Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(13):1824-1832. doi:10.7150/jca.15622 This issue Cite
Research Paper
Potential Function of Exogenous Vimentin on the Activation of Wnt Signaling Pathway in Cancer Cells
Arun Satelli1*, Jiemiao Hu1*, Xueqing Xia1, Shulin Li1,2 ![]()
1. Department of Pediatrics, The University of Texas MD Anderson Cancer Center, Houston, Texas;
2. The University of Texas Graduate School of Biomedical Sciences, Houston, Texas.
* Equal contributors
Received 2016-3-23; Accepted 2016-6-30; Published 2016-8-12
Abstract
Cancer cell signaling, growth, morphology, proliferation and tumorigenic potential are largely depending on the signaling molecules present naturally in the tumor microenvironment and the identification of key molecules that drive the tumor progression is critical for the development of new modalities for the prevention of tumor progression. High concentrations of vimentin in the blood of cancer patients have been reported, however the function of blood circulating vimentin remains unknown. Here, we investigated the functional role of exogenously supplemented vimentin on colon cancer cells and examined the Wnt Signaling activation and cancer cell invasion. Vimentin when supplemented to the cancer cells remained bound to the surface of the cancer cells. Furthermore, bound vimentin activates Wnt signaling pathway as detectable by increased β-catenin accumulation in the nucleus with concomitant activation of β-catenin-dependent transcription of Wnt signaling downstream targets. Functionally, there was an increase in the rate of cellular invasion in these cancer cells upon binding with vimentin. Our results thus suggest that free vimentin in the tumor microenvironment acts as a positive regulator of the β-catenin signaling pathway, thus providing a basis for cancer invasive properties.
Keywords: Vimentin, Wnt Signaling, EMT, invasion, β-catenin.
Introduction
The tumor microenvironment has emerged as a key mediator for tumor progression and serves as an important target for cancer therapy [1]. Tumor progression induction molecules present in the tumor environment are either secreted from the surrounding infiltrating cells or from circulating proteins. The concentrations of various signaling molecules in the surrounding environment influence cancer cell growth and tumorigenic potential including cell proliferation, migration, invasion and other critical cancer biological processes. These properties thus allow the cancer cells to sustain and expand in the immune cell rich microenvironment. For example, the presence of EGFR ligands in the tumor microenvironment provides sustained EGFR signaling in breast cancer cells that supports the growth of cancer cells and inhibiting the availability of these ligands thus affects the growth of cancer cells thereby prevents tumor progression [2].
Vimentin although considered to be strictly cytoskeletal in nature has also been reported to be transported to the cell surface in certain cell types [3]. Also, vimentin has shown to be secreted by certain cell types in the blood [4-6] and its presence in the tumor stroma of colorectal cancer patients was correlated to a higher malignant potential and associated with a shorter survival rate [7]; however, the function of secreted vimentin still remains elusive. Garg et al. have previously shown the function of externally secreted vimentin on immune cells, wherein vimentin released by mycobacterium infected monocytes binds to natural killer (NK) cells through Nkp46 receptor and reduces the cytolytic activity of NK cells [8]. However, there are no reports on the ability of vimentin to bind cancer cells and its ability to serve as a ligand to induce any signaling pathway. Here in this study for the first time we investigate and dissect the functional role of exogenously added vimentin on cancer cells.
Materials and Methods
Cell lines and culture conditions
Human colorectal cancer (CRC) cell lines DLD-1, GEO, SW-480, SW-620, HT-29, HCT-116, and Caco-2 were obtained from the American Type Culture Collection (Vanassas, MD) and cultured according to the supplier's instructions. HPC1 (human primary colon cancer cells) and HLM3A (human colon cancer cells metastasized to liver) were isolated from surgical specimens and cultured in our laboratory [9]. NCM-356 normal colonic epithelial cells were used as control cell line. These cell lines were The M.D. Anderson Cancer Center (MDACC) Institutional Review Board approved human specimen procurement, and informed consents were obtained.
Flow cytometry
In general, a total of 5 x 105 cells were detached with a non-enzymatic dissociation buffer, washed, and used for flow cytometry. To detect if externally added vimentin binds to the cancer cells; cells were incubated with physiologically relevant levels of rhVim (250 ng/mL) [6] (recombinant human vimentin R&D Systems CF2105-VI-100 with C-terminal 6-His tag) or with serum albumin (AbCAM 94020 with 6-His tag) (250 ng/mL) as control for 15 min on ice. Later the cells were washed twice in phosphate-buffered saline (PBS). For CSV analysis, cells were stained with the 84-1 monoclonal antibody developed in our lab [10](1:100); mouse primary antibody (Invitrogen) was used as an isotype control. Later, cells were rinsed twice in PBS and labeled for secondary antibody using Alexa Fluor-488 secondary antibody (Invitrogen). Cells were then washed twice in PBS and immediately used for data acquisition using an Attune flow cytometer (Applied Biosystems) and detected binding using excitation/emission maxima of 495/519 nm. For the analysis, 50,000 cells were counted. The data were analyzed using FlowJo software (Treestar). Y axis denotes cell count and x axis indicates BL1A log fluorescence intensity.
