Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(13):1755-1771. doi:10.7150/jca.15170 This issue Cite
Research Paper
Molecular Characterization and Enhancement of Anticancer Activity of Caffeic Acid Phenethyl Ester by γ Cyclodextrin
Renu Wadhwa1, Nupur Nigam1,2, Priyanshu Bhargava1,2, Jaspreet Kaur Dhanjal3, Sukriti Goyal4, Abhinav Grover4, Durai Sundar3, Yoshiyuki Ishida5, Keiji Terao5,6 ![]() , Sunil C Kaul1
, Sunil C Kaul1 ![]()
1. DBT-AIST International Laboratory for Advanced Biomedicine (DAILAB), National Institute of Advanced Industrial Science & Technology (AIST), Central 5-41, 1-1-1 Higashi, Tsukuba - 305 8565, Japan;
2. Graduate School of Life & Environmental Sciences, University of Tsukuba, Ibaraki - 305 8575, Japan;
3. Department of Biochemical Engineering & Biotechnology, Indian Institute of Technology Delhi, New Delhi - 110 016, India;
4. School of Biotechnology, Jawaharlal Nehru University, New Delhi - 110 067, India.
5. CycloChem Co., Ltd., 7-4-5 Minatojima-minamimachi, Chuo-ku, Kobe - 650 0047, Japan;
6. Graduate School of Medicine, Kobe University, 7-5-1, Kusunoki-cho, Chuo-ku, Kobe - 650 0017, Japan.
Received 2016-2-1; Accepted 2016-6-29; Published 2016-8-11
Abstract
Caffeic Acid Phenethyl Ester (CAPE) is a key component in New Zealand propolis, known for a variety of health promoting and therapeutic potentials. We investigated the molecular mechanism of anticancer and anti-metastasis activities of CAPE. cDNA array performed on the control and CAPE-treated breast cancer cells revealed activation of DNA damage signaling involving upregulation of GADD45α and p53 tumor suppressor proteins. Molecular docking analysis revealed that CAPE is capable of disrupting mortalin-p53 complexes. We provide experimental evidence and demonstrate that CAPE induced disruption of mortalin-p53 complexes led to nuclear translocation and activation of p53 resulting in growth arrest in cancer cells. Furthermore, CAPE-treated cells exhibited downregulation of mortalin and several other key regulators of cell migration accountable for its anti-metastasis activity. Of note, we found that whereas CAPE was unstable in the culture medium (as it gets degraded into caffeic acid by secreted esterases), its complex with gamma cyclodextrin (γCD) showed high efficacy in anti-tumor and anti-metastasis assays in vitro and in vivo (when administered through either intraperitoneal or oral route). The data proposes that CAPE-γCD complex is a potent anti-cancer and anti-metastasis reagent.
Keywords: Propolis, CAPE, γCD, complex, upregulation, p53, anticancer, anti-metastasis.
Introduction
Recent advances in cancer diagnostic and therapeutic tools have increased the success rate and survival of cancer patients. However, treatment of metastatic cancers continues to be a challenge. Cancer metastasis involves dissemination of cancer cells from their primary site and establishment at either the surrounding or distant tissues by invasion through the extracellular matrix (ECM) or the blood/lymph streams, respectively. It is a complex multi-step phenomenon and involves interplay of several factors that enable cancer cells to acquire invasiveness and intravasate into circulatory system, survive in the blood or lymph circulation, form microvasculature and extravasate to form tumor mass at distant organs [1-3]. These involve activation of several processes at cellular level including epithelial-mesenchymal transition (EMT), motility, adhesion and invasiveness that are regulated by activation of oncogenes, proteases and other tumor-microenvironment components and inactivation of tumor suppressor proteins and apoptosis-mediating factors [4-7]. Chemotherapeutic drugs target these pro-cancer signaling pathways. However, most of the available drugs also target the noncancerous normal dividing cells in the body resulting in undesired adverse side-effects including low-immune response, hair loss, fatigue, nausea and hormone-imbalance. Furthermore, most of the anticancer drugs show low efficacy for metastatic cancer cells resulting in either treatment failure or relapse. Chemoresistance and radioresistance of cancer cells and nonspecific toxicity of chemical drugs towards normal cells are the major hurdles the cancer treatment. Several epidemiological studies have suggested that the consumption of fruits and vegetables lower cancer risk. Several plant-derived polyphenols including curcumin, genistein, silymarin, caffeic acid phenethyl ester, resveratrol, green tea polyphenols, flavopiridol, emodin and piperine have hence been investigated for their chemo-therapeutic and -preventive properties and for sensitization of tumor cells to chemo- and radio-therapeutic agents [8-14]. Owing to their safety, availability and economic aspects, natural chemicals are gaining increasing focus in research aiming to understand their cancer-preventive and -curative activities, and their mechanism(s) of action.
Propolis is a natural amalgam produced by honeybees by mixing plant materials with wax and resin to build their hives. In folk medicine, it has been used for anti-bacterial, anti-inflammatory, wound healing, skin infections, gastro-intestinal disorders, immunomodulation and cardiovascular benefits. CAPE (Caffeic Acid Phenethyl Ester) is an active phenolic compound isolated from propolis. It was ascribed several activities, previously detected in propolis, and has been known as an anti-mitogenic and anticancer natural drug [15,16]. Several studies have shown that CAPE and its derivative compounds exhibit selective toxicity to transformed/cancer cells [17-23]. CAPE- and Artepillin C (ARC)- rich propolis extracts were suggested as natural anti-PAK1 inhibitors and NF1/NF2 remedies [24,25]. CAPE was shown to inhibit TPA (12-O-tetradecanoylphorbol-13-acetate)-induced tumors [26,27], and suggested as an anticancer drug for treatment of colorectal carcinoma [28], hepatoma [29] and gastric carcinoma [30]. CAPE-induced cell cycle arrest and apoptosis are mediated by down-regulation of beta-catenin [31,32] and p38MAPK [33] signaling. It was shown to inhibit NF-kappaB, COX-2 [34,35] and stimulate human anti-oxidative response element-mediated expression of the NAD (P) H: quinone oxidoreductase (NQO1) gene [36]. CAPE was shown to (i) restore intercellular communication through gap junctions, regulated by phosphorylation of connexin 43 [35], (ii) cause rearrangement of actin cytoskeleton and loss of focal adhesion plaques causing reduced cell invasion [37]. It inhibited growth of cancer cells in soft agar through downregulation of p21ras protein [38]. Consistent with the in vitro reports, anticancer activity of CAPE was endorsed by in vivo studies in mice [39-41]. CAPE and its derivative, caffeic acid phenylpropyl ester (CAPE), were shown to inhibit PI3-K/Akt, AMPK and m-TOR signaling cascades both in vitro and in vivo [41]. Although such reports proposed CAPE as a natural drug for cancer prevention and treatment, molecular studies are warranted to clarify its mechanism of action and rational designing of its derivatives.
Cyclodextrin (CD) is produced from starch by the process of enzymatic conversion. It is widely used in food, agriculture, pharmaceutical, chemical and environmental engineering. It is also used in drug delivery, because of its structure with hydrophobic inside and a hydrophilic outside that enhances the solubility and bioavailability of compounds. γCD, widely accepted for use in food, pharmaceutical, agricultural and chemical industry constitutes of 8 glucose monomers arranged in the form of a cyclic ring. It has been reported to enhance the bioavailability of hydrophobic ingredients such as coenzyme Q10 [42-44].
In the present study, we generated conjugate of CAPE and γCD and investigated their anticancer and anti-metastasis potential using a variety of cancer cells in vitro and in vivo.
Materials and methods
Cell culture
Human lung carcinoma (A549), fibrosarcoma (HT1080), melanoma (G361), osteosarcoma (U2OS), breast carcinoma (MCF-7 and MDA-MB-231), were obtained from Japanese Collection of Research Bioresources. Mortalin overexpressing metastatic derivatives were obtained by retrovirus driven overexpression of mortalin as described earlier [45]. Cells were cultured in DMEM (Life Technologies, Carlsbad, CA, USA)-supplemented with 10% fetal bovine serum at 37°C, in an atmosphere of 5% CO2 and 95% air in a humidified incubator. CAPE (Caffeic Acid Phenethyl Ester) and CAPE-γCD (Caffeic Acid Phenethyl Ester-Gamma Cyclodextrin complex) were dissolved in dimethylsulfoxide (DMSO) to make 1 mM stocks and added to the complete cell culture medium to obtain the working concentrations as indicated. All the biochemical and imaging assays were performed on sub-confluent (60-70%) cultures.
