-6 and -15 induced a significant loss in mitochondrial membrane potential and subsequent release of cytochrome c (3, 21, 24, 55). Cytosolic cytochrome c is a critical component of the apoptosome complex which in turn signals caspase activation (56). The initiator caspase, caspase-9 and its effector caspases, caspase-3 and -7 have been shown to be activated by PBOX-6 and -15 (Table 2). However, the balance between pro- and anti-apoptotic members of the Bcl-2 family will ultimately determine whether or not a cell commits suicide or survives.
The cell death signal can also be triggered outside the cell by the extrinsic pathway. Extracellular death ligands such as TNF-related apoptosis-inducing ligand (TRAIL) interact with its corresponding death receptor/s (DR4, DR5) leading to the formation of the death-inducing signalling complex and subsequent activation of caspase-8 (50). PBOX-15 can also trigger apoptosis, albeit to a lesser extent, by the extrinsic pathway (21, 28). PBOX-15 increased DR5 expression in malignant myeloma and T-cell acute lymphoblastic leukemia (T-ALL) cells facilitating enhanced activation of the extrinsic pathway (21, 28). Accordingly, activation of the extrinsic pathway initiator caspase, caspase-8 by PBOX-15 was identified in CLL, malignant myeloma and T-ALL cells (21, 28, 32). The addition of the DR5 ligand TRAIL to cells further enhanced PBOX-15-induced activation of the extrinsic pathway (21, 28). However, only a partial dependence on the extrinsic pathway during PBOX-15-induced apoptosis was identified in these cells indicating a co-dependence on the intrinsic pathway.
Autophagy
Autophagy is the most recently characterised biochemical response to stress induced by the PBOXs and has much scope for further development (23). This trait is not unique to the PBOXs and is shared with most anti-cancer drugs (57). Autophagy is a cellular process of degradation and recycling triggered in response to normal physiological processes and adverse stress. The consequence of stress-induced autophagy can be either cell death or prolonged cell survival. Autophagic cell death is defined as cell death associated with the appearance of autophagic vesicles and can be inhibited by specific inhibitors of autophagy. Pharmacological inhibition of late stage autophagy enhanced PBOX-6-induced apoptosis in colon cancer cells (23). This result would suggest that PBOX-6-induced cytoprotective autophagy as opposed to autophagic cell death. It is noteworthy that further experiments demonstrating an increased sensitivity to autophagy-inducing treatments while inhibiting autophagy would ultimately establish a cytoprotective function.
Autophagy can facilitate tumour cell survival in two ways; by providing an energy source to fuel the increased metabolic demands of highly proliferative tumour cells and to confer stress tolerance, mediating resistance to most anti-cancer approaches. As such, autophagy is now a validated therapeutic target under clinical evaluation across 32 ongoing clinical trials (58). It would be relevant to determine any potential therapeutic benefit in combining the PBOXs with hydroxychloroquine, the predominant autophagy inhibitor under clinical evaluation.
Other anticancer properties
Apart from targeting tubulin in mitotic cells, the tubulin targeting properties of the PBOXs can also impede other functions involved in tumour progression such as cell adhesion, spreading, migration and angiogenesis (19, 22, 36). Tumour neovascularisation occurs by non-sprouting and sprouting angiogenesis, although the latter is more prominent (59). PBOX-6 and -16 inhibited several factors involved in the sprouting of new capillaries including the migration, proliferation and differentiation of endothelial cells into tube like structures (19). PBOX-15-mediated depolymerisation and post-translational modification of tubulin can dramatically impair T-cell migration (36). Furthermore, PBOX-15 downregulated the expression of β1-, β2-, α4-integrins in human ALL cells (22). Integrins are crucial for cell migration and invasion (60). Microtubules control integrin arrangement and hence influence integrin-associated physiological and pathological functions. Therefore, PBOX-15 can severely compromise integrin-mediated adhesion of malignant lymphocytes by a dual mechanism; by reducing available integrins and by disrupting the structural support provided by the microtubules that is required for proper integrin arrangement. Furthermore, PBOX-15-mediated inhibition of T-cell migration could be adapted to treat other pathologies such as autoimmune disorders and graft rejection.