Proteome Profiler Human Phospho-Kinase Array Kit
Proteome profiler array kit (ARY003B) was purchased from R&D Systems. Each array contains 4 sets of 2 membranes - each spotted in duplicate with antibodies against 43 different phosphorylated proteins and 2 related total proteins. Assay was performed according to manufacturer's instructions. Cell lysates were prepared from HPC1 or NCM356 cells for control serum albumin (250 ng/mL) treated or rhVim (250 ng/mL) treated for 24 hr. Cell lysates were loaded at 400 µg per array. After recommended incubations and washes, the membranes were exposed to X-ray film at different exposure times. The intensities of the relative expression levels of proteins were quantified by densitometry (VersaDoc imaging system, Bio-Rad).
Immunocytochemistry
For immunocytochemical characterization, cells growing on chamber slides (after respective treatments) were washed in PBS and fixed using 4% paraformaldehyde (PFA) at room temperature (RT) for 20 min. To detect intracellular proteins, fixed cells were then blocked in PBS containing 0.25% Nonidet-P40 and 1% FBS for 60 min. Cells were then washed and incubated with primary antibody [IgG control (1:250), β-catenin (1:250), E-Cadherin (1:250), cyclin D1 (1:100), c-myc (1:250) and vimentin (1:250) overnight at 4oC. Later cells were rinsed in PBS 2x. Cells were then rinsed in PBS and stained with Alexa Fluor-555 secondary antibodies (Invitrogen) (1:250) for respective antibodies. For nuclei staining, Sytox Green (Invitrogen) (1:1000) was incorporated along with secondary antibody for 60 min. The cells were then washed with PBS three times for 15 min each and mounted in SlowFade antifade reagent (Invitrogen). For confocal analysis, images were acquired in 8 bits with the Zeiss LSM 510 confocal microscope using LSM5 image capture and analysis software, version 3.2 (Zeiss). A 63x water-immersion objective lens (NA, 1.0) was used with digital zoom for image capture. All images were acquired by the same operator using the same intensity and photo detector gain in order to allow quantitative comparisons of relative levels of immunoreactivity between different samples.
Western blot analysis
Immunoblotting was performed as described in detail before [11]. Antibodies were against β-catenin (8480), β-catenin S552 (5651), β-catenin S675 (4176), Axin1 (2087), GSK-3 β, LRP-6 (3395), Dvl-3 (3218), cyclin D1 (2978), c-myc (5605) and β-actin (4967). All antibodies were purchased from Cell Signaling Technologies and the catalog numbers are included next to protein name. Primary and secondary antibodies were diluted in 5% nonfat milk/TBST or 5% bovine serum albumin/TBST and incubated overnight at 4°C, and blots were developed with Enhanced Chemiluminescence or Enhanced Chemiluminescence Plus (GE Biosciences). The blots were quantified using a Bio-Rad VersaDoc. The densities of the bands were normalized with respect to β-actin.
TOP FLASH/ FOP FLASH Reporter Assay
TOPFLASH and FOPFLASH plasmids were provided Dr. Hans Clevers. Briefly, 100,000 cells per well were plated onto six-well plates. Either TOPFLASH or FOPFLASH (1 μg/well) was transfected using LipofectAMINE Plus (Invitrogen) according to the manufacturer's instructions. Per well, 0.5 ng Renilla control luciferase plasmid was cotransfected to normalize for transfection efficiency. Forty-eight hours after transfection, cells were lysed with 1× passive lysis buffer (Promega), and the luciferase assay was done using the Dual-Luciferase Reporter Assay Kit (Promega). TOPFLASH and FOPFLASH values were normalized to Renilla, and fold induction for each cell line was calculated as normalized relative light units of TOPFLASH divided by normalized relative light units of FOPFLASH. For Wnt activation by rhVim, transfected cells were incubated with 250 ng/mL rhVim in DMEM/F-12 medium for 24 h before lysis or serum albumin was used as a control.
Quantitative real-time RT-qPCR
Total RNA was isolated from cells using TRIzol Reagent (Life Technologies). Residual genomic DNA was removed from total RNA using the TURBO DNA-free kit (Life Technologies).