Preparation of CAPE-γCD complex
To an aqueous solution of γCD, a solution of CAPE and ethanol in equal molar ratio was added gradually at 25oC. The mixture was incubated for 20 h with continuous stirring following which CAPE-γCD as the precipitation was generated. After removing supernatant from the resulting mixture by centrifugation, crude CAPE-γCD was washed with water and chloroform and dried in vacuo. CAPE-γCD thus obtained was used in the present study.
Cytotoxicity and growth inhibition assay
The effect of CAPE and CAPE-γCD on cell viability was determined using quantitative colorimetric MTT [3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay dependent on the mitochondrial activity of living cells (Sigma Aldrich, Japan). Cells (5 X 103)/well were plated in 96-well plates. After overnight incubation, the cells were treated with CAPE and CAPE-γCD as indicated. MTT (0.5 mg/ml) was added to the cell culture medium at the end of treatments and incubated for 4 h in a humidified incubator (37ºC and 5% CO2). MTT-containing medium was replaced with DMSO (100 µL) in each well to dissolve purple formazan crystals. Absorbance was measured at 550 nm using spectrophotometer (TECAN, Switzerland). Experiments were done in triplicates. The standard deviation & statistical significance of the data were determined by unpaired t-test using GraphPad software.
Morphological observations
The cells were cultured in 12-well plates and upon reaching 60% confluency they were treated with different concentrations of CAPE, CAPE-γCD and γCD. After 48-72 h, morphological changes were recorded with a phase contrast microscope.
Colony-forming assays
500 cells were plated in a six-well plate. After overnight incubation, the cells were treated with media-supplemented with CAPE. The treated cells were then left to form colonies for the next 10-15 days with a regular change in medium on every third day. Colonies were fixed in methanol, stained with 0.1% crystal violet solution, photographed and counted.
Cell cycle analysis
MCF-7 and MDA-MB-231 cells were plated in 6 well plates at density of 2 X 105 cells/well. After 24 h of seeding, cells were treated with CAPE for 24-36 h followed by harvesting by trypsin. The cell pellets were fixed with ice-cold 70% ethanol and stored at -200C until further use. The fixed cells were centrifuged at 500xg for 5 min and washed twice with cold PBS to remove ethanol followed by re-suspension 0.25 ml PBS with RNase A (5 μL of 10 mg/ml) to avoid false DNA-PI staining. The cells were incubated for 1 h at 370C followed by centrifugation at 500x g for 5 min. The pellet was re-suspended in Guava cell cycle reagent (EMD Millipore Corporation) and incubated in dark for 5 min. The stained cells were analyzed using Guava PCA flow cytometer (Millipore) and Flow Jo software (Version 7.6, Flow Jo, LLC, USA).
Apoptosis assay
MCF-7 and MDA-MB-231 cells were seeded at a density of 2 X 105 cells/well in 6 well plates for 24 h followed by treatment of CAPE for 24-36 h. Apoptosis assay was examined using Guava Nexin Reagent (EMD Millipore Corporation) using procedure recommended by the manufacturer. Apoptotic cells were quantitated by Flow Jo Software.
cDNA array
Total RNA was prepared using TRIzol reagent (Gibco BRL, Grand Island, NY) following manufacturer's instructions, and was labeled with Cy3 or Cy5 using Low Input Quick Amp Labeling Kit (Agilent Technologies), hybridized to human cDNA array slides and analyzed using Agilent 2100 BioAnalyzer series II (Hokkaido-System Science, Japan). cDNA array data on total of 22932 genes, upregulated more than 2-fold in CAPE-treated cells as compared to the control, were used to identify the most potential pathways using Gene Annotation Tool to Help Explain Relationships (Gather) [46] and Advaita Bio's iPathway Guide (http://www.advaitabio.com/ipathwayguide). The software analysis tool implemented the 'Impact Analysis' approach that took into consideration the direction and type of all signals on a pathway, the position, role and type of the genes [47]. The genes were plotted in KEGG pathway database [48].
Docking studies and molecular dynamics simulation of the obtained complex
AutoDock 4.2 suite was utilized for molecular-docking of CAPE with mortalin. The three dimensional structure of caffeic acid phenethyl ester (CAPE) was retrieved from PubChem database (CID: 5281787). Roots were detected for CAPE, 6 bonds were made rotatable and the coordinates were saved in PDBQT file format (an extended PDB format that was utilized for storing coordinates and atomic partial charges) using AutoDock Tools. The crystal structure of mortalin [PDB ID: 4KBO] was obtained from PDB (http://www.rcsb.org/pdb/home/home.do). AutoDock Tools were utilized for preparation of crystal structure by addition of all hydrogen atoms in order to ensure correct calculation of partial atomic charges followed by calculation of Gasteiger charges. A three dimensional grid map with 60 x 60 x 60 points was generated around p53 binding site on mortalin (residues 253-282). Lamarckian Genetic Algorithm (GA) was employed with a population size of 150 dockings and energy evaluation of 2.5 x 106 for performing docking step. The most stable or lowest energy conformation of the docked complex amongst all the docked structures was selected for further analysis.
To comment on the dynamic stability of CAPE within the active site of mortalin, molecular dynamics simulations were performed on the docked complex using Desmond module of Schrodinger's Suite and Maestro as interface [49,50]. MD simulations were performed by applying OPLS 2005 molecular mechanics force field. At first, the protein was prepared to fix erroneous atomic representations in the complex structure. Preparation steps involved addition and optimization of hydrogen atoms, generation of disulfide bonds and capping of end terminals. The prepared protein was then solvated in a triclinic periodic box of SPC water molecules and neutralized by addition of appropriate number of counter ions. In order to eliminate geometric problems such as unrealistic bond distances, bond angles and torsion angles, energy minimization was performed. Steepest descent method with restraints was used to minimize the system until a gradient threshold of 25 kcal/mol/Å was not reached. MD simulations were performed on the equilibrated systems for a time period of 40 ns at a constant pressure and temperature with a time step of 2 fs. Root mean square deviations were calculated for the mortalin-CAPE complex during the entire simulation trajectory with reference to the first frame.
MM-GBSA analysis for the docked complex pre- and post-molecular dynamics simulations
Various computational approaches are often used to calculate binding-free energies in complex bio-molecular systems. Use of scoring function of molecular docking for predicting the protein-ligand interaction and binding-free energies to identify potential lead compounds for a specific molecular target is one such approach [51]. Even though they are fast, some drawbacks like lack of treatment of protein flexibility and water molecules, improperly accounting for solvation, entropy, and polarizability need to be improved to achieve high scoring accuracy. MM-GBSA method that uses a single minimized protein-ligand structure instead of various frames derived from MD trajectories is an efficient method to refine and rescore docking results and calculate free binding energies [52]. ΔGbinding was calculated for the mortalin-CAPE complex using MM-GBSA analysis available in Prime module of Glide [53].
Prediction of pharmacokinetics of CAPE in human body
In order to determine the drug-likeness and pharmacokinetics of CAPE, its 3D structure was analyzed to assess various physicochemical properties. Its absorption, distribution, metabolism, excretion and toxicity (ADMET) were calculated in silico using Qikprop module of Schrodinger suite and admetSAR, an online open access server [54].
Western blotting
Control and treated cells were lysed in RIPA lysis buffer (Thermo Fisher Scientific Inc., IL, USA) containing complete protease inhibitor cocktail (Roche Applied Science, Mannheim, Germany) for 20 min on ice. Lysates were centrifuged at 15000 rpm for 15 min, and the supernatant was used for Western blotting. The protein concentrations of cell lysates were determined by BCA (Bicinchonic Acid) assay (Thermo Fisher Scientific Inc., IL, USA). The lysates (20 µg protein) were resolved on a 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, MA) using a semidry transfer blotter (ATTO Corporation, Japan). Membranes were blocked with 3% BSA/TBST and then probed with indicated antibodies: mortalin [45], GADD45α (H-165), p53 (DO-1), MMP-2 (H-76), vimentin (H-84) (Santa Cruz Biotechnology, CA) for overnight at 4oC followed by incubation with HRP conjugated-secondary antibodies (Santa Cruz Biotechnology, CA) and detection by ECL PLUS (GE Healthcare, UK). The membranes were probed by anti β-actin antibody (Abcam, UK) as an internal loading control. Protein signals were quantitated using the ImageJ software (NIH, USA). Standard deviation and statistical significance of the data were calculated by Microsoft office excel (Microsoft Corporation, WA, USA) and QuickCals t-test calculator (GraphPad Software, Inc., CA, USA), respectively. Statistical significance of the data was represented by *P<0.05, **P<0.01 and ***P<0.001 for significant, very significant, highly significant, respectively.
