Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(13):1907-1914. doi:10.7150/jca.15823 This issue Cite
Research Paper
Detecting Circulating Tumor DNA in Hepatocellular Carcinoma Patients Using Droplet Digital PCR Is Feasible and Reflects Intratumoral Heterogeneity
Ao Huang1*, Xin Zhang1*, Shao-Lai Zhou1, Ya Cao2, Xiao-Wu Huang1, Jia Fan1,3, Xin-Rong Yang1, Jian Zhou1,4 ![]()
1. Liver Surgery Department, Liver Cancer Institute, Zhongshan Hospital, Fudan University
Key Laboratory of Carcinogenesis and Cancer Invasion (Fudan University), Ministry of Education, No.180, Fenglin Rd, Shanghai, 200032, China.
2. Cancer Research Institute, Central South University
Key Laboratory of Carcinogenesis and Cancer Invasion, Ministry of Education, No.110, Xiangya Rd, Changsha, 410078, China.
3. Institute of Biomedical Sciences, Fudan University, Shanghai, No.130, Dong'an Rd, 200032, China.
4. State Key Laboratory of Genetic Engineering Fudan University, No.220, Handan Rd, Shanghai, 200433, China.
* co-first author
Received 2016-4-12; Accepted 2016-7-24; Published 2016-9-13
Abstract
Purpose: Circulating tumor DNA (ctDNA) is increasingly recognized as liquid biopsy to profile tumor genome. Droplet digital PCR (ddPCR) is a highly sensitive and easily operable platform for mutant detection. Here, we tried to detect ctDNA in hepatocellular carcinoma (HCC) patients using ddPCR.
Methods: Studies sequencing the genome of HCCs and COSMIC (Catalogue of Somatic Mutations in Cancer) database were reviewed to identify hotspot mutations. Circulating cell-free DNAs (cfDNAs) extracted from 1 ml preoperative plasma sample were analyzed to detect circulating mutants using ddPCR. The DNAs from matched tumor and adjacent liver tissues or peripheral blood mononuclear cells (PBMCs) were sequenced to identify the origin of circulating mutants.
Results: Forty-eight HCC patients were enrolled and four gene loci, TP53 (c.747G>T), CTNNB1 (c.121A>G, c.133T>C), and TERT (c.1-124C>T) were chosen as targets for ddPCR assay. Serial dilution demonstrated the detection limit of ddPCR to be 0.01%. Twenty-seven patients (56.3%, 27/48) were found to have at least one kind of circulating mutants, with the mutant allele frequency ranging from 0.33% to 23.7%. Six patients (22.2%, 6/27) also had matched mutants in tumor tissues while none of the mutants were detected in adjacent liver tissues or PBMCs in all patients, which excluded the nonneoplastic origin of these circulating mutants and qualified them as ctDNA.
Conclusions: ctDNA could be readily detected in HCC patients by targeting hotspot mutations using ddPCR and might reflect intratumoral heterogeneity. ctDNA detecting may serve as a promising liquid biopsy in HCC management.
Keywords: circulating tumor DNA, hepatocellular carcinoma, liquid biopsy, droplet digital PCR, intratumoral heterogeneity.
Introduction
Hepatocellular carcinoma (HCC) is the third leading cause of cancer-related death worldwide and its incidence is still on the rise [1]. In China, endemic hepatitis B virus (HBV) infection results in greater morbidity and mortality [2]. Although the overall survival of HCC has increased in the past decades, the multistep and multicentric carcinogenesis of HCC frequently leads to recurrence and impedes further improve of long-term prognosis [3]. Thus, early detection of HCC and its recurrence might provide additional chance to prolong the survival.
Currently, different biomarkers are used in the continuum of care of HCC, from diagnosis to disease surveillance [4]. Alpha fetoprotein (AFP) is the best-studied and the only serum biomarker undergone all five phases of biomarker development for HCC; other biomarkers including Lens culinaris agglutinin-reactive AFP (AFP-L3), Des-γ-carboxyprothrombin (DCP), Golgi Protein-73 (GP73), and Glypican 3 (GPC3) are still under investigation and require further validation [5]. However, the diagnostic performance of these biomarkers is unsatisfying: the cutoff value at 20 ng/ml of AFP for HCC detection yields sensitivity and specificity ranging from 41% to 65% and 80% to 94% respectively [6]. Therefore, novel biomarkers for early diagnosis and identification of recurrence are needed.
Recently, circulating tumor DNA (ctDNA) has attracted extensive attention for its wide utility in cancer research. ctDNAs are mutant DNAs released to the circulation by tumor cells and constitute part of circulating cell-free DNA (cfDNA) in different types of cancer [7]. With aberrantly genetic information harbored, ctDNA has been reported as liquid biopsy to profile the genome of tumor more comprehensively than conventional sampling method, thus qualifying it as a better vehicle to provide information for guiding targeted therapy [8], unveiling drug resistance [9], and monitoring treatment response [10]. Moreover, ctDNA is highly specific and could be detected at extremely low concentration, making it ideal for early diagnosis [11]. Analysis of ctDNA enabled efficient temporal assessment of disease status and early detection of incipient recurrence, providing an average of 10 months' lead time on detection of metastatic recurrence than traditional modalities [12]; detectable ctDNA after resection could identify cancer patients at high risk of recurrence [13] and dynamic ctDNA change predicts clinical relapse [14].