Application of in vivo and ex vivo models
The therapeutic potential of the PBOXs has also been assessed in a panel of in vivo and ex vivo cancer models and normal counterparts. Removal of the bone marrow, tumour or a blood sample from donors can facilitate the analysis of experimental drugs in ex vivo biological assays. Administration of conventional chemotherapeutics at clinically efficacious doses often associates with unwanted toxic side effects, frequently manifesting in the bone marrow. The granulocyte-macrophage colony-forming unit (CFU-GM) assay is routinely used to assess potential bone marrow toxicity. It is noteworthy that murine bone marrow is less sensitive than that of human to many chemotherapy-induced toxicities. For this reason, human donor samples were used to determine the effects of a selected potent member of the PBOX series, PBOX-15. Whilst marginal reduction in colonies was detected in some donor samples, reflective of donor heterogeneity, no significant overall reduction in colonies among the cohort of eight donors exposed to PBOX-15 [2 x IC50] for 72 h was observed (Table 3) (32). This result is promising and suggests a probability that PBOXs may reach therapeutic doses in vivo without causing adverse side effects. Furthermore, no lasting changes in haematological parameters including red and white blood cell count and platelets were noted in mice following eight consecutive daily doses of PBOX-6 [35 mg/kg/day] (4). Similarly, neither PBOX-6- nor PBOX-15-induced cell death in normal peripheral blood cells derived from healthy donors (12, 32).
More importantly, PBOX-15 is very effective in ex vivo CLL cells irrespective of high risk prognostic markers (Table 3) (32). PBOX-15 surpassed the current first line treatment fludarabine by inducing significantly more apoptosis in ex vivo CLL cells (Table 3) (32). Both PBOX-6 and -15 effectively induced apoptosis in ex vivo chronic myelogenous leukaemia (CML) cells including those harbouring the T315I mutation, which ultimately predisposes to resistance to tyrosine kinase inhibitors. Mice bearing breast 4T1 or CML BAF/3 T315I tumours were developed as mouse tumour models, inclusive of a clinically relevant model of resistance, to determine the ability of PBOX-6 to inhibit tumour growth. PBOX-6 [7.5 mg/kg] significantly inhibited tumour growth in both models without any adverse side effects (16, 25). It is worth noting that, the dose administered was almost 5-fold below the dose confirmed to have no lasting side effects, indicating a wide therapeutic window, with scope for improvement.
Like many anti-cancer agents, the PBOXs may not be curative as a single agent but may act more favourably as an adjuvant therapy. Given the low toxicity profile of the PBOXs, combination with other established chemotherapeutics, may be more beneficial by increasing therapeutic efficacy, whilst reducing the levels of unwanted toxicity.
Experimental PBOX-containing drug combinations
The introduction of combinational drug therapy just over two decades ago has been a clinical success, leading to a progressive decline in cancer mortality rates. Table 4 clearly depicts that combining the PBOXs with established chemotherapeutics or radiation is therapeutically advantageous. The therapeutic outcome of drug combinations can be quantified using the combination index (CI) as defined by Chou (61). Many PBOX combination strategies were deemed to be synergistic or favourable by the pinnacle of isobologram equations to define synergy, the CI (Table 4). Whilst the mechanism of action for single drugs are often clearly defined, it can be challenging to decipher the mechanism of synergistic drug combinations. It would appear that the molecular signature of PBOX combinations seems to predominantly amplify that observed with the PBOXs alone (Tables 2 and 4). It would be of interest to clarify if these synergistic outcomes can be translated to in vivo models.