Immunofluorescence
For immunofluorescence, cells (1 X 103/well) were plated on glass coverslips placed in 12-well culture dish. Control and treated cells were washed twice with PBS, fixed in pre-chilled methanol:acetone (1:1) for 5 min on ice, and were then permeabilized by 0.1% Triton X-100 in PBS for 15 min. The samples were blocked with 1% bovine serum albumin in PBS, and followed by incubation in anti-mortalin [45] and p53 (DO-1) and GADD45α (H-165), MMP-2 (H-76), vimentin (H-84) (Santa Cruz Biotechnology, CA) p53BP-1 primary antibodies. Immunostaining was visualized by secondary staining with either Alexa-488 or Alexa-594 conjugated antibodies (Molecular probes). After three to four washings with 0.2% Triton X-100 in PBS (PBST), cells were counter stained with Hoechst for nuclear staining, overlaid with Fluoromount (Difco). Cells were examined on a Zeiss Axiovert 200 M microscope and analyzed by AxioVision 4.6 software (Carl Zeiss).
Immunoprecipitation assay
Cell lysates (500 μg) of control and CAPE treated cells were incubated with dyna beads®Protein A conjugated with anti-mortalin polyclonal antibody [45] for overnight at 4o C (Thermofisher Scientific, Life Technologies AS Oslo). The immunocomplexes (IC) were washed with PBST (PBS with 0.2% Triton X100) three times followed by elution in denaturing SDS loading buffer (25 μL) by heating at 70oC for 10 min. IC were resolved on 7.5% SDS-PAGE along with controls including and cell lysate (20 μg), IgG and Dyna beads, and were electro-blotted onto a PVDF membrane (Millipore Corporation) using a semi-dry transfer system (Bio-Rad). The membrane was probed with anti-p53 (DO-1; SC126) and anti-mortalin monoclonal antibodies. Proteins were detected using ECL (Enhanced Chemi Luminescence; Amersham Pharmacia Biotech). Amount of p53 in mortalin-IC was quantitated by normalizing with the amount of IgG and mortalin controls in IC.
Wound-scratch assay
The in vitro migration capability of cells in response to CAPE and CAPE-γCD treatment was examined with a wound-scratch assay. The cells were grown in a monolayer and wounded by uniformly scratching the cells in a line with a 200 µl pipette tip. Cells were washed with PBS to remove cell debris or floating cells, and cultured on either the control or CAPE or CAPE-γCD-supplemented medium, respectively. The time of wound scratching was designated as time 0. Movement of cells into the wounded area was recorded during next 24 h under a phase contrast microscope with a 10 X phase objective. Migration capacity was calculated by measuring the percent of open area in 6-10 randomly captured images.
In vitro chemotaxis assay
Cells at 60-70% confluency were washed with cold PBS, trypsinized, and re-suspended in DMEM-supplemented with 0.5% bovine serum albumin (Sigma) at 5X104 cells/ml. 2.5 X 104 cells were plated in BioCoatTM MatrigelTM Invasion Chambers (8-mm pore, BD Biosciences), in the presence or absence of the drugs, and the invasion assay was performed following the manufacturer's instructions. Cells that had invaded through the Matrigel and filters were extracted with 10% acetic acid and quantitated by absorbance at 590 nm using a microplate reader (TECAN, Switzerland).
In vivo anti-tumor and anti-metastasis assays
The tumor- and metastasis-inhibitory effect of CAPE and CAPE-γCD was assessed using nude mice subcutaneous xenografts and tail vein injection models, respectively. Balb/c nude mice (4-weeks old, female) were bought from Nihon Clea (Tokyo, Japan). Animals were acclimatized in the laboratory for one week. Cells were injected subcutaneously (1 X 107 suspended in 0.2 ml of growth medium) into the abdomen of nude mice and into tail vein (1 X 106 suspended in 0.2 ml of growth medium). CAPE or CAPE-γCD was administered (either intraperitoneal or oral route, as indicated) every alternate day starting one day after injection of cells. Tumor formation and body weight of mice were monitored every alternate day. Volume of the subcutaneous tumors was calculated as V = L x W2/2, where L was length and W was the width of the tumor, respectively. Statistical significance of the data was calculated from three independent experiments (n=3 per experiment). For metastasis assay, the mice were sacrificed 30 days post-injection and screened for the presence of tumors. Number of lung tumors in control and treated mice were recorded. All procedures were carried out in accordance with the Animal Experiment and Ethics Committee, Safety and Environment Management Division, National Institute of Advanced Industrial Science & Technology (AIST), Tsukuba, Japan.
Results
Anticancer and anti-metastasis activity of CAPE and CAPE-gamma CD conjugate
We examined the cytotoxicity of CAPE to a variety of human cancer cell lines including A549, HT1080, G361, U2OS, MCF-7 and MDA-MB-231, and found that it is cytotoxic to all cancer cells in a range of 5-100 μM (Fig. 1A, B). Whereas most of them showed IC50 of 10-20 μM, A549 showed remarkable resistance (IC50-100 μM) and MCF-7 showed high sensitivity (IC50-5 μM) (Fig. 1B). In order to determine if CAPE could be used for treatment of aggressive and metastatic cancers, we used genetically isogenic breast cancer cells that differed in their cell migration and metastasis potentials.
Cytotoxicity of CAPE to human cancer cells. Dose dependent loss in cell viability in cells treated with CAPE is shown. IC50 for A549, HT1080, G361 and U20S were ~100, 5, 20, 60 μM respectively (A). Effect of CAPE on cell viability (B) and colony forming efficiency (C) of MCF-7, MDA-MB-231 cells and their mortalin-overexpressing metastatic deriatives showing that CAPE inhibited cell proliferation in both short (A) and long term (B) viability assays. (D) Cell cycle analysis of control and CAPE-treated cells showing G1 arrest both in MCF-7 and MDA-MB-231 treated with low dose (2 μM and 10 μM CAPE, respectively) of CAPE. (E) High dose (10 μM and 40 μM CAPE, respectively) caused apoptosis.
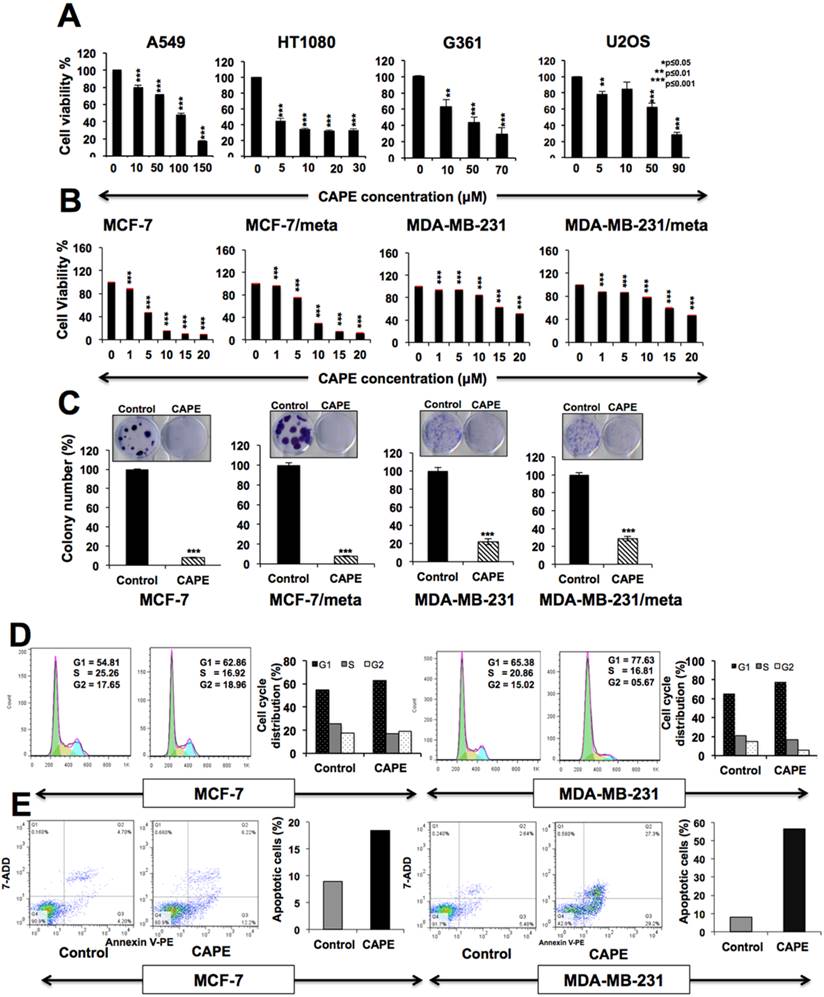
These were generated by retrovirus-mediated overexpression of stress chaperone mortalin [45,55]. In short- and long-term survival assays, we found that the CAPE inhibited the growth of parent as well as their metastatic-derivatives to a comparable level (Figs. 1B and 1C). Furthermore, we subjected control and CAPE (low and high dose) treated MCF-7 and MDA-MB-231cells to FACS analysis and found that whereas low dose (1-2 μM and 5-10 μM, respectively) of CAPE caused G1 arrest of cell cycle, high dose (5-10 μM and 25-50 μM, respectively) treatment resulted in apoptosis (Figs. 1D and 1E). Similar results were obtained in both parent and metastatic breast cancer cell lines suggesting that CAPE may also be used for treatment of aggressive and malignant cells.