Till now, few studies have evaluated the presence or features of ctDNA in HCC patients, let alone its potential translational significance. Hereby, we tried to investigate whether ctDNA could be detected by targeting hotspot mutations using droplet digital PCR (ddPCR) in HCC patients. Mutations in the plasma were first detected and then the tumor and adjacent liver tissues/peripheral blood mononuclear cells (PBMCs) were sequenced to identify the origin of these mutants. The evidence presented here demonstrated the feasibility of detecting ctDNA using ddPCR in HCC patients and provided evidence to support the clinical utility of ctDNA in the management of HCC.
Materials and Methods
Patients and sample collection
Patients treated at Zhongshan Hospital, Fudan University between October 2014 and March 2015 were enrolled if they had: 1, no previous histories of or synchronous malignancies in other organs; 2, no anti-tumor treatments of any forms prior to surgery; 3, histopathologically verified HCC. Ten milliliter blood was drawn from ulnar vein preoperatively and collected in EDTA tube (BD, Plymouth, UK). The primary tumor and matched adjacent liver tissues were dissected during operation and stored at -80 oC until use. The clinicopathological information was retrieved from medical records. This study was approved by the Institutional Review Board of Zhongshan Hospital, Fudan University. Informed consent was obtained from individual patient.
Plasma separation and cfDNA extraction
Blood samples were processed within 3 h after venipuncture by a two-step centrifugation method: first spun at 3,000 rpm for 10 min to remove the majority of blood cells and a second spin at 14,000 rpm for another 10 min to remove the cellular debris. The plasma was subpackaged in aliquots of 1 ml and stored at -80 oC until use. The blood cells after first centrifugation were used to separate PBMCs using Ficoll-Paque PLUS (GE Healthcare, Uppsala, Sweden). The cfDNA was extracted using the QIAamp Circulating Nucleic Acid Kit (QIAGEN, Hilden, Germany) with the QIAvac 24 Plus vacuum manifold, according to the manufacturer's instructions.
ddPCR
The QX200 droplet digital PCR system (Bio-Rad, Hercules, USA) was used for mutant detection according to the manufacturer's instructions. The ddPCR reactions were carried out in 20 µl volumes using 10 µl 2x ddPCR SuperMix (Bio-Rad), 5 µl cfDNA template, 1 µl (20x) wild type probe/primer mix, 1µl (20x) mutant probe/primer mix, and 3µl deionized distilled water. Then each 20 µl reaction volume was carefully loaded into the well of Droplet Generator Cartridge (Bio-Rad) and 70 µl droplet generation oil (Bio-Rad) was subsequently loaded to generate droplets. The cartridge was covered with Droplet Generator Gasket (Bio-Rad) and transferred into QX200 Droplet Generator (Bio-Rad) to generate a maximum of 20,000 droplets from each sample. Then droplets from each sample were transferred into a 96-well PCR plate for amplification, using the following program: 95 ˚C for 10 min, followed by 40 cycles of 94 ˚C for 30 s and 60 ˚C (TERT/TP53/CTNNB1:121A>G) or 62 ˚C (CTNNB1:133T>C) for 60 s, followed by 98 ˚C for 10 min, and holding at 4 ˚C. The rate of temperature rise was set at 2.5 ˚C/s. When the amplification was done, the sealed 96-well plate was read with QX200 Droplet Reader (Bio-Rad) using QuantaSoft (Bio-Rad). Mutant allele concentration (copies/µl, CMUT) and wild-type allele concentration (copies/µl, CWT) were calculated and mutant allele frequency (MAF) was calculated as: MAF = CMUT/(CMUT + CWT). The primers and probes were synthesized by Sangon Biotech (Shanghai, China) and sequence information was in Table 1.
Sequence information of the primers and probes for the ddPCR assays.
| Mutation | Primer and probe | Sequence |
|---|---|---|
| TERT (c.1-124C>T) | Forward primer | 5'-GTCCTGCCCCTTCACCTT-3' |
| Reverse primer | 5'-CAGCGCTGCCTGAAACTC-3' | |
| Mutant probe | 5'-FAM-AGCCCCCTCTGGG-MGB-3' | |
| Wild type probe | 5'-VIC-AGCCCCCTCCGGG-MGB-3' | |
| CTNNB1 (121A>G) | Forward primer | 5'-TCACTGGCAGCAACAGTCTT-3' |
| Reverse primer | 5'-CAGGACTTGGGAGGTATCCA-3' | |
| Mutant probe | 5'-FAM-GTGCCACTGCCAC-MGB-3' | |
| Wild type probe | 5'-VIC-GGTGCCACTACCAC-MGB-3' | |
| CTNNB1 (133 T>C) | Forward primer | 5'-TCACTGGCAGCAACAGTCTT-3' |
| Reverse primer | 5'-CAGGACTTGGGAGGTATCCA-3' | |
| Mutant probe | 5'-FAM-GTGCCACTGCCAC-MGB-3' | |
| Wild type probe | 5'-VIC-GGTGCCACTACCAC-MGB-3' | |
| TP53 (c.747G>T) | Forward primer | 5'-TGTAACAGTTCCTGCATGG-3' |
| Reverse primer | 5'-TCAGAGGCAAGCAGAGG-3' | |
| Mutant probe | 5'-FAM-CATGAACCGGAGTCCCATC-MGB-3' | |
| Wild type probe | 5'-VIC-CATGAACCGGAGGCCCATC-MGB-3' |
Sanger sequencing
The frozen primary tumor and matched adjacent liver tissues were obtained in 10 HCC patients and used to extract DNA using DNeasy Blood & Tissue Kit (QIAGEN). For another 31 HCC patients, the formalin-fixed paraffin-embedded (FFPE) tumor and adjacent liver tissues were retrieved and a 8x3 mm cylindrical tissue block was acquired from each FFPE sample to purify DNA using QIAamp DNA FFPE Tissue Kit (QIAGEN). Both the frozen and FFPE tumor tissues were confirmed pathologically to contain more than 50% tumor cell content. Germline DNA was extracted from PBMCs using DNeasy Blood & Tissue Kit in the left seven patients. The PCR products were purified with a MinElute PCR Purification Kit (QIAGEN). The sequencing reactions were set up with 1 μl of purified PCR products using the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems, Foster, USA) and sequencing was performed on ABI 3730XL DNA sequencer (Applied Biosystems). The investigators who performed Sanger sequencing were blinded to the ddPCR results.