Summary of preclinical in vitro evaluations of the pro-apoptotic PBOXs with established chemotherapeutics and radiation.
| Drug Combination | Cancer type | Outcome | Marker of Apoptosis | Molecular targets | Ref |
|---|---|---|---|---|---|
| Imatinib + PBOX-6/-15 | CML | Significant enhancement | PARP cleavage | BcrAbl↓, Bcl-XL↓, Mcl-1 ↓ | 14 |
| Imatinib + PBOX-21 | CML | Significant enhancement | PARP cleavage, activation caspase-8, partial caspase-8 dependence | Bcr-Abl↓, Bim↑, Bcl-XL↓, Mcl-1 ↓, cleavage Bcl-2↑ | 9 |
| PBOX-6/-15 (18 h); flavopiridol (24 h) | CML | synergistic | Cyclin B1↓, CDK1↓, survivin↓ | 16 | |
| PBOX-6/-15 (18 h); flavopiridol (24 h) | prostate | synergistic | 18 | ||
| PBOX-15 + TRAIL | Multiple myeloma | synergistic | Annexin V + | 21 | |
| PBOX-15 + 2 Gy | Prostate*, NSCLC, glioma | PBOX-15 radiosentises cancer cells | Caspase 3/7* activation | 20 | |
| PBOX-6 + BAF-A1 | Colon | synergistic | Caspase-dependent, PARP cleavage, cleavage caspase-3 | Mcl-1↓ | 23 |
| PBOX-6 + carboplatin | neuroblastoma | synergistic | PARP cleavage↑, MMP↓, Cytosolic cytochrome c↑ | Cleaved Bcl-2↑, activated Bak↑, Mcl-1↓ | 24 |
| PBOX-15 + TRAIL | T-ALL | Synergistic | PARP cleavage↑, active caspase-3,-8,-9↑, MMP↓,caspase-dependent | DR5↑, Bcl-XL↓, Mcl-1↓, Bim↓, BidEL↓, C-FLIP↓, CIAP2↓, XIAP↓, livin↓, Akt↓, Akt-P↓, c-raf-P↓, PTEN-P↓ | 28 |
| PBOX-6 + imatinib | GIST | Synergistic | PARP cleavage↑, active caspase-3↑ | CDC37↓, CDC37-P↓, Mcl-1↓, c-kit-P↓, AKT↓, AKT-P↓, HSP90↓, CKIIα↓ | 27 |
PARP, poly (ADP-ribose) polymerase; Bcl-xL, B-cell lymphoma-extra large; Mcl-1, Myeloid cell leukaemia 1; Bcl-2, B-cell lymphoma 2; CDK1, cyclin-dependent kinase 1; NSCLC, non-small cell lung carcinoma; BAF-A1, bafilomycin A1; MMP, mitochondrial membrane potential; BAK, Bcl-2-antagonist/killer 1; JNK, c-Jun n-terminal kinase; MAPK, mitogen-activated protein kinase; TRAIL, TNF-related apoptosis-inducing ligand; DR5, death receptor 5; c-FLIP, cellular FADD-like IL-1β-converting enzyme-inhibitory protein; CIAP2, cellular inhibitor of apoptosis 2; XIAP, x-linked inhibitor of apoptosis; -P, phosphorylated; PTEN-P, phosphatase and tensin homolog -phosphorylated; GIST, gastrointestinal stromal tumour; HSP90, heat shock protein 90, CKIIα, casein kinase 2. *, links molecular targets to cell line.
Comparison with other anti-cancer agents with particular emphasis on chemoresistance
MTAs as a group are structurally diverse and complex (33). Taxol posed a structure so complex that a strategy for chemical synthesis spanned across two decades (62). Structural complexity can significantly delay the clinical development of a therapeutic agent. The microtubule targeting combretastatins, on the other hand, are structurally simple, a property that allowed the rapid development of superior synthetic water soluble derivatives and prompt clinical evaluation (63). Like the combretastatins, the PBOXs are simple in structure, easily synthesised and readily modified. Properties advantageous to mass production and rapid clinical development. PBOX-15 and -16, the most potent of the series, are active in cancer cells in the mid nanomolar range, falling slightly behind the clinically relevant taxanes and vinca alkaloids, in terms of general potency. However, once the exact binding site of the PBOXs on tubulin is clearly identified by means of x-ray studies, the structural simplicity of the core PBOXs will readily allow the design and synthesis of therapeutically superior analogues.