In order to investigate the molecular mechanism of CAPE-induced cytotoxicity, we took time-lapse observations of control and CAPE-treated cells. MCF-7 cells when treated with CAPE (10 μM) showed apoptosis at about 24 h. However, dividing cells appeared at about 48-60 h and overtook the apoptotic cell population (Fig. 2A and data not shown) suggesting a short-term effect of CAPE that might be due to its unstable characteristic [56-58]. We confirmed this by regular (every 24 h) change of cell culture medium and with fresh addition of CAPE. With this treatment regime, growth arrest/apoptosis (10 μM was sustained (Fig. 2A and data not shown). The data confirmed that short-term effect of CAPE was due to, at least in part, its unstable characteristic. CAPE possesses reactive sites such as ester bond, α and β-unsaturated carbonyl group and catechol group that accounts for its instability in physiological conditions causing its hydrolysis, Michael addition and oxidation (Fig. 2B).
Instability of CAPE in cell culture medium and its complex with γCD. (A) Phase contrast images of MCF-7 cells treated with CAPE either one time or regularly (every 24 h) for total of 72 h. Whereas healthy dividing cells appeared in culures treated for only once, the regularly treated cultures showed growth arrest/apoptosis. (B) Reactive sites of CAPE, such as ester bond, α, β-unsaturated carbonyl and catechol group, that accounts for its unstability are shown. (C) The binding constant (K) for the complexation of CAPE with βCD, as by a UV/Vis-spectroscopic-titration method is shown. The analysis of the titration curve revealed 1:1 complexation provided a K value of (2.52 ± 0.04) x 103 M-1 (inset). (D) Binding constant (K) values of complex of CAPE with αCD or γCD.
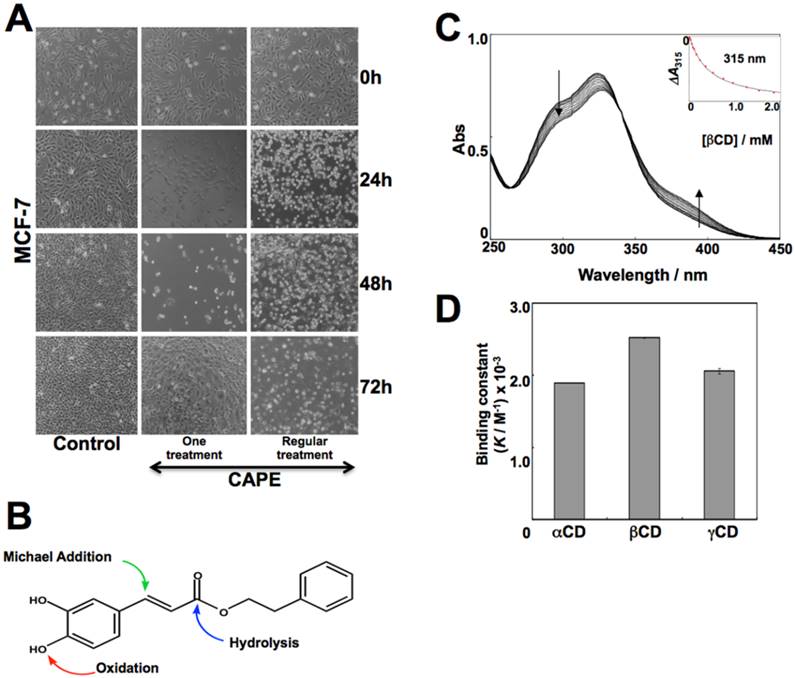
In order to increase the stability of CAPE, we made its complex with γCD in molar ratios of 1:1. The binding constant (K) for the CAPE-γCD complex was determined in 1 X 10-1 M phosphate buffer at pH 7.4 and 25oC by UV/Vis-spectroscopic-titration method. The spectral change in CAPE, endorsing formation of its complex with γCD, was first recorded (Fig. 2C). The analysis of the titration curve with a theoretical equation for the 1:1 conjugation provided a K value of (2.52 ± 0.04) x 103 M-1 (Fig. 2C inset). Similar K values were obtained for the conjugation of CAPE and αCD or γCD (Fig. 2D). The thermostability of CAPE and CAPE-γCD was examined by incubating them at 140oC for 24 h. As expected, color change and melting were observed in the case of CAPE. On the other hand, CAPE-γCD retained its solid state and showed only a minor change in color. The enhanced thermostability of the CAPE-γCD was also confirmed by HPLC-based quantitation of CAPE in heat-treated samples (Fig. 3A).
Characterization of CAPE-γCD complex and its stability to various adverse conditions. (A) Thermostability analyis at 140oC at 24 h showing color change and melting of CAPE, but not CAPE-γCD. The enhanced thermostability was also observed from the analysis of contents of CAPE in heat-treated samples by HPLC. (B) Stability of CAPE and CAPE-γCD against hydrolysis by α-Chymotrypsin (ChT) for 3 h at pH 7.4 and 25oC. Decrease in CAPE and increase in caffeic acid were concurrently observed (a). CAPE-γCD indicated much higher stability than CAPE. Stabilization of CAPE against Michael 1,4-addition reaction with cysteine by γCD (is shown (b). (C) Stability of CAPE and CAPE-γCD against oxidation as monitored by addition of potassium iodate and UV/Vis-spectroscopy. Large proportion of CAPE (80%) reacted with potassium iodate in incubation. In case of CAPE-γCD, 40% CAPE reacted and 60% was protected in the same condition.
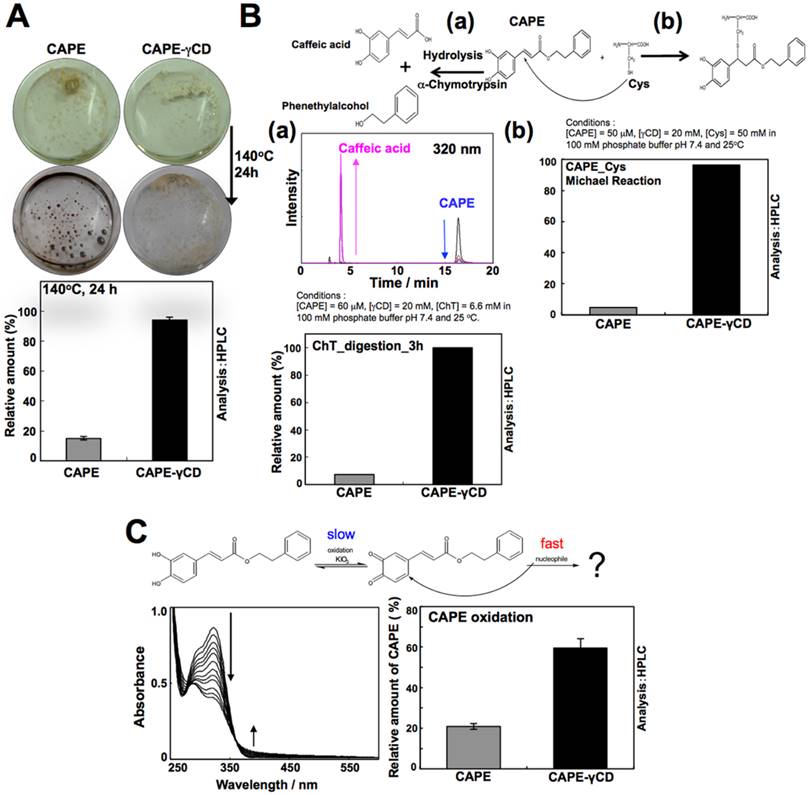
To examine the stability of CAPE and CAPE-γCD against hydrolysis by α-Chymotrypsin (ChT), a protease secreted by pancreas, CAPE (50 μM) and ChT were incubated for 3 h at pH 7.4 and 25oC in the absence or presence of γCD (20 μM). As shown in Fig. 3B, decrease in CAPE and increase in caffeic acid were monitored concurrently by HPLC (Fig. 3B-a). The data showed that CAPE undergoes hydrolysis by α-Chymotrypsin (digestive enzyme present in gut). Of note, CAPE-γCD showed resistance to such digestion by ChT indicating its stability. Similar results were observed in assays for testing the stability of CAPE and CAPE-γCD in Michael 1,4-addition reaction with cysteine (Fig. 3B-b). To examine the stability of CAPE and CAPE-γCD against oxidation, potassium iodate (an established oxidant) was added to CAPE (50 μM) solution in the absence or presence of γCD (20 mM). The oxidation reaction was monitored by UV/Vis-spectroscopy (Fig. 3C). Generation of quinone derivative, as usually indicated by increase in the peak (around 400 nm), was not observed. Therefore, observed time-dependent spectral change in CAPE in this condition presumably indicated rapid Michael 1,4-addition reaction of quinone derivative with some nucleophile, for example H2O or OH- following oxidation reaction as rate limiting step. Noticeably, whereas 80% of CAPE in the free form, reacted with potassium iodate in 3 h, only 40% reacted when in complex with γCD under the same condition of experiment suggesting that γCD could stabilize CAPE against oxidation (Fig. 3C).