Statistical analysis
The mutation concordance rate between plasma and tumor tissues, and the sensitivity and specificity of ddPCR were calculated respectively. The concordance rate was calculated as the number of individuals having the same mutations in both plasma and tumor tissues plus the number of individuals without mutations in neither plasma nor tumor tissues out of the total number of individuals with matched plasma and tumor tissues. The sensitivity, specificity and 95% confidence interval (CI) of ddPCR were calculated by comparing plasma DNA results with those of the paired tumor tissues using Cohen's kappa test, as previously reported [15]. The statistical analysis was conducted with SPSS version 19.0.
Results
Patients' demographics
Totally, 48 newly diagnosed HCC patients were included in this study. Table 2 summarized the clinicopathological and epidemiological information. Most of the HCC patients were male (43/48, 89.6%) with the median age being 58 (range, 40-78) years. HBsAg was positive in 39 patients and 40 patients had liver cirrhosis. Before surgery, 26 patients had AFP level above 20 ng/ml and the Child-Pugh Score of all patients were rated A. All patients successfully underwent hepatectomy with curative intention and the Edmonson grades of their tumors were I (1), II (31), III (14), and IV (2); most patients (47) were classified as BCLC (Barcelona Clinic Liver Cancer) stage A.
Clinical characteristics of enrolled patients.
| Clinical characteristics | No. of patients |
|---|---|
| Age, years | |
| ≥ 50 | 38 |
| < 50 | 10 |
| Gender | |
| Male | 43 |
| Female | 5 |
| HBsAg | |
| Positive | 39 |
| Negative | 9 |
| Liver cirrhosis | |
| Yes | 40 |
| No | 8 |
| AFP, ng/ml | |
| ≥ 20 | 26 |
| < 20 | 22 |
| ALT, U/L | |
| ≥ 75 | 16 |
| < 75 | 32 |
| No. of tumors | |
| Single | 38 |
| Multiple | 10 |
| Tumor size | |
| Small HCC | 25 |
| Large HCC | 23 |
| Child-Pugh score | |
| A | 48 |
| B | 0 |
| BCLC stage | |
| A | 47 |
| B | 1 |
Detection of circulating mutant DNA in the plasma of HCC patients using ddPCR
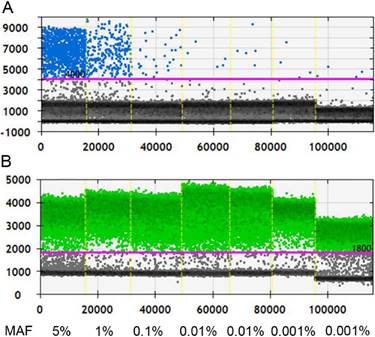
Before applying ddPCR to detect ctDNA, we first evaluated the lower of detection (LOD) and working stability of the platform. Through serial dilution of the cfDNA from a patient with known KRAS G12D mutation (MAF, 5%; starting concentration, 10 ng/µl), we were able to stably detect this mutant at 1:500 dilution. Although positive signals had also been detected at further dilution of 1:5000, the detected MAF was 0.015%, higher than the theoretical value and it was unknown whether this was caused by dilution error or the inherent limit of the platform (Figure 1). Therefore, the LOD of ddPCR platform was set at 0.01% with good reproducibility.
Measurement the LOD of ddPCR platform. A, Positive signals (blue dots) from mutant type could be stably detected at 1:500 dilution, at which level the MAF is 0.01%; B, Positive signals (green dots) of wild type DNA at different dilution. Black dots are non-amplification signals. (MAF, mutant allele frequency).
Unlike breast cancer, colorectal cancer, and lung cancer which have hotspot mutations enriched in genes such as KRAS, PIK3CA, and EGFR, HCC has a wide spectrum of genes harboring multiple mutation sites and types. To this regard, we analyzed the results of studies which conducted whole genome/exome sequencing and searched the COSMIC (Catalogue of somatic mutations in cancer) database to find frequently reported mutations. Four mutant sites within three genes, TP53 (c.747G>T), TERT (c.1-124C>T), and CTNNB1 (c.121A>G) and (c.133T>C) were identified as recurrent mutant events. These four mutants were chosen as targets in following ddPCR analysis.