Two micrograms of RNA were used for cDNA synthesis using the High-Capacity RNA-to-cDNA Kit (Life Technologies). The relative gene expression levels were determined using RT- qPCR and SYBR Green labeling method in a StepOnePlus™ Real-Time PCR System (Life Technologies). The reaction contained 2 µL cDNA, 12.5 µL SYBR Green PCR Master Mix (Life Technologies), and 200 µM primer in a total volume of 25 µL. The PCR cycling conditions were as follows: 40 cycles of 15 s at 95C and 60 s at 60C. All samples were run in duplicates. The CT value of each sample was acquired, and the relative level of gene expression was calculated by the delta CT method, which was normalized to the endogenous control of GAPDH. Data were expressed as n-fold relative to control. The primer sequences for human genes are:
Fibronectin: CCATCGCAAACCGCTGCCAT and AACACTTCTCAGCTATGGGCTT
N-cadherin: TGATCGAGAAAAAGTGCAACAGTATAC and GGCTGTGTTTGAAAGGCCATA
Slug: AAGCATTTCAACGCCTCCAAA and GGATCTCTGGTTGTGGTATGACA
Snail: TCGGAAGCCTAACTACAGCGA and AGATGAGCATTGGCAGCGAG
Twist: CCGGAGACCTAGATGTCATTG and CCACGCCCTGTTTCTTTG
Vimentin: GCAGGCTCAGATTCAGGAACA and GTGAGGTCAGGCTTGGAAACA
Gapdh: ATGGAAATCCCATCACCATCTT and CATCGCCCCACTTGATTTTG
FluoroBlok invasion Assay
Normal colon cell line NCM-356 and colon cancer cell lines HPC1, HLM3, Caco-2, DLD-1, HT-29, SW480 and SW620 were trypsinized and counted. Cells were seeded on matrigel-coated FluoroBlok inserts or non-coated FluoroBlok inserts and allowed to invade or migrate in presence of Aphidicolin (Sigma Aldrich), a cell proliferation inhibitor. 24 or 48 hours later cells that migrated or invaded to the underside of the insert were quantified on a spectrophotometer at excitation 485 and emission 520.
Statistical Analysis
Data are presented as mean ± SEM. Unless stated otherwise, data were analyzed using the Student t test, and considered statistically significant when the P value was less than 0.05.
Results
Externally added vimentin binds to cell surface of cancer cells
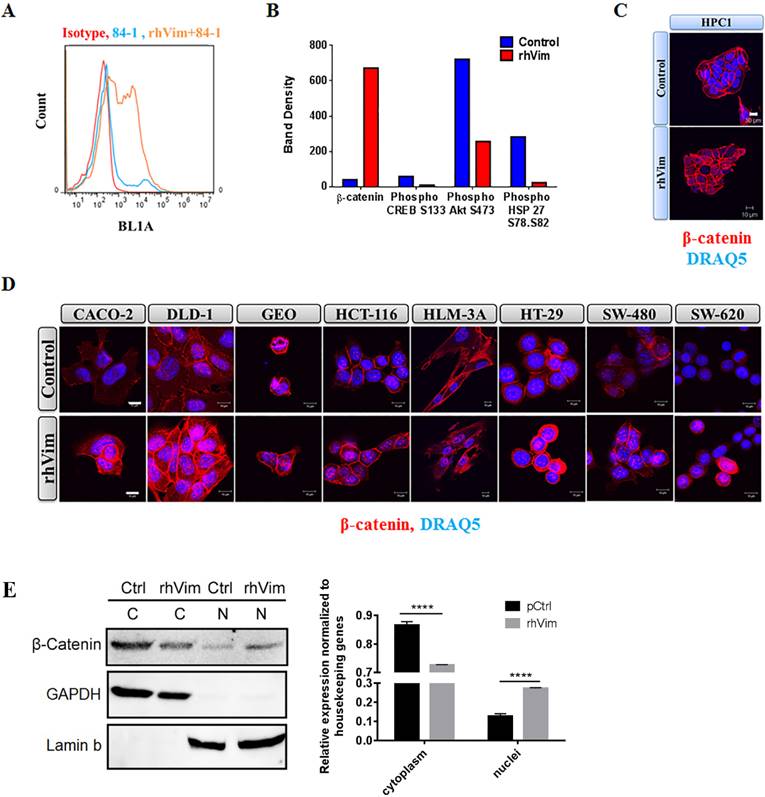
To assess if external vimentin found circulating in the blood of cancer patients binds to the surface of cancer cells, we tested the binding ability of externally added recombinant human vimentin (rhVim) to cancer cells using 84-1, a specific antibody against CSV and analyzed binding using flow cytometry. We utilized HPC1 colon cancer cells for this analysis and from the data shown in Fig. 1a, it is interesting to note that the cancer cells express basal levels of CSV depending on the metastatic phenotype of the cancers and upon external addition of rhVim; there was a dramatic increase in the levels of vimentin bound to the surface of cancer cells. We also observed this binding in several other cancer cell lines that were tested (representative are presented in Supplementary Table 1). Normal NCM-356 cells were tested as negative control and vimentin doesn't bind to these cells indicating the presence of specific receptors on surface of cancer cells. This data thus shows that externally added vimentin binds to the cell surface of cancer cells.