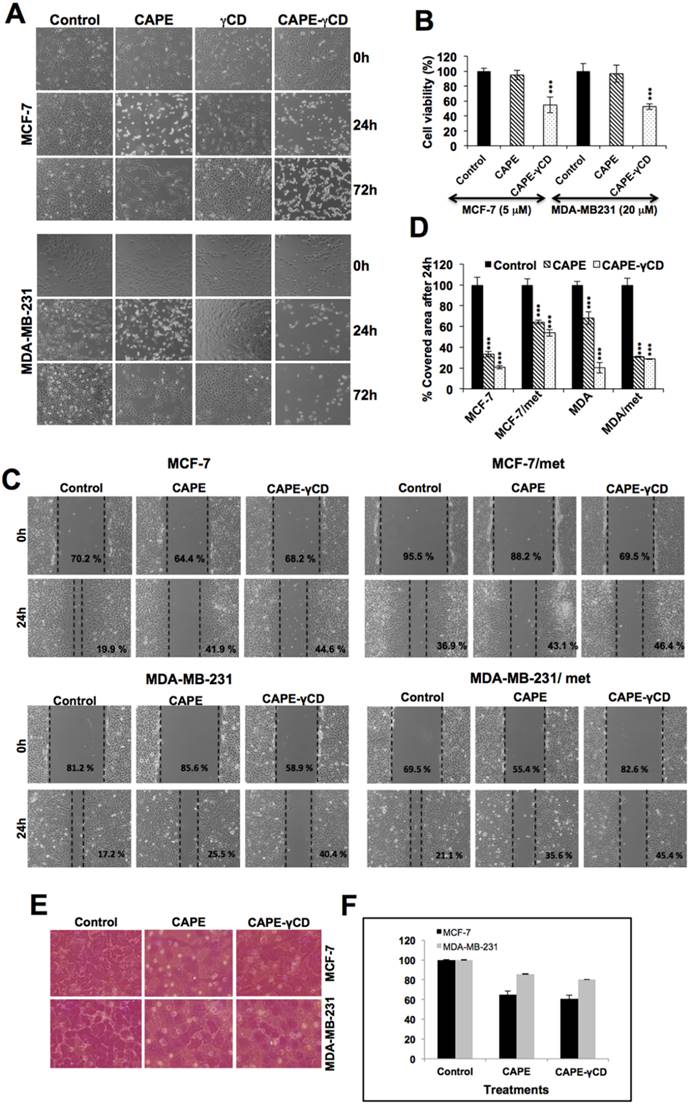
Equivalent doses of CAPE in its free form and in complex with γCD were next used for cell viability studies. As shown in Fig. 4A, wherein γCD by itself was non-toxic to cells, CAPE showed short-term toxicity, CAPE-γCD complex showed sustained growth arrest or apoptosis suggesting that CAPE-γCD is potent than CAPE. Quantitative cell viability analysis also revealed that the equivalent dose of CAPE in its complex with γCD was more cytotoxic as compared to its free form. Similar results were obtained in the breast cancer cell lines and their metastatic derivatives (Figs. 4A and 4B, and data not shown). We next used sub-toxic doses of CAPE and examined their effect on cell migration (Fig. 4C). Wound-scratch and invasion assays on MCF-7 and MDA-MB-231 cells and their metastatic mortalin-overexpressing derivatives revealed that CAPE-γCD complex possessed stronger anti-migration activity (Figs. 4C and 4D). Similar result was obtained in cell invasion assays (Figs. 4E and 4F).
Anticancer potential of CAPE and CAPE-γCD complex. (A) Effect of CAPE, γCD and CAPE-γCD on viability, migration and invasion of MCF7 and MDA-MB-231 cells. Phase contrast images (A), cell viability (B) migration in wound scratch (C&D) and cell invasion assay (E&F) are shown.
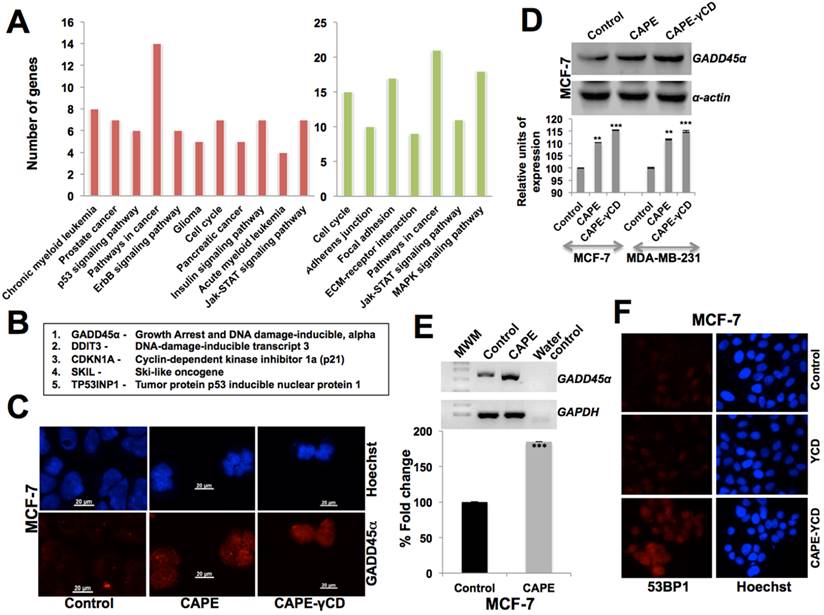
CAPE causes upregulation of DNA damage response
In order to determine the molecular mechanism of CAPE-induced growth arrest and anti-migration activity, we subjected the control and CAPE (10 μM)-treated MCF-7 cells to cDNA array analysis. We found that the CAPE caused upregulation and downregulation of several proteins at the transcription level. The top upregulated signaling pathways were involved in cell cycle arrest and negative regulation of cell cycle that included upregulation of GADD45α (Growth Arrest and DNA Damage-inducible, alpha), CDKN1A (Cyclin-dependent kinase inhibitor 1a (p21), DDIT3 (DNA-Damage-Inducible Transcript 3), SKIL (Ski-Like oncogene, TP53INP1 (Tumor protein p53 inducible nuclear protein 1, GATA3 (Gata binding protein 3) and RHOB (Ras homolog family member b) (Figs. 5A and 5B). In light of this data, we performed expression analysis of GADD45α both at protein and mRNA level. Western blotting and immunostaining revealed an upregulation of GADD45α in CAPE-treated cells (Fig. 5C and 5D). Furthermore, there was two-fold increase in GADD45α transcript in treated cells, suggesting that CAPE may trigger the DNA damage response in cancer cells (Fig. 5E). It was further confirmed by 53BP1 and γΗ2ΑX staining that revealed upregulation in CAPE, CAPE-γCD, but not in the γCD treated cells (Fig. 5F and data not shown).
Characterization of CAPE cytotoxicity by cDNA array analysis. (A) Pathways derived from cDNA array analysis; genes upregulated (Red) and downregulated (green) in CAPE treated as compared to the control cells. (B) Genes upreguleated in CAPE treated cells as determined by cDNA array analysis. (C) Immunostaining of GADD45α in control and CAPE treated cells showing increase in the latter. Increase in GADD45α in CAPE and CAPE-γCD treated cells detected by Western blotting (D) and RT-PCR. (E) 53BP1 immunostaining showing increase in CAPE-γCD tretaed cells.