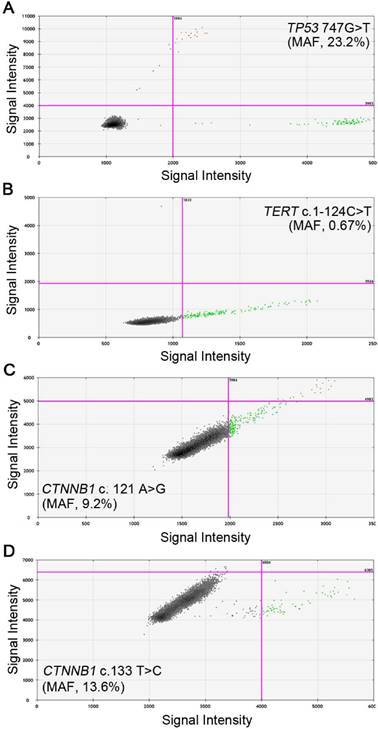
We then evaluated the feasibility of detecting these four mutants in cfDNA using ddPCR. The preoperative blood samples were prospectively collected and 1 ml plasma was used to isolate cfDNA. The cfDNA was successfully extracted from each sample and the amount of cfDNA ranged from 0.4 to 3.1 milligram, which was above the detection threshold of concentration. The wild and mutant DNA copies of the four sites in cfDNA were detected and positive reactions were identified by fluorescent intensities; the concentrations of mutant and wild type DNA copies were calculated respectively. Examples of positive ddPCR assays were shown in Figure 2. In general, 27 (56.3%) out of the 48 patients were found to have at least one mutant in the plasma: 7 patients were positive for TP53 (c.747G>T), with the MAF ranging from 0.33% to 23.2%; 11 patients had detectable TERT (c.1-124C>T) mutants, from 0.55% to 14.3%; CTNNB1 (c.121A>G) and (c.133T>C) were separately identified in 6 patients, ranging from 9.2% to 23.7% and 0.82% to 23.2%, respectively (Table 3). The MAFs were all higher than the LOD of the ddPCR platform, therefore ruling out the possibility of false positive. In three patients, two mutants were simultaneously detected: TP53 (c.747G>T) and TERT (c.1-124C>T) in patient H04 and H41, and TERT (c.1-124C>T), CTNNB1 (c.121A>G) in patient H43.
Representative images of ddPCR analysis results. A, Positive signals of TP53 747 G>T from patient H66 with MAF of 23.2%; B, Positive signals of TERT c.1-124 C>T from patient H32 with MAF of 0.67%; C, Positive signals of CTNNB1 c.121 A>G from patient H09 with MAF of 9.2%; D, Positive signals of CTNNB1 c.133 T>C from patient H62 with MAF of 13.6%. Green dots are signals from wild type DNA copies, blue dots are signals from mutant DNA copies, orange dots were mixed signals from wild type and mutant DNA copies, and black dots are non-amplification signals. (MAF, mutant allele frequency).
Analysis of mutation status in plasma with ddPCR.
| Patient | Mutation fraction (mutant/wild type, copies/μl) | |||
|---|---|---|---|---|
| TP53 (c.747G>T) | TERT (c.1-124C>T) | CTNNB1 (c.121A>G) | CTNNB1 (c.133T>C) | |
| H04 | 0.33% (0.1/30.3) | 7.0% (4.3/61.8) | Negative | Negative |
| H06 | Negative | Negative | 11.5% (1.9/16.5) | Negative |
| H09 | Negative | Negative | 9.2% (1.8/19.6) | Negative |
| H10 | 1.0% (0.1/9.6) | Negative | Negative | Negative |
| H20 | Negative | 0.55% (0.07/12.8) | Negative | Negative |
| H23 | Negative | 7.2% (1.2/16.6) | Negative | Negative |
| H32 | Negative | 0.67% (0.07/10.4) | Negative | Negative |
| H35 | 1.8% (0.11/6.2) | Negative | Negative | Negative |
| H41 | 3.2% (0.1/3.1) | 0.89% (0.07/7.9) | Negative | Negative |
| H43 | Negative | 0.63% (0.08/12.8) | 21.6% (3/13.9) | Negative |
| H47 | Negative | 1.4% (0.07/4.9) | Negative | Negative |
| H50 | Negative | Negative | Negative | 2.3% (0.34/14.6) |
| H51 | Negative | Negative | 18.9% (1.7/9) | Negative |
| H52 | Negative | 3.3% (0.08/2.4) | Negative | Negative |
| H53 | 0.55% (0.11/20.1) | Negative | Negative | Negative |
| H57 | Negative | Negative | Negative | 0.82% (0.1/12.2) |
| H58 | Negative | 1.3% (0.07/5.6) | Negative | Negative |
| H59 | 10% (0.6/6) | Negative | Negative | Negative |
| H61 | Negative | Negative | Negative | 16.5% (1.7/10.3) |
| H62 | Negative | Negative | Negative | 13.6% (0.6/4.4) |
| H63 | Negative | Negative | Negative | 20.3% (1.6/7.9) |
| H65 | Negative | Negative | Negative | 23.2% (2.2/9.5) |
| H66 | 23.2% (1.9/8.2) | Negative | Negative | Negative |
| H67 | Negative | Negative | 16.8% (6.8/40.4) | Negative |
| H68 | Negative | 0.92% (0.08/8.7) | Negative | Negative |
| H78 | Negative | 14.3% (0.77/5.4) | Negative | Negative |
| H79 | Negative | Negative | 23.7% (11/46.5) | Negative |
Identification of the circulating mutants' origin with Sanger sequencing