Vimentin binds to cancer cell surface and induces nuclear β-catenin accumulation: (A) Immunological assessment of CSV expression and external addition of rhVim to HPC1 cells using flow cytometry. Basal levels of CSV were detected in HPC1 cells and upon addition of vimentin; there is an increase in the levels of vimentin bound to the surface of the cells as observed in the histogram. Isotype controls were used as negative controls. (B) Proteomic array profile in HPC1 cells. HPC1 cells were treated with serum albumin (control) and with rhVim 250 ng/mL and cell lysates were incubated with arrays and images were collected and analyzed using Biorad imaging system. Columns indicate band density of different proteins that showed the highest level of changes. (C) Detection of β-catenin in HPC1 cancer cell line using confocal microscopy. β-catenin (red) was detectable in nucleus of cells treated with external rhVim (250 ng/mL). DRAQ5 was used to demarcate the nucleus of the cells. Scale bar, 10 µm. (D) Detection of β-catenin in various colon cancer cell lines using confocal microscopy. β-catenin (red) was detectable in nucleus of cells treated with rhVim (250 ng/mL). DRAQ5 was used to demarcate the nucleus of the cells. Scale bar, 10 µm. (E) Immunoblots of β-catenin in nuclear and cytoplasmic proteins from HPC1 cells. HPC1 cells treated with serum albumin or with rhVim (250 ng/mL) for ON were lysed to harvest cytoplasmic and nuclear proteins, respectively, and western blot was developed using respective antibodies. GAPDH was used as a loading control for cytoplasmic proteins, and Lamin b was used as a loading control for nuclear proteins. Intensity quantification (intensity of β-catenin/intensity of loading control) shown on the right panel represents the mean intensity of three repeated experiments.
Externally added vimentin induces β-catenin expression
Analyzing the phosphorylation profiles of kinases and protein substrates is one of the approaches of understanding the effect of the microenvironment on cancer cells. Since external vimentin binds to cell surface of cancer cells, we tested if this binding alters any kinases and substrates status. To evaluate the effect of externally added vimentin we utilized the human phospho-kinase array from R&D systems that is an economical tool to simultaneously detect the relative site-specific phosphorylation of 43 different proteins and 2 related total proteins. This method thus provides an easy and efficient way of detecting changes in the cancer cells after treating with rhVim. HPC1 cells were treated with rhVim (250 ng/mL) and serum albumin (250 ng/mL) overnight. After incubating the arrays with treated and control lysates, the results showed that there were four targets that altered significantly when compared to control (Fig. 1b). These changes were observed in β-catenin, phospho Akt, CREB and phospho Hsp27. Given that Wnt signaling is very prominent in colon cancer cells, we pursued β-catenin status in these cells. Also as an additional control to show that external vimentin specific binding occurs only in cancer cells, we used NCM356 cells as control cells, which were also treated with rhVim and serum albumin respectively. There was no change observed in any of the target genes in this cell line (data not shown).
Externally added vimentin increases nuclear accumulation of β-catenin
To test if β-catenin translocates to nucleus upon addition of external rhVim, we treated human primary colon cancer cells HPC-1 with rhVim at 250-ng/mL concentration overnight and immunostained cells for β-catenin expression to visualize changes in its localization. From the results in Fig. 1c it can be seen that externally added rhVim potentiated the accumulation of β-catenin in the nucleus of the cells in comparison to that of the control cells. This observation was also examined by immunofluorescence in a set of seven human colon cancer cell lines that include Caco-2, DLD-1, GEO, HCT-116, HT-29, SW480, SW620 and one liver metastasized cell line HLM3A (Fig. 1d). Importantly, this nuclear accumulation of β-catenin was observed in ~70% of the cells tested, indicating that externally added vimentin is a potent activator of β-catenin signaling pathway. Quantifications of β-catenin nuclear accumulation are included in Supplement Figure 1. As we indicated, in most human colon cancer cell lines, exogenous vimentin leads to nuclear accumulation of β-catenin. Further, to validate this result, Western blotting of β-catenin in nuclear and cytoplasmic proteins of HPC1 cells was performed (Fig. 1e). Our data showed that overnight incubation with vimentin significantly promoted nuclear localization of β-catenin.
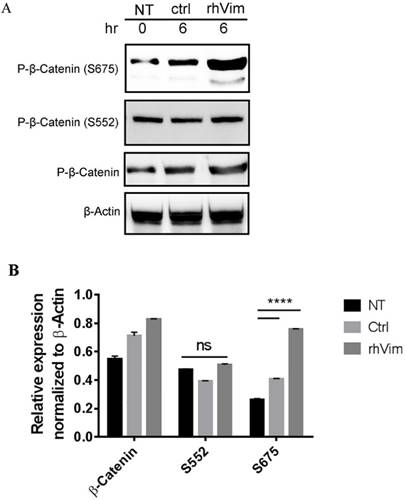
Externally added vimentin increased phosphorylation of β-catenin at Serine 675
It has been previously reported that increased phosphorylation of β-catenin at Serine 675 is associated with increased nuclear accumulation of β-catenin [13]. Thus we tested the phosphorylation status of β-catenin S552 and S675 phosphorylation using externally added rhVim in HPC1 cells using western blotting. From the Fig. 2a it is observed that externally added vimentin increases the phosphorylation of β-catenin at S675, but not of phosphor-β-catenin (S552) as assessed by densitometric analysis (Fig. 2b). Interestingly, the NCM-356 cells treated with rhVim did not show an induction in the levels of phosphorylated β-catenin at S675 (Data not shown).
β-catenin phosphorylation status in HPC1 cells treated with control serum albumin or with rhVim: (A) HPC1 cells treated with serum albumin or with rhVim (250 ng/mL) at 6 hours were lysed and western blot was developed using respective antibodies. β-actin was used as a loading control. (B) Intensity quantification (intensity of indicated protein/intensity of β-actin) represents the mean intensity of three repeated experiments.