Apart from structural simplicity, other factors that increase the clinical appeal of novel MTAs include a reduced toxicity and activity against drug resistant cells. Unlike taxol, the PBOXs displayed no sign of toxicity to normal blood cells and were active in multi-drug resistant cells (12, 32, 64). Long term palliation or cure is the ultimate goal of cancer therapy. Multi-drug resistance is the predominant obstacle preventing this goal. The energy-dependent multi-drug efflux pump P-glycoprotein (P-gp) is a major player in mediating drug resistance to lead chemotherapeutics including the taxanes and vinca alkaloids (65). Resistance can be reversed by pre-treating with pump inhibitors such as verapamil (66). However, this is costly, often ineffective and can be associated with increased toxicity (67). A more favourable approach to overcome P-gp-mediated resistance is to administer agents which are not substrates for P-gp, such as the PBOXs (6, 24). Furthermore, PBOXs can bypass mutations in Bcr-Abl, p53 and various genetic anomalies in CLL and also, overexpression of Bcl-2, c-kit, P-gp, breast cancer resistance protein (BCRP) and v-myc avian myelocytomatosis viral oncogene (MYCN); to potently induce apoptosis in chemotherapy resistant cells (6, 12, 14, 17, 24, 27, 32). In summary, the PBOXs offer many advantages over current MTAs used within the clinic including structural simplicity, increased tumour specificity and insensitivity to a variety of mechanisms of chemoresistance.
Conclusion and future perspectives
To conclude, the pro-apoptotic PBOXs represent a novel class of anti-mitotic agents that prevent microtubule assembly. The PBOXs exert their anti-tumour effects by interfering with microtubule dynamics resulting in cell cycle arrest and tumour cell death. To date, we have collected no evidence to suggest that the influence of the pro-apoptotic PBOXs on the molecular pathways discussed within the review are independent of their effects on tubulin but more a direct consequence of tubulin depolymerisation.
There is no universal cure for cancer. Given the complexity of cancer, broad spectrum anti-cancer approaches such as radiation, DNA-targeting and anti-mitotic agents are more frequently used. Some scientists argue that more specific approaches such as genetic profiling of tumours together with administration of a tailored therapeutic strategy may be a more effective approach. However, it is both costly and time consuming and may never be achievable for the 14 million people diagnosed with cancer each year. Due to the urgency of cancer treatment and wide spread occurrence, broadly active cancer agents, in particular tubulin-targeting agents, remain central to current anti-cancer therapy. PBOXs are active across all five of the most common cancer types namely; lung, colon, breast, prostate and stomach. Furthermore, when used in combination, the PBOXs enhance the therapeutic efficacy of many frontline chemotherapeutics. Importantly, the PBOXs are therapeutically advantageous over established chemotherapeutics by overcoming multidrug resistance whilst displaying a low toxicity profile. A prospective clinical use of the PBOXs may be as an adjuvant therapy to target sub-populations of multidrug resistant cancer cells.
Future research objectives include; identifying the tubulin binding site, increasing water solubility, identification of metabolic profiles, applied use of artificial delivery systems and more extensive in vivo data; all of which will ultimately improve the clinical appeal of the PBOXs. However, the cost of bringing new pharmaceutical agents to the market remains the ultimate rate limiting step.