The positive mutants detected in the plasma should not be rashly claimed to be ctDNAs since germline mutations might also be detected in cfDNA. Thus, we sequenced the DNAs of corresponding tumor tissues and matched adjacent liver tissues or PBMCs to identify the origin of these circulating mutants. Sanger sequencing was performed in all samples regardless of mutant status in the cfDNA. Somehow surprisingly, sequencing of the tumor DNA only detected positive mutations in six patients and they all had one mutant validated each, including the patient having two circulating mutants. In two patients (H16 and H19) whose cfDNAs were mutants negative, heterozygous genotype of TERT (c.1-124C>T) and TP53 (c.747G>T) were respectively detected in tumor tissues; in patient H6 and H50 who had circulating CTNNB1 mutants, the tumor tissues were found TERT (c.1-124C>T) positive whereas the genotype of CTNNB1 were demonstrated to be wild type. Interestingly, none of the mutants were detected in DNAs extracted from adjacent liver tissues or PBMCs, thus excluding germline mutations as the source of these circulating mutants. The concordance rate between plasma and tumor tissues, sensitivity, specificity, and 95% CI of ddPCR assay were shown in Table 4. Combining together, we could conclude that the mutants detected in the plasma of HCC patients were not derived from germline DNA and were very likely to be acquired as somatic events during the carcinogenesis of liver, which qualified them as ctDNA.
Concordance of mutants detected in plasma by ddPCR and their status in paired tumor tissues.
| Plasma DNA | Tumor tissue | Total | Concordance (95% CI) | |
|---|---|---|---|---|
| Sensitivity (95% CI) | ||||
| + | - | Specificity (95% CI) | ||
| TP53 (747G>T) | ||||
| + | 1 | 5 | 6 | 85.4% * (71.6%-93.1% ) |
| - | 1 | 34 | 35 | 50% (9.5%-90.6%) |
| Total | 2 | 39 | 41 | 87.2% (73.3%-94.4%) |
| TERT (c.1-124C>T) | ||||
| + | 5 | 5 | 10 | 80.5% * (66.0%-89.8%) |
| - | 3 | 28 | 31 | 62.5% (30.6%-86.3%) |
| Total | 8 | 33 | 41 | 84.9% (69.1%-93.4%) |
| CTNNB1 (c.121A>G) | ||||
| + | 0 | 5 | 5 | 87.8% (74.5%-94.7%) |
| - | 0 | 36 | 36 | NA |
| Total | 0 | 41 | 41 | 87.8% (74.5%-94.7%) |
| CTNNB1 (c.133T>C) | ||||
| + | 0 | 5 | 5 | 87.8% (74.5%-94.7%) |
| - | 0 | 36 | 36 | NA |
| Total | 0 | 41 | 41 | 87.8% (74.5%-94.7%) |
95% CI, 95% confidence interval; *, P < 0.05; NA, not applicable.
Discussion
Recent evidences demonstrated that ctDNA could be used as liquid biopsy with broad application including early diagnosis, disease progression prediction, and personalized treatment [16]. Indeed, ctDNA has been widely reported in different malignancies whereas, on the contrary, it was rarely studied in HCC. Considering the status quo of HCC management, which requires novel strategies for early diagnosis and disease surveillance [17], and the trend of precision medicine in oncology [18], it's necessary to investigate the role of ctDNA in HCC. To the best of our knowledge, this study is the first to evaluate the presence of ctDNA in HCC patients by detecting multiple point mutations using ddPCR.
ctDNAs generally only account for a small fraction (could be lower than 0.01%) of cfDNAs [19], which necessitates the employment of highly sensitive detection methods. ddPCR and next generation sequencing (NGS) are currently the most commonly used mutation detection methods. Although there existed limitations including low throughput and the optimization of reaction condition for each mutation of interest, the advantages including low cost, in-house operation, and quick turnaround time of ddPCR made it easily accessible to researchers and clinicians. Moreover, each reaction of ddPCR was performed in individual droplet containing no more than one template, thus enabling absolute quantification of nucleic acids and avoiding the dependency on DNA calibrant in qPCR. Specially, optimal template dilution and droplet partitioning could reduce competitive amplification effects, allowing detection of mutant as low as 0.001%, 1,000 times lower than that in qPCR [20].