Externally added vimentin elevated β-catenin/TCF Transcriptional activity
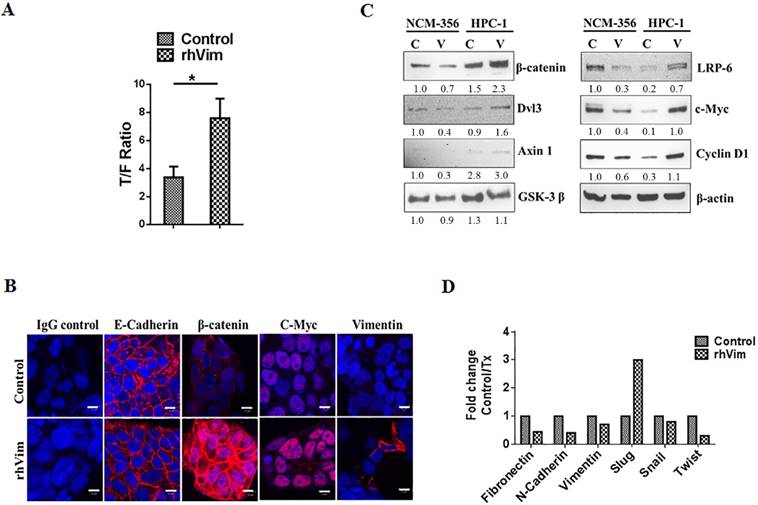
Since β-catenin accumulates in the nucleus upon addition of external rhVim, we then tested the ability of rhVim to transactivate a luciferase reporter plasmid containing wild-type (TOP-FLASH) or mutated (FOP-FLASH) β-catenin/TCF binding sites as regulatory elements. For this purpose, we examined HPC1 cells by treating with external rhVim and control serum albumin. Our results showed that rhVim supplementation displayed a higher ability to transactivate the TOP-FLASH reporter plasmid in comparison to the control treated (Fig. 3a). There was also decreased activity of FOP-FLASH observed in rhVim treated sample. In accordance with our findings showing an increasing cytoplasmic and nuclear localization of β-catenin in rhVim treated cells, these data clearly suggested that rhVim treatment have a stronger β-catenin/TCF transcriptional activity in comparison to the control serum albumin treated samples.
β-catenin transcribes the expression of Wnt signaling downstream targets: (A) rhVim addition affects β-catenin-mediated transcription in HPC1 cells. Graph represents β-catenin/TCF/LEF-mediated transcriptional activity. Bars, SE of two independent experiments done in triplicate. (B) Detection of E-Cadherin, C-Myc, β-catenin and vimentin expression in HPC1 cancer cell line using confocal microscopy. Respective protein (red) was detectable in nucleus of cells treated with rhVim (250 ng/mL). DRAQ5 was used to demarcate the nucleus of the cells. Scale bar, 10 µm. (C) immunoblot analysis of various Wnt signaling molecules in HPC1 and NCM-356 cells treated with either rhVim (250 ng/mL) or serum albumin (control) for 24 h. Actin was used as a loading control. Intensity quantification (intensity of indicated protein/intensity of β-Actin) shown under each blot represents the mean intensity of three repeated experiments. (D) RT-qPCR was performed in HPC1 cells that were treated with rhVim to assess the levels of different epithelial-mesenchymal genes. Data is represented as fold change control to that of treated.
Externally added vimentin regulates expression of Wnt target genes
To examine if external rhVim mediated β-catenin accumulation in the nucleus leads to increased expression of Wnt target genes, HPC1 cells were treated with rhVim or serum albumin and analyzed for the expression of different proteins that are direct targets of Wnt signaling activation. From immunocytochemical analysis of these cells after treatment as seen in Fig. 3b it is evident that c-myc was up-regulated upon rhVim treatment, while there was no significant change in the expression of E-Cadherin protein. Interestingly, few of the cells that were treated with rhVim started expressing vimentin, which could be attributed to the ability of β-catenin to transcribe vimentin expression [14]. It is suggested that disheveled (Dsh or Dvl) play a key role in the stabilization of β-catenin [15] and from our analysis of western blot in Fig. 3c we observed a slight increase in the expression of Dvl3 protein indicating that external rhVim could be stabilizing β-catenin expression via Dvl3. Also, our western blot data further show an increase in the expression of cyclin D1, c-myc, axin-1 and LRP-6 which are target genes for the β-catenin/ TCF-LEF pathway activation [12]. However, there was no change in the levels of GSK-3 β. These changes were not observed in normal colon epithelial cells since there was no binding to external vimentin in these cells.
Given the ability of external rhVim to induce the levels of vimentin in epithelial cells, we tested the possibility of epithelial mesenchymal transition phenomenon in these cancer cells by utilizing an RT-PCR analysis for EMT specific markers that include Fibronectin, N-Cadherin, Slug, Snail and Twist proteins. From Fig. 3d it is evident that there is a drastic increase in the levels of slug [16] protein in cells treated with external rhVim indicating the process of EMT in these cells.