Abbreviations
MTAs, microtubule targeting agents; PBOXs, pyrrolo-1, 5-benzoxazepines; PBR, peripheral-type benzodiazepine receptor; CDKs, cyclin-dependent kinases; SAC, spindle assembly checkpoint; CLL, chronic lymphocytic leukaemia; JNK, c-Jun n-terminal kinase; Bcl-2, B-cell lymphoma 2; Bcl-xL, B-cell lymphoma-extra large; Mcl-1, Myeloid cell leukaemia 1; P13K, phosphoinositide 3-kinase; AKT, protein kinase B; mTOR, mechanistic target of rapamycin; IAPs, inhibitors apoptosis proteins; MMP, mitochondrial membrane potential; BAX, Bcl-2-associated X protein; BAK, Bcl-2-antagonist/killer 1; MOMP, mitochondrial outer membrane permeability; DR5, death receptor 5; T-ALL, T-cell acute lymphoblastic leukemia; CFU-GM, granulocyte-macrophage colony-forming unit; CML, chronic myelogenous leukaemia, CI, combination index; P-gp, P-glycoprotein; BCRP, breast cancer resistance protein; MYCN, v-myc avian myelocytomatosis viral oncogene; TRAIL, TNF-related apoptosis-inducing ligand.
Acknowledgements
Lisa M Greene was supported by a grant obtained from Children's Leukaemia Research Project (Dublin, Ireland).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Campiani G, Nacci V, Fiorini I. et al. Synthesis, biological activity, and SARs of pyrrolobenzoxazepine derivatives, a new class of specific "peripheral-type" benzodiazepine receptor ligands. J Med Chem. 1996;39:3435-45
2. Zisterer DM, Hance N, Campiani G. et al. Antiproliferative action of pyrrolobenzoxazepine derivatives in cultured cells: absence of correlation with binding to the peripheral-type benzodiazepine binding site. Biochem Pharmacol. 1998;55:397-403
3. Zisterer DM, Campiani G, Nacci V, Williams DC. Pyrrolo-1, 5-benzoxazepines induce apoptosis in HL-60, Jurkat, and Hut-78 cells: a new class of apoptotic agents. J Pharmacol Exp Ther. 2000;293:48-59
4. Mc Gee MM, Gemma S, Butini S. et al. Pyrrolo[1,5]benzoxa(thia)zepines as a new class of potent apoptotic agents. Biological studies and identification of an intracellular location of their drug target. J Med Chem. 2005;48:4367-77
5. Mc Gee MM, Campiani G, Ramunno A. et al. Pyrrolo-1, 5-benzoxazepines induce apoptosis in chronic myelogenous leukemia (CML) cells by bypassing the apoptotic suppressor bcr-abl. J Pharmacol Exp Ther. 2001;296:31-40
6. Nathwani SM, Butler S, Fayne D. et al. Novel microtubule-targeting agents, pyrrolo-1, 5-benzoxazepines, induce apoptosis in multi-drug-resistant cancer cells. Cancer Chemother Pharmacol. 2010;66:585-96
7. Pozarowski P, Darzynkiewicz Z. Analysis of cell cycle by flow cytometry. Methods Mol Biol. 2004;281:301-11
8. Mulligan JM, Campiani G, Ramunno A. et al. Inhibition of G1 cyclin-dependent kinase activity during growth arrest of human astrocytoma cells by the pyrrolo-1, 5-benzoxazepine, PBOX-21. Biochim Biophys Acta. 2003;1639:43-52
9. Bright SA, Greene LM, Greene TF. et al. The novel pyrrolo-1, 5-benzoxazepine, PBOX-21, potentiates the apoptotic efficacy of STI571 (imatinib mesylate) in human chronic myeloid leukaemia cells. Biochem Pharmacol. 2009;77:310-21
10. Zisterer DM, McGee MM, Campiani G. et al. Pyrrolo-1, 5-benzoxazepines: a new class of apoptotic agents. Biochem Soc Trans. 2001;29:704-6
11. Mc Gee MM, Hyland E, Campiani G. et al. Caspase-3 is not essential for DNA fragmentation in MCF-7 cells during apoptosis induced by the pyrrolo-1, 5-benzoxazepine, PBOX-6. FEBS Lett. 2002;515:66-70