Successful ctDNA detection also necessitates the selection of tumor-specific gene aberrations. This would be unnecessary for NGS-based methods; however, ddPCR-based methods could only target limited mutants per reaction, even for multiplex ddPCR [21]. Thus, hotspot mutations are preferred for identification of ctDNA using ddPCR. Although mutant loci within gene KRAS, PIK3CA, and EGFR have been widely used to identify ctDNA in breast, colorectal and lung cancer [21-23], they were less frequently mutated in HCC [24, 25]. Therefore, studies which sequenced whole genome/exome and COSMIC database were reviewed to identify genes frequently mutated in HCC [26-31]. Despite discrepancies, three genes, TP53, CTNNB1, and TERT were identified as the top repeatedly mutated ones, with multiple gene loci and mutant type reported, and four sites were selected as candidate targets. The TP53 is the most commonly mutated gene in HCC and c.747G>T (R249S) is highly related with HBV infection and Aflatoxin B1 exposure; CTNNB1 is found mutually exclusive with TP53 mutation [27] and thus would increase mutant detection rate in combination with TP53. TERT promoter mutation is an early event in liver carcinogenesis and -124C>T mutation was found positive in about 50% HCC cases [32, 33]. We found 7 patients were TP53 mutation positive, and these patients were exclusive of CTNNB1 mutants as reported. Compared to previous study using serum DNA to detect TP53 c.747G>T mutation [34], we acquired higher positive rate and this might be explained by the contamination of genomic DNA from blood cells in serum [35], which lowered the MAF, and the high sensitivity of ddPCR, which reduced false negative. The result was quite encouraging: circulating plasma mutants could be readily detected by targeting point mutations using ddPCR in HCC and with a panel composed of multiple mutant loci in different genes, more patients might be identified circulating mutant positive.
However, it's reasonable to doubt that the circulating mutants are not all derived from tumor cells since germline mutation might also be detected in plasma. We thus screened the tumor samples by Sanger sequencing to identify their origin. Surprisingly, nearly half of the circulating mutants were not detected in tumor tissues while, not unexpectedly, the mutants were not present in any adjacent liver tissues or PBMCs. Considering the sensitivity of our ddPCR platform, the high MAF of circulating mutants, the absence of mutants in germline DNA, and no histories of previous or synchronous malignancy, we suggested that the positive signals in cfDNA were neither false positives nor derived from the nonneoplastic origin, which qualified them as ctDNAs from liver tumor cells. As to the discordance of mutant status between plasma and tumor tissues, intratumor heterogeneity (ITH) might possibly be the underlying reason. Genetically, ITH means different regions within the tumor do not share the same mutational landscape and it's widely existed in cancers [36-39], including HCC [40]. Under such condition, it was hard to get a full view of the genomics of tumor by single-site sampling [41]. Supporting this, studies have reported detection of invalidated mutants in primary tumors by analyzing plasma DNA with ddPCR [15] and ctDNA proved to be a potential tool for de novo mutation identification, namely capturing ITH [42]. Herein, the tumor DNA was only extracted from 25 mg tissues, which accounted for a very small fraction of the whole tumor and would surely simplify the mutant profile. Although four false negative events suggested this ddPCR should be further improved, these results suggested that detecting ctDNA might bear the potential to overcome the limit of ITH and identify rare mutations missed by single-site sampling.
Several limitations should be recognized. The etiology of HCC patients in this study was mainly HBV infection and thus it's unclear whether our ddPCR strategy could achieve similar sensitivity to detect ctDNA in HCC patients of other causes. In addition, only four mutations were selected to screen ctDNA and negative results were noted in approximately half of the patients. Multiplex ddPCR might be a feasible solution. Finally, the clinical significance of ctDNA as diagnostic and predictive biomarker in HCC patients should be further evaluated. Further studies aiming at improving the assay sensitivity by using multiple mutations and unveiling the utility of ctDNA using larger sample size and sequential blood sampling are warranted.
Conclusively, we presented herein initial evidence demonstrating the presence of ctDNA in HCC patients and the feasibility of detecting ctDNA using ddPCR. Although the detection rate needs to be increased and the clinical significance should be further evaluated, detection of ctDNA using ddPCR in HCC patients shows the potential as liquid biopsy and might help disease diagnosis and surveillance.
Acknowledgements