Externally added vimentin regulates cell invasion
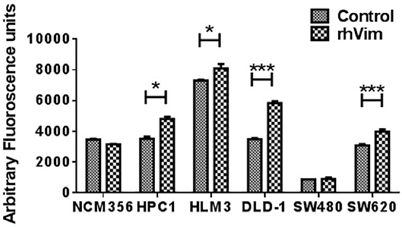
Since Wnt signaling is involved in a variety of cellular processes and given the ability of external vimentin to activate EMT phenotype, we tested if there is an increase in invasion rate of the cells. Invasion was evaluated using a FluoroBlok invasion assay. Cells were treated with external rhVim or serum albumin and analyzed for invasion. The results (Fig. 4) indicated a significant increase in the invasion of cancer cells (except SW480 cells) when compared to that of the serum albumin control. Normal cell line NCM-356 did not show any difference in invasion upon addition of rhVim. There was no significant change observed in the migration ability of these cells. Taken together these results suggest that vimentin presence in the extracellular milieu promotes tumor cell invasion.
rhVim increases invasion potential of colon cancer cells: Invasion of colon cancer cells treated with rhVim or serum albumin was quantified using FluoroBlok chamber assay. Data are representative of two independent experiments with three replicates in each experiment. (*p<0.01)
Discussion
During metastasis, epithelial cells acquire the ability to invade the surrounding tissue and travel to surrounding organs. This ability to invade is mainly associated with the gain of mesenchymal characteristics and loss of epithelial characteristics in the cancer cells. This gain of mesenchymal characteristic is associated with an increase in intracellular vimentin expression levels [3]. Vimentin is a cytosolic protein that can be localized on the tumor cell surface by several researchers including us [17,9,10,18-21]. Also, vimentin is shown to be secreted into the surrounding environment by different cell types [4-6] and the function of this secreted vimentin remains unknown.
This study for the first time reveals the binding of external vimentin to the surface of cancer cells but not to normal cells. This binding thus shows the presence of specific receptors in different cancer cell lines that could interact with vimentin. This receptor needs to be elucidated in the future study. To elucidate the signaling pathway that activates upon vimentin binding, we did a proteomic array analysis. From proteomic analysis it is evident that there is an increase in the levels of β-catenin, while there was a decrease in the phosphorylation of Akt which is an indication of reduction in apoptosis and increased tumor progression [22]. While phosphorylation of HSP27 has many subsequent effects [23]. Given the extensive role of Wnt signaling pathway in colorectal tumorigenesis, we pursued the role of vimentin in Wnt signaling pathway.
In normal epithelial cells, β-catenin is cytoplasmic in nature that forms complexes with E-cadherin which contributes to cell-cell adhesion properties of the cells. Under appropriate stimulus, β-catenin accumulates in the cytosol and then translocates to the nucleus where it binds to TCF/LEF-1 domain, thus transactivating several genes that have been identified as targets of β-catenin/ transcriptional regulation. Some of the prominent genes that are transcribed include cyclin D1, c-myc, TCF-1, slug, vimentin and CD44. Here in our study we show the transactivation of β-catenin pathway by external addition of vimentin that induces the expression of various Wnt specific down-stream targets. This involved an increase in the phosphorylation of S675 residue on β-catenin, which causes an accumulation in the nucleus. It is known that S675 residue in β-catenin is phosphorylated by PKA [24] and this phosphorylation does not affect the GSK3-dependent phosphorylation of β-catenin, its stability or intracellular localization; and further phosphorylation at Ser-675 site promotes the binding of β-catenin to its transcriptional coactivator, CREB-binding protein (CBP). Although from our data it is evident that there is an increased S675 phosphorylation which explains the Wnt downstream targets over expression, it is very intriguing to understand the activation of Wnt signaling pathway using external vimentin treatment.
To dissect the Wnt signaling activation in rhVim treated cells, we utilized the online HitPredict algorithm that has a database of high confidence protein-protein interactions [25]. Using this algorithm, we screened for possible interacting partners of vimentin that play a critical role in Wnt signaling. From our analysis we found Ryk protein, a prominent molecule involved in Wnt signaling pathway [26] as a possible interacting partner of vimentin as analyzed by the algorithm (http://hintdb.hgc.jp/htp/interactions/222175.html). On the basis of the association of vimentin with Ryk protein and the role of Ryk protein in the Wnt signaling pathway (schematic representation in Figure 5), we hypothesize that vimentin could serve as a ligand to Ryk receptor or would associate with Ryk receptor and enhance the Wnt signaling activation, a possibility which shall be investigated in the future.
Schematic representation of Wnt signaling pathway: We hypothesize, externally added vimentin binds to Ryk receptor that activates Wnt signaling pathway by enhancing S675 phosphorylation of β-catenin that increases its accumulation in the nucleus thereby inducing Wnt downstream targets through TCF/LEF factors. In the absence of ligand, β-catenin gets degraded.