12. Mc Gee MM, Greene LM, Ledwidge S. et al. Selective induction of apoptosis by the pyrrolo-1, 5-benzoxazepine 7-[[dimethylcarbamoyl]oxy]-6-(2-naphthyl)pyrrolo-[2,1-d] (1,5)-benzoxazepine (PBOX-6) in Leukemia cells occurs via the c-Jun NH2-terminal kinase-dependent phosphorylation and inactivation of Bcl-2 and Bcl-XL. J Pharmacol Exp Ther. 2004;310:1084-95
13. Mulligan JM, Greene LM, Cloonan S. et al. Identification of tubulin as the molecular target of proapoptotic pyrrolo-1,5-benzoxazepines. Mol Pharmacol. 2006;70:60-70
14. Greene LM, Kelly L, Onnis V. et al. STI-571 (imatinib mesylate) enhances the apoptotic efficacy of pyrrolo-1,5-benzoxazepine-6, a novel microtubule-targeting agent, in both STI-571-sensitive and -resistant Bcr-Abl-positive human chronic myeloid leukemia cells. J Pharmacol Exp Ther. 2007;321:288-297
15. Greene LM, Campiani G, Lawler M. et al. BubR1 is required for a sustained mitotic spindle checkpoint arrest in human cancer cells treated with tubulin-targeting pyrrolo-1, 5-benzoxazepines. Mol Pharmacol. 2008;73:419-30
16. Bright SA, Campiani G, Deininger MW. et al. Sequential treatment with flavopiridol synergistically enhances pyrrolo-1,5-benzoxazepine-induced apoptosis in human chronic myeloid leukaemia cells including those resistant to imatinib treatment. Biochem Pharmacol. 2010;80:31-38
17. Bright SA, McElligott AM, O'Connell JW. et al. Novel pyrrolo-1, 5-benzoxazepine compounds display significant activity against resistant chronic myeloid leukaemia cells in vitro, in ex vivo patient samples and in vivo. Br J Cancer. 2010;102:1474-82
18. Nathwani SM, Cloonan SM, Stronach M. et al. Novel microtubule-targeting agents, pyrrolo-1, 5-benzoxazepines, induce cell cycle arrest and apoptosis in prostate cancer cells. Oncol Rep. 2010;24:1499-507
19. Nathwani SM, Butler S, Meegan MJ. et al. Dual targeting of tumour cells and host endothelial cells by novel microtubule-targeting agents, pyrrolo-1,5-benzoxazepines. Cancer Chemother Pharmacol. 2010;65:289-300
20. Forde JC, Maginn EN, McNamara G. et al. Microtubule-targeting-compound PBOX-15 radiosensitizes cancer cells in vitro. Cancer Biol Ther. 2011;11:421-28
21. Maginn EN, Browne PV, Hayden P. et al. PBOX-15, a novel microtubule targeting agent, induces apoptosis, upregulates death receptors, and potentiates TRAIL-mediated apoptosis in multiple myeloma cells. Br J Cancer. 2011;104:281-89
22. Lysaght J, Verma NK, Maginn EN. et al. The microtubule targeting agent PBOX-15 inhibits integrin-mediated cell adhesion and induces apoptosis in acute lymphoblastic leukaemia cells. Int J Oncol. 2013;42:239-46
23. Greene LM, Nolan DP, Regan-Komito D. et al. Inhibition of late-stage autophagy synergistically enhances pyrrolo-1, 5-benzoxazepine-6-induced apoptotic cell death in human colon cancer cells. Int J Oncol. 2013;43:927-35
24. Lennon JC, Bright SA, Carroll E. et al. The novel pyrrolo-1, 5-benzoxazepine, PBOX-6, synergistically enhances the apoptotic effects of carboplatin in drug sensitive and multidrug resistant neuroblastoma cells. Biochem Pharmacol. 2014;87:611-24
25. Greene LM, Fleeton M, Mulligan J. et al. The pyrrolo-1, 5-benzoxazepine, PBOX-6, inhibits the growth of breast cancer cells in vitro independent of estrogen receptor status and inhibits breast tumour growth in vivo. Oncol Rep. 2005;14:1357-63