CAPE causes upregulation of p53 function by targeting mortalin-p53 interactions
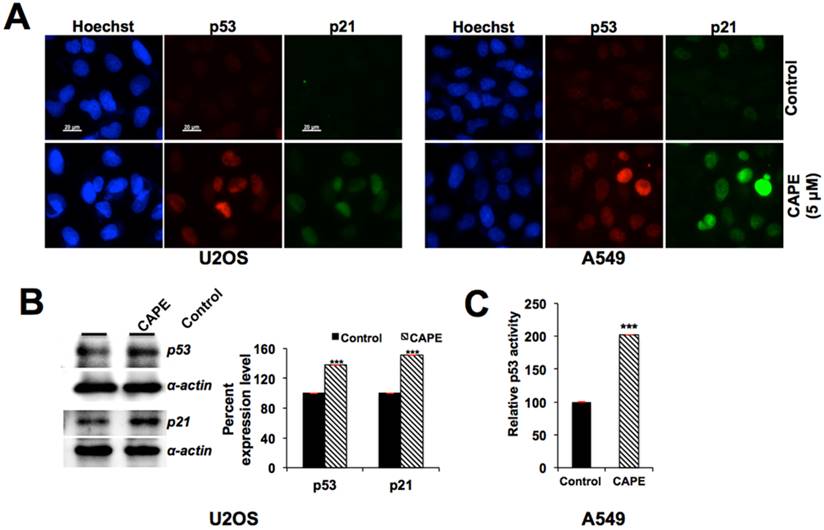
Based on the fact that GADD45α is a downstream effector of p53 tumor suppressor, involved in growth arrest of cells in response to DNA damage, we hypothesized that CAPE-induced increase in GADD45α might be mediated by activation of p53. Examination of p53 in control and CAPE-treated cells indeed revealed its upregulation and nuclear translocation of p53 and increase in its downstream effector p21WAF1 in MCF-7, U2OS as well as A549 cells (Fig. 6A and data not shown). Increase in p53 and p21WAF1 in treated cells was confirmed by Western blotting (Fig. 6B). Furthermore, p53-dependent reporter assays revealed increase in transcriptional activation function of p53 in CAPE-treated cells (Fig. 6C). We next predicted that the increase in p53 might be due to targeting of mortalin, a stress chaperone known to inhibit p53 activities [45,55,59-62] by CAPE. Using bioinformatics and molecular docking tools, we examined whether CAPE could bind to mortalin and interfere with mortalin-p53 interaction as follows.
Activation of p53-p21 pathway by CAPE. (A) Immunostaining of p53 and p21 in control and CAPE treated cells showing their increase. (B) Increase in p53 and p21 as determined by Western blotting with specific antibodies. (C) p53-dependent reporter assay showing increase in transcriptional activation function of p53 in CAPE-treated than in control cells.
The 3D structure of CAPE was docked at the desired grid coordinates of the protein molecule using Lamarckian Genetic Algorithm (GA) of AutoDock Tools. The compound showed good binding affinity with the p53-binding domain of mortalin possessing a docking score of -5.72 kcal/mol, i.e. binding energy of -23.93 kJ/mol approximately. The residues from 258 to 282 of mortalin are involved in interaction with p53. CAPE was found interacting with many of these important residues via hydrogen bonds and hydrophobic interactions (Fig. 7A). Nitrogen (NZ) of residue Lys 135 formed two hydrogen bonds of 2.59 Å and 3.32 Å with third and fourth oxygen atom of CAPE respectively while oxygen atom (OG1) of Thr 271 formed one H-bond of 2.61 Å with third oxygen of CAPE. The residues participating in maintaining a hydrophobic contact with CAPE involved Tyr 196, Phe 250, Asp 251, Thr 267, Asn 268, Gly 269, Asp 270 and Phe 272. Since the ligand molecule CAPE was occupying the mortalin cavity where p53 binds, it was strongly suggestive that mortalin-CAPE interaction would hinder the mortalin-p53 interface and thereby inhibit sequestration of p53 in the cytoplasm resulting into its nuclear translocation and activation. The maestro pose viewer file obtained after docking of CAPE with mortalin was used as an input for MM-GBSA analysis. Using the MM-GBSA method on the docked complex, free energy of binding (ΔGbinding) was calculated using the following formula: ΔGbinding = Energy of the minimized complex - (Energy of the minimized ligand + Energy of the minimized receptor). The free energy of binding for the docked complex by MM-GBSA method was estimated to be -9.74 kcal/mol suggesting that binding of CAPE with mortalin is a thermodynamically favorable process.
Molecular docking analysis of CAPE and mortalin. (A) Amino acid residues of mortalin participating in hydrogen bond (pink), hydrophobic and van der Waal interactions (orange) wih CAPE after molecular docking. (B) Graph showing deviation of the docked complex from its initial conformation during the complete simulation run. (C) Molecular interaction pattern between mortalin and CAPE after simulation. Residues were mainly involved in hydrophobic and van der Waal interactions (shown in orange). Change in position of CAPE can be observed. The ligand showed variations in the orientation and hence an average representative structure from the stable time frame was used for interaction analysis. (D) p53 immunostaining of MCF7 control and CAPE treated cells showing intense nuclear staining in the later. (E) Relative amount of p53 in mortalin-p53 complex in control and CAPE-treated cells showing decrease in mortalin-p53 complex in the latter.
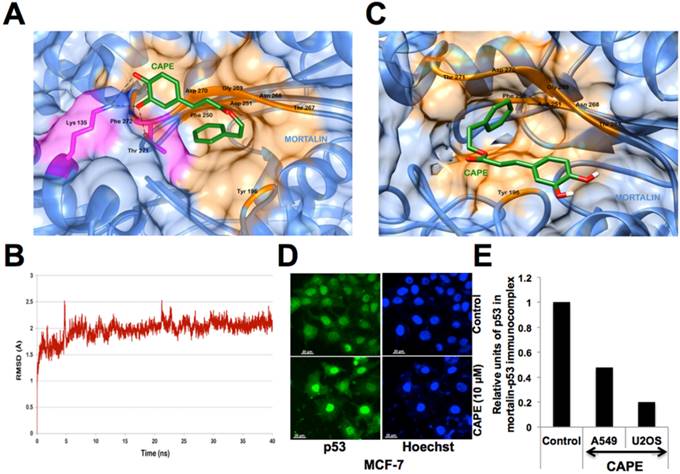
Changes in interaction pattern were revealed by thermodynamic stability analysis of mortalin-CAPE complex in a 40 ns long MD simulation run. The protein backbone in the complex deviated about 2.0 Å from its initial state during the first 5 ns of the simulation run and then attained a stable conformation for the rest 35 ns (Fig. 7B). Figure 7C illustrates the change in position of CAPE during the course of a 40 ns simulation run. Even though the location of ligand was same, it attained different conformations to reach its final energetically stable state. The frame representing the average of all the frames for the last 30 ns was obtained and used for further analysis. It was observed that the ligand moved deeper into one of the buried cavity of the protein structure. CAPE was stably interacting with the protein with the help of hydrophobic and van der Waal interactions. The amino acid residues of mortalin involved in this binding included Tyr 196, Phe 250, Asp 251, Thr 267, Asn 268, Gly 269, Asp 270, Thr 271 (Fig. 7C). Many of these residues were interacting even before the MD simulations and are a part of crucial p53 binding domain of mortalin. The free energy of binding was calculated and estimated to be -17.87 kcal/mol for post MD complex endorsing that binding of CAPE with mortalin protein is a thermodynamically favorable process. Based on this data, it was strongly predicted that CAPE targets mortalin-p53 complex and causes reactivation of p53 function as shown in Fig. 6. Furthermore, we performed validation of this prediction by co-immunostaining and co-immunoprecipitation of mortalin and p53 in control and CAPE-treated cells. As compared to the control, CAPE treated cells showed intense nuclear staining (Fig. 7D) and possessed decreased amount of p53 in mortalin-p53 complexes (Fig. 7E and data not shown).
CAPE causes downregulation of metastasis signaling
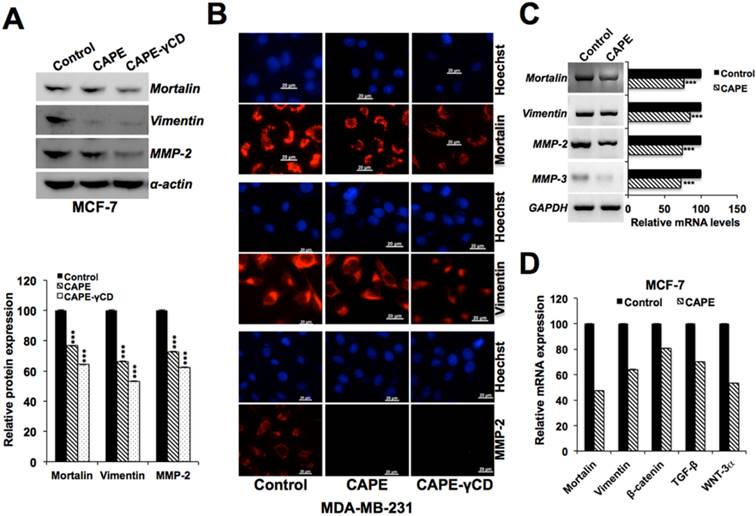
Mortalin is enriched in cancer cells, and has been shown to promote carcinogenesis and metastasis signaling [45,55,59-62]. We next examined the expression level of mortalin in control and CAPE-treated cells. As shown in Fig. 8A-C, there was a decrease in mortalin at both protein and transcript level in CAPE-treated cells. We examined the expression level of key regulators of cell migration in these cells and found decrease in vimentin, MMP-2, MMP-3, β-catenin, TGF-β and WNT-3α transcripts (Figs. 8C and 8D). Of note, CAPE-γCD treated cells showed higher decrease (Figs. 8A and 8B).