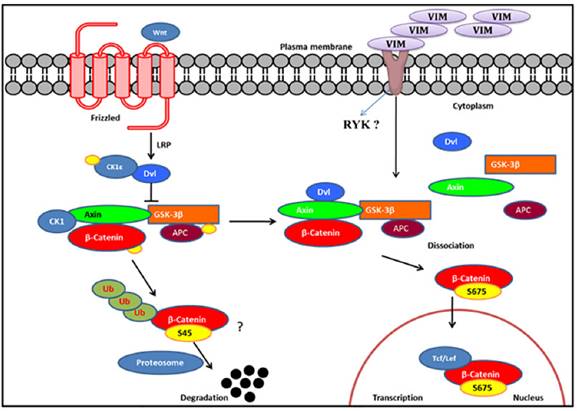
From our observations reported here, we speculate the following possible roles of secreted vimentin in cancer patients; circulating vimentin induces Wnt signaling pathway which is involved in a variety of tumorigenic events; Since externally added vimentin induces mesenchymal phenotype in the epithelial HPC1 cells, secreted vimentin detected in cancer patient serum may impart mesenchymal nature to the epithelial cells either at the primary tumor site or circulating tumor cells in the blood. Recently, Par-4 protein was shown to interact with vimentin and arylquin was able to disrupt the interaction and secrete Par-4 which induced apoptosis in cancer cells [27] indicating another possible role of secreted vimentin to interact with Par-4 in extracellular milieu so as to negate its anti-apoptotic effects.
There are a number of gene mutations involving tumor suppressor genes during the gradual progression of sporadic colorectal cancers (CRCs). Inactivating somatic mutations in the adenomatous polyposis coli (APC) has been found in majority (~70%) CRCs as an initial event of histological changes [28]. Germline mutation of APC is responsible for familial adenomatous polyposis (FAP) [29]. Normally, APC functions as a regulator of β-catenin in Wnt pathway. In the absence of Wnt ligands, APC facilitates the degradation of cytoplasmic β-catenin through Axin and GSK3β [30]. By contrast, inactivating mutation of APC impaired its regulation of β-catenin, resulting in the activation of Wnt pathway [30]. In our study, most of the human colon cancer cell lines (Caco2, DLD1, HCT116, HT29, SW480 and SW620) were characterized with mutations in APC. HCT116 cells carry wild type APC, and HPC1, GEO and HLM3A are uncertain. Our results showed that exogenous vimentin enhanced nuclear accumulation of β-catenin in spite of APC gene status. Nevertheless, whether or not mutated APC has any influence in the effect of exogenous vimentin in Wnt signaling pathway remains unclear. In our future studies, it could be interesting to investigate the differences in physiological levels of vimentin and downstream Wnt-related gene status between wildtype and mutant APC-carrying CRCs.
In conclusion, exogenously supplemented vimentin was shown to promote tumor cell invasion in vitro. Together, these results suggest the circulating vimentin secreted into the blood as a potential target to control the rate of invasion of cancer cells and these studies thus provide a deeper look into the unknown roles of vimentin. We envision that the blockade of freely available vimentin in the blood of cancer patients might be a useful tool in preventing the activation of Wnt signaling pathway that is known to activate a multitude of tumorigenic events.
Supplementary Material
Supplementary table 1 and figure S1.
Abbreviations
rhVim: Recombinant human vimentin; CSV: Cell Surface Vimentin.
Acknowledgements
Work in the author's laboratory was supported by Grants from the National Institutes of Health to Dr. Shulin Li (NIH RO1CA120895).
Competing Interests
None.
References
1. Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer cell. 2005;7:513-20
2. Knudsen SL, Mac AS, Henriksen L, van Deurs B, Grovdal LM. EGFR signaling patterns are regulated by its different ligands. Growth factors. 2014;32:155-63
3. Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cellular and molecular life sciences: CMLS. 2011;68:3033-46
4. Teshigawara K, Kuboyama T, Shigyo M, Nagata A, Sugimoto K, Matsuya Y. et al. A novel compound, denosomin, ameliorates spinal cord injury via axonal growth associated with astrocyte-secreted vimentin. Br J Pharmacol. 2013;168:903-19
5. Xu B, deWaal RM, Mor-Vaknin N, Hibbard C, Markovitz DM, Kahn ML. The endothelial cell-specific antibody PAL-E identifies a secreted form of vimentin in the blood vasculature. Mol Cell Biol. 2004;24:9198-206
6. Mor-Vaknin N, Punturieri A, Sitwala K, Markovitz DM. Vimentin is secreted by activated macrophages. Nat Cell Biol. 2003;5:59-63
7. Ngan CY, Yamamoto H, Seshimo I, Tsujino T, Man-i M, Ikeda JI. et al. Quantitative evaluation of vimentin expression in tumour stroma of colorectal cancer. British journal of cancer. 2007;96:986-92
8. Garg A, Barnes PF, Porgador A, Roy S, Wu S, Nanda JS. et al. Vimentin expressed on Mycobacterium tuberculosis-infected human monocytes is involved in binding to the NKp46 receptor. Journal of immunology. 2006;177:6192-8
9. Satelli A, Mitra A, Brownlee Z, Xeuqing X, Bellister S, Overman MJ. et al. Epithelial-mesenchymal transitioned circulating tumor cells capture for detecting tumor progression. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014