26. O'Callaghan K, Palagano E, Butini S. et al. Induction of apoptosis in oral squamous carcinoma cells by pyrrolo-1, 5-benzoxazepines. Mol Med Rep. 2015;12:3748-54
27. Kinsella P, Greene LM, Bright SA. et al. The novel pyrrolo-1, 5-benzoxazepine, PBOX-15, synergistically enhances the apoptotic efficacy of imatinib in gastrointestinal stromal tumours; suggested mechanism of action of PBOX-15. Invest New Drugs. 2016;34:159-67
28. Nathwani SM, Greene LM, Butini S. et al. The pyrrolo-1, 5-benzoxazepine, PBOX-15, enhances TRAIL-induced apoptosis by upregulation of DR5 and downregulation of core cell survival proteins in acute lymphoblastic leukaemia cells. Int J Oncol. 2016;49:74-88
29. Bane FT, Bannon JH, Pennington SR. et al. The microtubule-targeting agents, PBOX-6 [pyrrolobenzoxazepine 7-[(dimethylcarbamoyl)oxy]-6-(2-naphthyl)pyrrolo-[2,1-d] (1,5)-benzoxazepine] and paclitaxel, induce nucleocytoplasmic redistribution of the peptidyl-prolyl isomerases, cyclophilin A and pin1, in malignant hematopoietic cells. J Pharmacol Exp Ther. 2009;329:38-47
30. Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol. 2012;22:R966-80
31. Hu M, Liu Q, Song P. et al. Abnormal expression of the mitotic checkpoint protein BubR1 contributes to the anti-microtubule drug resistance of esophageal squamous cell carcinoma cells. Oncol Rep. 2013;29:185-92
32. McElligott AM, Maginn EN, Greene LM. et al. The novel tubulin-targeting agent pyrrolo-1, 5-benzoxazepine-15 induces apoptosis in poor prognostic subgroups of chronic lymphocytic leukemia. Cancer Res. 2009;69:8366-75
33. Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9:790-803
34. Mukhtar E, Adhami VM, Mukhtar H. Targeting microtubules by natural agents for cancer therapy. Mol Cancer Ther. 2014;13:275-84
35. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253-65
36. Verma NK, Dempsey E, Conroy J. et al. A new microtubule-targeting compound PBOX-15 inhibits T-cell migration via post-translational modifications of tubulin. J Mol Med (Berl). 2008;86:457-69
37. Lu Y, Chen J, Xiao M. et al. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res. 2012;29:2943-71
38. Sternlicht H, Yaffe MB, Farr GW. A model of the nucleotide-binding site in tubulin. FEBS Lett. 1987;214:226-35
39. Rai SS, Wolff J. Localization of the vinblastine-binding site on beta-tubulin. J Biol Chem. 1996;271:14707-11
40. Rao S, He L, Chakravarty S. et al. Characterization of the Taxol binding site on the microtubule. Identification of Arg (282) in beta-tubulin as the site of photoincorporation of a 7-benzophenone analogue of Taxol. J Biol Chem. 1999;274:37990-4
41. Prota AE, Bargsten K, Diaz JF. et al. A new tubulin-binding site and pharmacophore for microtubule-destabilizing anticancer drugs. Proc Natl Acad Sci U S A. 2014;111:13817-21
42. Prota AE, Bargsten K, Northcote PT. et al. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew Chem Int Ed Engl. 2014;53:1621-25
43. Kanakkanthara A, Rowe MR, Field JJ. et al. βI-tubulin mutations in the laulimalide/peloruside binding site mediate drug sensitivity by altering drug-tubulin interactions and microtubule stability. Cancer Lett. 2015;365:251-60
44. Yenjerla M, Cox C, Wilson L, Jordan MA. Carbendazim inhibits cancer cell proliferation by suppressing microtubule dynamics. J Pharmacol Exp Ther. 2009;328:390-98