Downregulation of mortalin and other metastasis-regulatory proteins by CAPE. Effect of CAPE on mortalin and cell migration regulatory proteins. Decrease in mortalin, vimentin, MMP-2 was obserevd by Western blotting (A), immunostaining (B) and RT-PCR (C). Decrease in mortalin, vimentin, β-catenin, TGF-β and Wnt-3α as determined by qPCR is shown (D).
In vivo tumor suppressor and anti-metastasis activities of CAPE and CAPE-γCD complex
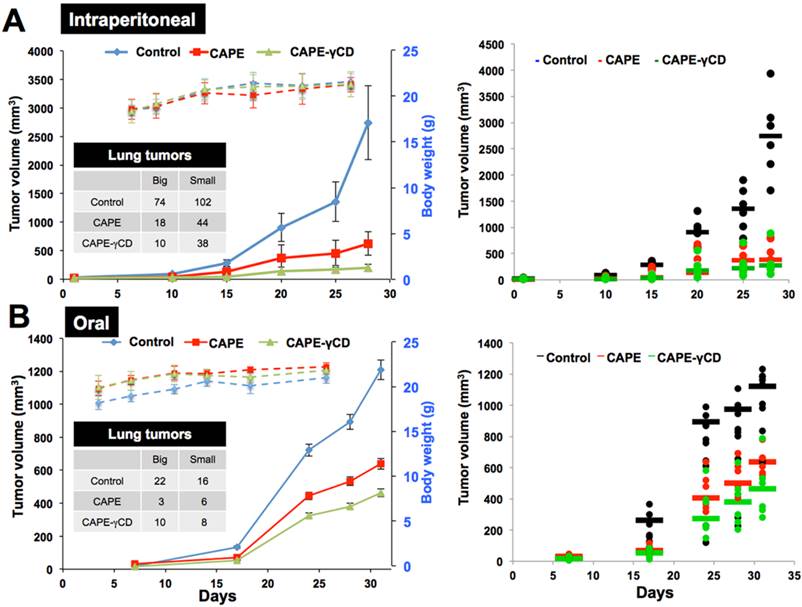
We finally investigated anticancer and anti-metastasis potential of CAPE and CAPE-γCD complex in in vivo tumor assays in nude mice. An established model cell line (human fibrosarcoma, HT1080) was used for subcutaneous xenografts (for tumor growth) and tail vein injections (lung metastasis) (Fig. 9). CAPE showed significant tumor suppression in subcutaneous xenografts. Whereas γCD itself did not show any effect, CAPE-γCD showed stronger activity than CAPE when administration either by intraperitonial injections or gavage. Examination of lung metastasis in control, CAPE and CAPE-γCD treated mice revealed stronger anti-metastasis activity by CAPE-γCD complex. In both routes of administration, whereas CAPE-treated mice showed significant reduction in lung metastasis there was significantly enhanced effect in response to treatment with CAPE-γCD.
In vivo anti-tumor and anti-metastasis activties of CAPE and CAPE-γCD complex. Effect of CAPE and CAPE-γCD on in vivo progression of tumors generating either by subcutaneous xenografts or tail vein injections of HT1080 in nude mice. Tumor volumes showed decrease in mice treated with CAPE and CAPE-γCD complex (either peritoneal injections or gavage). CAPE-γCD treated mice showed higher degree of tumor suppression in both models. No effect on the body weight was observed.
Physicochemical properties and pharmacokinetics of CAPE
QikProp predicts physically significant descriptors and pharmaceutically relevant properties of organic molecules. It also provides significant ranges for comparing these molecular properties with those of 95% of already known pharmaceutical drugs. It gives a descriptor “#star” which denotes the number of properties of the organic molecule which do not fall within the range of values for same properties of already known drugs. According to this predictive algorithm, lower the number better is the drug-likeness of the small molecule. The #star value for CAPE was 0. Hence, none of the computed property lied outside the required range and was very similar to that of the known drugs. Lipinski's rule of five is a thumb rule that determines the likeliness of being a drug using four molecular properties. CAPE with a molecular mass of 284.31 g/mol, 2 hydrogen bond donors, 3.5 hydrogen bond acceptors and an octanol/water partition coefficient of 3.18 full-filled all the conditions of rule of five.
Solvent accessible surface area (SASA) and especially polar surface area (PSA) is known to have a good correlation with the passive molecular transport through membranes and therefore allows estimation of transport properties for drugs. The total SASA for CAPE was 607.45 Å2 with hydrophobic component of 97.271 Å2 and hydrophilic component of 162.901 Å2. These values were well within the range given by QuickProp. It also calculates the percentage probability of the drug getting orally absorbed in the human body using some knowledge based set of rules. This value has been shown to correlate well with the human oral absorption. CAPE showed high oral absorption with a percentage value of 89.30%. It was also predicted to be inactive against the central nervous system. Some other toxicity related effects were predicted using an online free platform, AdmetSAR. Blood brain barrier (BBB) is a regulatory system that separates the brain environment from the direct contact of circulatory system thereby protecting the brain for unwanted solute particles. CAPE was shown to be BBB negative ensuring its administration safe for the brain. Induction of the P-glycoprotein (P-gp), an efflux transporter, leads to reduced bioavailability of orally administered drugs and can therefore decrease is therapeutic efficacy. Alternatively, its inhibition can also result in increased bioavailability, thus leading to an increased risk of adverse side effects. Thus, in general a drug should be neither a substrate nor an inhibitor of P-gp. It is expected that P-gp would not act as a barrier for the systemic exposure of CAPE as it was predicted to be a non-substrate and non-inhibitor of P-gp. It was also shown to be non-carcinogenic and readily biodegradable with some levels of toxicity in Tetrahymena pyriformis, fish and honeybee.
Discussion
Anti-tumor and anti-metastatic potency of CAPE was increased by γCD
The in vitro cytotoxicity assays of CAPE on a variety of cancer cell lines revealed that it possesses anticancer activity; the range of cytotoxic doses for different cancer cells lines was consistent with other reports [15,18,26,29,41,58]. Taken together, it was suggestive that the toxicity of CAPE is widespread rather than being specific to certain cancer cell lines. Furthermore, by using metastatic cells, we found that CAPE is effective not only for inhibiting the growth of cancer cells but also for metastasis (Figs. 4, 8 and 9). Computational and molecular analyses revealed that CAPE targets mortalin-p53 interactions that are specific to cancer cells [45,46] and hence may account for, at least in part, for selective toxicity of CAPE to cancer cells in several earlier studies [17-20]. It has been earlier shown that mortalin interacts and inactivates wild as well as mutant p53 proteins. Abrogation of these interactions resulted in activation of growth arrest and apoptotic activities of wild and mutant p53 proteins, respectively [60,61]. Taken together, it is suggestive that CAPE could be used for treatment of both wild type as well as mutant p53 harboring cancer cells. Furthermore, CAPE-treated cells showed down-regulation of mortalin, that is enriched in cancer cells and has been shown to contribute to carcinogenesis and metastasis, suggesting that it is an anti-mortalin molecule suitable for cancer and metastatic cell therapy [55,59-62].
Based on the reports that the bioavailability of several lipophilic ingredients, such as coenzyme Q10, tocotrienol and curcumin, could be enhanced by their conjugation/complex with γCD, Terao and co-workers reported that such enhancement of bioavailability was due to increase in water solubility of the lipophilic ingredients by γCD in micelle solution [63]. Sodium taurocholate (Na TCA), a major component of bile acid, in small intestine was also shown to increase of water solubility of CAPE-γCD by similar mechanism [63]. Taken together, these data revealed that the instability of CAPE could be compensated by generation of its complex with γCD leading to its increased solubilization, protection and enhanced bioavailability. We next investigated the hypothesis by in vitro and in vivo experiments.