10. Satelli A, Mitra A, Cutrera JJ, Devarie M, Xia X, Ingram DR. et al. Universal marker and detection tool for human sarcoma circulating tumor cells. Cancer research. 2014;74:1645-50
11. Satelli A, Rao PS, Gupta PK, Lockman PR, Srivenugopal KS, Rao US. Varied expression and localization of multiple galectins in different cancer cell lines. Oncology reports. 2008;19:587-94
12. Herbst A, Jurinovic V, Krebs S, Thieme SE, Blum H, Goke B. et al. Comprehensive analysis of beta-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/beta-catenin signaling. BMC genomics. 2014;15:74
13. Zhu G, Wang Y, Huang B, Liang J, Ding Y, Xu A. et al. A Rac1/PAK1 cascade controls beta-catenin activation in colon cancer cells. Oncogene. 2012;31:1001-12
14. Gilles C, Polette M, Mestdagt M, Nawrocki-Raby B, Ruggeri P, Birembaut P. et al. Transactivation of vimentin by beta-catenin in human breast cancer cells. Cancer research. 2003;63:2658-64
15. Gonzalez-Sancho JM, Brennan KR, Castelo-Soccio LA, Brown AM. Wnt proteins induce dishevelled phosphorylation via an LRP5/6- independent mechanism, irrespective of their ability to stabilize beta-catenin. Molecular and cellular biology. 2004;24:4757-68
16. Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Molecular biology of the cell. 2008;19:4875-87
17. Satelli A, Brownlee Z, Mitra A, Meng QH, Li S. Circulating tumor cell enumeration with a combination of epithelial cell adhesion molecule- and cell-surface vimentin-based methods for monitoring breast cancer therapeutic response. Clinical chemistry. 2015;61:259-66
18. Mitra A, Satelli A, Xia X, Cutrera J, Mishra L, Li S. Cell-surface Vimentin: A mislocalized protein for isolating csVimentin CD133 novel stem-like hepatocellular carcinoma cells expressing EMT markers. International journal of cancer Journal international du cancer. 2014
19. Plummer EM, Thomas D, Destito G, Shriver LP, Manchester M. Interaction of cowpea mosaic virus nanoparticles with surface vimentin and inflammatory cells in atherosclerotic lesions. Nanomedicine. 2012;7:877-88
20. Steinmetz NF, Cho CF, Ablack A, Lewis JD, Manchester M. Cowpea mosaic virus nanoparticles target surface vimentin on cancer cells. Nanomedicine. 2011;6:351-64
21. Steinmetz NF, Maurer J, Sheng H, Bensussan A, Maricic I, Kumar V. et al. Two Domains of Vimentin Are Expressed on the Surface of Lymph Node, Bone and Brain Metastatic Prostate Cancer Lines along with the Putative Stem Cell Marker Proteins CD44 and CD133. Cancers. 2011;3:2870-85
22. Itoh N, Semba S, Ito M, Takeda H, Kawata S, Yamakawa M. Phosphorylation of Akt/PKB is required for suppression of cancer cell apoptosis and tumor progression in human colorectal carcinoma. Cancer. 2002;94:3127-34
23. Vidyasagar A, Wilson NA, Djamali A. Heat shock protein 27 (HSP27): biomarker of disease and therapeutic target. Fibrogenesis & tissue repair. 2012;5:7
24. Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. The Journal of biological chemistry. 2006;281:9971-6
25. Patil A, Nakai K, Nakamura H. HitPredict: a database of quality assessed protein-protein interactions in nine species. Nucleic acids research. 2011;39:D744-9
26. Green J, Nusse R, van Amerongen R. The role of Ryk and Ror receptor tyrosine kinases in Wnt signal transduction. Cold Spring Harbor perspectives in biology. 2014:6
27. Burikhanov R, Sviripa VM, Hebbar N, Zhang W, Layton WJ, Hamza A. et al. Arylquins target vimentin to trigger Par-4 secretion for tumor cell apoptosis. Nature chemical biology. 2014;10:924-6
28. Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256(5057):668-70
29. Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M. et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66(3):589-600
30. Yang J, Zhang W, Evans PM, Chen X, He X, Liu C. Adenomatous polyposis coli (APC) differentially regulates beta-catenin phosphorylation and ubiquitination in colon cancer cells. J Biol Chem. 2006;281(26):17751-7
Author contact
![]() Corresponding author: Dr. Shulin Li, Department of Pediatrics, Unit 853, The University of Texas Graduate School of Biomedical Sciences, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd. Houston, Texas 77030, USA. E-mail: sli4org Phone: 713-563-9608 Fax: 713-563-96.
Corresponding author: Dr. Shulin Li, Department of Pediatrics, Unit 853, The University of Texas Graduate School of Biomedical Sciences, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd. Houston, Texas 77030, USA. E-mail: sli4org Phone: 713-563-9608 Fax: 713-563-96.