This study was jointly funded by the National Science Foundation for Distinguished Young Scholars of China (No. 81225019), the National Natural Science Foundation of China (No. 81572823), the National Science and Technology Major Project (No.2013ZX10002007-005), and Shanghai Hospital Development Center (SHDC12015104). The authors thank Ms DING Feifei and Ms ZHANG Cheng (Shanghai Zhangjiang Center for Translational Medicine) for providing technical help.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-386
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016
3. Tang ZY. Hepatocellular carcinoma surgery-review of the past and prospects for the 21st century. J Surg Oncol. 2005;91:95-96
4. Wong KF, Xu Z, Chen J, Lee NP, Luk JM. Circulating markers for prognosis of hepatocellular carcinoma. Expert Opin Med Diagn. 2013;7:319-329
5. Rich N, Singal AG. Hepatocellular carcinoma tumour markers: current role and expectations. Best Pract Res Clin Gastroenterol. 2014;28:843-853
6. Lok AS, Sterling RK, Everhart JE, Wright EC, Hoefs JC, Di Bisceglie AM, Morgan TR, Kim HY, Lee WM, Bonkovsky HL, Dienstag JL, Group H-CT. Des-gamma-carboxy prothrombin and alpha-fetoprotein as biomarkers for the early detection of hepatocellular carcinoma. Gastroenterology. 2010;138:493-502
7. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, Brem H, Cameron JL, Lee CC, Fecher LA, Gallia GL, Gibbs P, Le D, Giuntoli RL, Goggins M, Hogarty MD, Holdhoff M, Hong SM, Jiao Y, Juhl HH, Kim JJ, Siravegna G, Laheru DA, Lauricella C, Lim M, Lipson EJ, Marie SK, Netto GJ, Oliner KS, Olivi A, Olsson L, Riggins GJ, Sartore-Bianchi A, Schmidt K, Shih l M, Oba-Shinjo SM, Siena S, Theodorescu D, Tie J, Harkins TT, Veronese S, Wang TL, Weingart JD, Wolfgang CL, Wood LD, Xing D, Hruban RH, Wu J, Allen PJ, Schmidt CM, Choti MA, Velculescu VE, Kinzler KW, Vogelstein B, Papadopoulos N, Diaz LA Jr. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra224
8. Luke JJ, Oxnard GR, Paweletz CP, Camidge DR, Heymach JV, Solit DB, Johnson BE, Cell Free DNAWG. Realizing the potential of plasma genotyping in an age of genotype-directed therapies. J Natl Cancer Inst. 2014:106
9. Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, Marass F, Humphray S, Hadfield J, Bentley D, Chin TM, Brenton JD, Caldas C, Rosenfeld N. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497:108-112
10. Montagut C, Siravegna G, Bardelli A. Liquid biopsies to evaluate early therapeutic response in colorectal cancer. Ann Oncol. 2015;26:1525-1527
11. Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem. 2015;61:112-123
12. Reinert T, Scholer LV, Thomsen R, Tobiasen H, Vang S, Nordentoft I, Lamy P, Kannerup AS, Mortensen FV, Stribolt K, Hamilton-Dutoit S, Nielsen HJ, Laurberg S, Pallisgaard N, Pedersen JS, Orntoft TF, Andersen CL. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut. 2015
13. Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, Anagnostou V, Parpart-Li S, Murphy D, Kay Li Q, Hruban CA, Scharpf R, White JR, O'Dwyer PJ, Allen PJ, Eshleman JR, Thompson CB, Klimstra DS, Linehan DC, Maitra A, Hruban RH, Diaz LA Jr, Von Hoff DD, Johansen JS, Drebin JA, Velculescu VE. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun. 2015;6:7686
14. Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, Cheang M, Osin P, Nerurkar A, Kozarewa I, Garrido JA, Dowsett M, Reis-Filho JS, Smith IE, Turner NC. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7:302ra133
15. Zhu G, Ye X, Dong Z, Lu YC, Sun Y, Liu Y, McCormack R, Gu Y, Liu X. Highly Sensitive Droplet Digital PCR Method for Detection of EGFR-Activating Mutations in Plasma Cell-Free DNA from Patients with Advanced Non-Small Cell Lung Cancer. J Mol Diagn. 2015;17:265-272
16. Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32:579-586
17. Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut. 2014;63:844-855
18. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793-795
19. Mouliere F, Robert B, Arnau Peyrotte E, Del Rio M, Ychou M, Molina F, Gongora C, Thierry AR. High fragmentation characterizes tumour-derived circulating DNA. PLoS One. 2011;6:e23418
20. Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, Kitano TK, Hodel MR, Petersen JF, Wyatt PW, Steenblock ER, Shah PH, Bousse LJ, Troup CB, Mellen JC, Wittmann DK, Erndt NG, Cauley TH, Koehler RT, So AP, Dube S, Rose KA, Montesclaros L, Wang S, Stumbo DP, Hodges SP, Romine S, Milanovich FP, White HE, Regan JF, Karlin-Neumann GA, Hindson CM, Saxonov S, Colston BW. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604-8610
21. Taly V, Pekin D, Benhaim L, Kotsopoulos SK, Le Corre D, Li X, Atochin I, Link DR, Griffiths AD, Pallier K, Blons H, Bouche O, Landi B, Hutchison JB, Laurent-Puig P. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin Chem. 2013;59:1722-1731
22. Higgins MJ, Jelovac D, Barnathan E, Blair B, Slater S, Powers P, Zorzi J, Jeter SC, Oliver GR, Fetting J, Emens L, Riley C, Stearns V, Diehl F, Angenendt P, Huang P, Cope L, Argani P, Murphy KM, Bachman KE, Greshock J, Wolff AC, Park BH. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin Cancer Res. 2012;18:3462-3469
23. Oxnard GR, Paweletz CP, Kuang Y, Mach SL, O'Connell A, Messineo MM, Luke JJ, Butaney M, Kirschmeier P, Jackman DM, Janne PA. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014;20:1698-1705