45. Waller CF. Imatinib mesylate. Recent Results Cancer Res. 2014;201:1-25
46. Mc Gee MM, Campiani G, Ramunno A. et al. Activation of the c-Jun N-terminal kinase (JNK) signaling pathway is essential during PBOX-6-induced apoptosis in chronic myelogenous leukemia (CML) cells. J Biol Chem. 2002;27:18383-9
47. Mayer IA, Arteaga CL. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu Rev Med. 2016;67:11-28
48. Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11:109-24
49. Tournier C. The 2 Faces of JNK Signaling in Cancer. Genes Cancer. 2013;4:397-400
50. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495-516
51. Tait SW, Ichim G, Green DR. Die another way-non-apoptotic mechanisms of cell death. J Cell Sci. 2014;127:2135-44
52. Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004-10
53. Danial NN. BCL-2 family proteins: critical checkpoints of apoptotic cell death. Clin Cancer Res. 2007;13:7254-63
54. Dewson G, Kluck RM. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci. 2009;122:2801-8
55. McGrath LB, Onnis V, Campiani G. et al. Caspase-activated DNase (CAD)-independent oligonucleosomal DNA fragmentation in chronic myeloid leukaemia cells; a requirement for serine protease and Mn2+-dependent acidic endonuclease activity. Apoptosis. 2006;11:1473-87
56. Cain K, Bratton SB, Langlais C. et al. Apaf-1 oligomerizes into biologically active approximately 700-kDa and inactive approximately 1.4-MDa apoptosome complexes. J Biol Chem. 2000;275:6067-70
57. Thorburn A, Thamm DH, Gustafson DL. Autophagy and cancer therapy. Mol Pharmacol. 2014;85:830-8
58. Duffy A, Le J, Sausville E, Emadi A. Autophagy modulation: a target for cancer development. Cancer Chemother Pharmacol. 2015;75:439-47
59. Hillen F, Griffioen AW. Tumour vascularization: sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007;26:489-502
60. Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2:91-100
61. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621-81
62. Nicolaou KC, Yang Z, Liu JJ, Ueno H, Nantermet PG, Guy RK, Claiborne CF, Renaud J, Couladouros EA, Paulvannan K. et al. Total synthesis of taxol. Nature. 1994;367:630-4
63. Greene LM, Meegan MJ, Zisterer DM. Combretastatins: more than just vascular targeting agents? J Pharmacol Exp Ther. 2015;355:212-27
64. Perez EA. Paclitaxel in Breast Cancer. Oncologist. 1998;3:373-389
65. Nobili S, Landini I, Mazzei T, Mini E. Overcoming tumor multidrug resistance using drugs able to evade P-glycoprotein or to exploit its expression. Med Res Rev. 2012;32:1220-62
66. Tolcher AW, Cowan KH, Solomon D. et al. Phase I crossover study of paclitaxel with r-verapamil in patients with metastatic breast cancer. J Clin Oncol. 1996;14:1173-84
67. Hudachek SF, Gustafson DL. Coadministration of lapatinib increases exposure to docetaxel but not doxorubicin in the small intestine of mice. Anticancer Drugs. 2013;24:958-68
Author contact
![]() Corresponding author: Dr. Lisa Greene, School of Biochemistry and Immunology, Trinity Biomedical Sciences. Institute, Trinity College Dublin, Dublin 2, Ireland. E-mail lisamariagreeneie Tel: 353-1- 8961855; Fax: 353-1-6772400
Corresponding author: Dr. Lisa Greene, School of Biochemistry and Immunology, Trinity Biomedical Sciences. Institute, Trinity College Dublin, Dublin 2, Ireland. E-mail lisamariagreeneie Tel: 353-1- 8961855; Fax: 353-1-6772400