24. Shibata T, Aburatani H. Exploration of liver cancer genomes. Nat Rev Gastroenterol Hepatol. 2014;11:340-349
25. Li X, Zhang Q, He W, Meng W, Yan J, Zhang L, Zhu X, Liu T, Li Y, Bai Z. Low frequency of PIK3CA gene mutations in hepatocellular carcinoma in Chinese population. Pathol Oncol Res. 2012;18:57-60
26. Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, Arai Y, Takahashi H, Shirakihara T, Nagasaki M, Shibuya T, Nakano K, Watanabe-Makino K, Tanaka H, Nakamura H, Kusuda J, Ojima H, Shimada K, Okusaka T, Ueno M, Shigekawa Y, Kawakami Y, Arihiro K, Ohdan H, Gotoh K, Ishikawa O, Ariizumi S, Yamamoto M, Yamada T, Chayama K, Kosuge T, Yamaue H, Kamatani N, Miyano S, Nakagama H, Nakamura Y, Tsunoda T, Shibata T, Nakagawa H. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760-764
27. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, Clement B, Balabaud C, Chevet E, Laurent A, Couchy G, Letouze E, Calvo F, Zucman-Rossi J. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694-698
28. Huang J, Deng Q, Wang Q, Li KY, Dai JH, Li N, Zhu ZD, Zhou B, Liu XY, Liu RF, Fei QL, Chen H, Cai B, Zhou B, Xiao HS, Qin LX, Han ZG. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat Genet. 2012;44:1117-1121
29. Cleary SP, Jeck WR, Zhao X, Chen K, Selitsky SR, Savich GL, Tan TX, Wu MC, Getz G, Lawrence MS, Parker JS, Li J, Powers S, Kim H, Fischer S, Guindi M, Ghanekar A, Chiang DY. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology. 2013;58:1693-1702
30. Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, Gao H, Hao K, Willard MD, Xu J, Hauptschein R, Rejto PA, Fernandez J, Wang G, Zhang Q, Wang B, Chen R, Wang J, Lee NP, Zhou W, Lin Z, Peng Z, Yi K, Chen S, Li L, Fan X, Yang J, Ye R, Ju J, Wang K, Estrella H, Deng S, Wei P, Qiu M, Wulur IH, Liu J, Ehsani ME, Zhang C, Loboda A, Sung WK, Aggarwal A, Poon RT, Fan ST, Wang J, Hardwick J, Reinhard C, Dai H, Li Y, Luk JM, Mao M. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422-1433
31. Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, Calatayud AL, Pinyol R, Pelletier L, Balabaud C, Laurent A, Blanc JF, Mazzaferro V, Calvo F, Villanueva A, Nault JC, Bioulac-Sage P, Stratton MR, Llovet JM, Zucman-Rossi J. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505-511
32. Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, Laurent A, Cherqui D, Balabaud C, Zucman-Rossi J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013;4:2218
33. Quaas A, Oldopp T, Tharun L, Klingenfeld C, Krech T, Sauter G, Grob TJ. Frequency of TERT promoter mutations in primary tumors of the liver. Virchows Arch. 2014;465:673-677
34. Hosny G, Farahat N, Tayel H, Hainaut P. Ser-249 TP53 and CTNNB1 mutations in circulating free DNA of Egyptian patients with hepatocellular carcinoma versus chronic liver diseases. Cancer Lett. 2008;264:201-208
35. El Messaoudi S, Rolet F, Mouliere F, Thierry AR. Circulating cell free DNA: Preanalytical considerations. Clin Chim Acta. 2013;424:222-230
36. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883-892
37. de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, Jamal-Hanjani M, Shafi S, Murugaesu N, Rowan AJ, Gronroos E, Muhammad MA, Horswell S, Gerlinger M, Varela I, Jones D, Marshall J, Voet T, Van Loo P, Rassl DM, Rintoul RC, Janes SM, Lee SM, Forster M, Ahmad T, Lawrence D, Falzon M, Capitanio A, Harkins TT, Lee CC, Tom W, Teefe E, Chen SC, Begum S, Rabinowitz A, Phillimore B, Spencer-Dene B, Stamp G, Szallasi Z, Matthews N, Stewart A, Campbell P, Swanton C. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251-256
38. Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, Martinez P, Phillimore B, Begum S, Rabinowitz A, Spencer-Dene B, Gulati S, Bates PA, Stamp G, Pickering L, Gore M, Nicol DL, Hazell S, Futreal PA, Stewart A, Swanton C. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46:225-233
39. Crona J, Backman S, Maharjan R, Mayrhofer M, Stalberg P, Isaksson A, Hellman P, Bjorklund P. Spatiotemporal Heterogeneity Characterizes the Genetic Landscape of Pheochromocytoma and Defines Early Events in Tumorigenesis. Clin Cancer Res. 2015;21:4451-4460
40. Friemel J, Rechsteiner M, Frick L, Bohm F, Struckmann K, Egger M, Moch H, Heikenwalder M, Weber A. Intratumor heterogeneity in hepatocellular carcinoma. Clin Cancer Res. 2015;21:1951-1961
41. Nault JC, Villanueva A. Intratumor molecular and phenotypic diversity in hepatocellular carcinoma. Clin Cancer Res. 2015;21:1786-1788
42. De Mattos-Arruda L, Weigelt B, Cortes J, Won HH, Ng CK, Nuciforo P, Bidard FC, Aura C, Saura C, Peg V, Piscuoglio S, Oliveira M, Smolders Y, Patel P, Norton L, Tabernero J, Berger MF, Seoane J, Reis-Filho JS. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol. 2014;25:1729-1735
Author contact
![]() Corresponding author: Jian Zhou, E-mail: zhou.jiansh.cn, Tel: 86-21-64041990; Fax: 86-21-64037181.; or Xin-Rong Yang, E-mail: yang.xinrongsh.cn, Tel: 86-21-64041990; Fax: 86-21-64037181.
Corresponding author: Jian Zhou, E-mail: zhou.jiansh.cn, Tel: 86-21-64041990; Fax: 86-21-64037181.; or Xin-Rong Yang, E-mail: yang.xinrongsh.cn, Tel: 86-21-64041990; Fax: 86-21-64037181.