The mechanisms of the anti-tumor activity in CAPE have not been clarified. It was shown to suppress 12-O-tetradecanoylphorbol-13-acetate-induced cell transformation and induce apoptosis in mouse epidermal cells through p53-dependent and p53-independent pathways [64]. It was shown to inhibit production of nitric oxide and iNOS protein expression (involved in inflammatory and autoimmune-mediated tissue destruction) induced by lipopolysaccharide (LPS) plus interferon-gamma (IFN-gamma), and cause anti-inflammatory, anti-viral and anticancer effects [65], CAPE treatment-induced cell cycle arrest and growth inhibition were shown to be mediated via activation of Skp2, p53, p21Cip1 and p27Kip1 proteins [66]. It was shown to inhibit NFkB, activate MAPK family proteins p38 and JNK resulting in apoptosis [67]. Anti-metastasis activity of CAPE (decreased expression of matrix metalloproteinases) was supported by earlier studies [68-71]. Taken together and in addition to these reports, we demonstrate that CAPE targets stress chaperone mortalin leading to (i) activation of growth arrest through p53-GADD45α-p21 signaling and (ii) suppression of cell migration through matrix metalloproteinases. Furthermore, such activities could significantly be enhanced by CAPE-γCD complex that protected it from degradation. In in vivo antitumor assays, CAPE-γCD complex (used either by intraperitoneal injections or by oral feeding) resulted in better anti-tumor outcomes in in vivo assays and hence it is proposed as a potent anticancer reagent.
Acknowledgements
The authors thank Navjot Shah, Tomoko Nakamoto and Tomomi Toda for technical assistance and Nashi Widodo for analysis of cDNA array data.
Authors' Contributions
Conception and design: RW, SCK, KT
Acquisition of data (provided animals, acquired, provided facilities, etc.): NN, YI, SG, PB, AG
Writing, review, and/or revision of the manuscript: RW, PB, YI, AG, JKG, DS, SCK,
Administrative, technical, or material support (i.e., reporting or organizing data): RW, NN, YI
Competing Interests
The authors have declared that no competing interest exists.
References
1. Han Y, Zhang Y, Jia T, Sun Y. Molecular mechanism underlying the tumor-promoting functions of carcinoma-associated fibroblasts. Tumour Biol. 2015;36:1385-94
2. Javelaud D, Alexaki VI, Dennler S, Mohammad KS, Guise TA, Mauviel A. TGF-beta/SMAD/GLI2 signaling axis in cancer progression and metastasis. Cancer Res. 2011;71:5606-10
3. Weissmueller S, Manchado E, Saborowski M, Morris JPt, Wagenblast E, Davis CA. et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell. 2014;157:382-94
4. Gao D, Vahdat LT, Wong S, Chang JC, Mittal V. Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer Res. 2012;72:4883-9
5. Saitoh M. The epithelial-mesenchymal transition is regulated at the post-transcriptional levels by TGF-beta signaling during tumor progression. Cancer Sci. 2015;106:481-8
6. Drake LE, Macleod KF. Tumour suppressor gene function in carcinoma-associated fibroblasts: from tumour cells via EMT and back again? J Pathol. 2014;232:283-8
7. Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW. et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nature Cell Biol. 2011;13:317-23
8. Jagtap S, Meganathan K, Wagh V, Winkler J, Hescheler J, Sachinidis A. Chemoprotective mechanism of the natural compounds, epigallocatechin-3-O-gallate, quercetin and curcumin against cancer and cardiovascular diseases. Current Med Chem. 2009;16:1451-62
9. Bilecova-Rabajdova M, Birkova A, Urban P, Gregova K, Durovcova E, Marekova M. Naturally occurring substances and their role in chemo-protective effects. Central Eur J Public Health. 2013;21:213-9
10. Nabekura T. Overcoming multidrug resistance in human cancer cells by natural compounds. Toxins (Basel). 2010;2:1207-24
11. Sinha D, Biswas J, Sung B, Aggarwal BB, Bishayee A. Chemopreventive and chemotherapeutic potential of curcumin in breast cancer. Current Drug Targets. 2012;13:1799-819
12. Desai AG, Qazi GN, Ganju RK, El-Tamer M, Singh J, Saxena AK. et al. Medicinal plants and cancer chemoprevention. Current Drug Metabol. 2008;9:581-91
13. Shukla Y, Singh R. Resveratrol and cellular mechanisms of cancer prevention. Ann N Y Acad Sci. 2011;1215:1-8
14. Singh M, Suman S, Shukla Y. New Enlightenment of Skin Cancer Chemoprevention through Phytochemicals: In Vitro and In Vivo Studies and the Underlying Mechanisms. Biomed Res Int. 2014:243452
15. Grunberger D, Banerjee R, Eisinger K, Oltz EM, Efros L, Caldwell M. et al. Preferential cytotoxicity on tumor cells by caffeic acid phenethyl ester isolated from propolis. Experientia. 1988;44:230-2
16. Mahmoud NN, Carothers AM, Grunberger D, Bilinski RT, Churchill MR, Martucci C. et al. Plant phenolics decrease intestinal tumors in an animal model of familial adenomatous polyposis. Carcinogenesis. 2000;21:921-7
17. Su ZZ, Lin J, Prewett M, Goldstein NI, Fisher PB. Apoptosis mediates the selective toxicity of caffeic acid phenethyl ester (CAPE) toward oncogene-transformed rat embryo fibroblast cells. Anticancer Res. 1995;15:1841-8
18. Lee YJ, Liao PH, Chen WK, Yang CY. Preferential cytotoxicity of caffeic acid phenethyl ester analogues on oral cancer cells. Cancer Lett. 2000;153:51-6
19. Lee YT, Don MJ, Hung PS, Shen YC, Lo YS, Chang KW. et al. Cytotoxicity of phenolic acid phenethyl esters on oral cancer cells. Cancer Lett. 2005;223:19-25
20. Su ZZ, Lin J, Grunberger D, Fisher PB. Growth suppression and toxicity induced by caffeic acid phenethyl ester (CAPE) in type 5 adenovirus-transformed rat embryo cells correlate directly with transformation progression. Cancer Res. 1994;54:1865-70
21. Burke TR Jr, Fesen MR, Mazumder A, Wang J, Carothers AM, Grunberger D. et al. Hydroxylated aromatic inhibitors of HIV-1 integrase. J Med Chem. 1995;38:4171-8
22. Chen CN, Wu CL, Lin JK. Propolin C from propolis induces apoptosis through activating caspases, Bid and cytochrome c release in human melanoma cells. Biochem Pharmacol. 2004;67:53-66
23. Lin J, Su Z, Grunberger D, Zimmer S, Fisher P. Expression of the transformed phenotype induced by diverse acting viral oncogenes mediates sensitivity to growth suppression induced by caffeic Acid phenethyl ester (cape). Int J Oncol. 1994;5:5-15
24. Demestre M, Messerli SM, Celli N, Shahhossini M, Kluwe L, Mautner V. et al. CAPE (caffeic acid phenethyl ester)-based propolis extract (Bio 30) suppresses the growth of human neurofibromatosis (NF) tumor xenografts in mice. Phytotherapy Res. 2009;23:226-30
25. Messerli SM, Ahn MR, Kunimasa K, Yanagihara M, Tatefuji T, Hashimoto K. et al. Artepillin C (ARC) in Brazilian green propolis selectively blocks oncogenic PAK1 signaling and suppresses the growth of NF tumors in mice. Phytotherapy Res. 2009;23:423-7
26. Frenkel K, Wei H, Bhimani R, Ye J, Zadunaisky JA, Huang MT. et al. Inhibition of tumor promoter-mediated processes in mouse skin and bovine lens by caffeic acid phenethyl ester. Cancer Res. 1993;53:1255-61
27. Huang MT, Ma W, Yen P, Xie JG, Han J, Frenkel K. et al. Inhibitory effects of caffeic acid phenethyl ester (CAPE) on 12-O-tetradecanoylphorbol-13-acetate-induced tumor promotion in mouse skin and the synthesis of DNA, RNA and protein in HeLa cells. Carcinogenesis. 1996;17:761-5
28. Xiang D, Wang D, He Y, Xie J, Zhong Z, Li Z. et al. Caffeic acid phenethyl ester induces growth arrest and apoptosis of colon cancer cells via the beta-catenin/T-cell factor signaling. Anticancer Drugs. 2006;17:753-62
29. Beltran-Ramirez O, Aleman-Lazarini L, Salcido-Neyoy M, Hernandez-Garcia S, Fattel-Fazenda S, Arce-Popoca E. et al. Evidence that the anticarcinogenic effect of caffeic acid phenethyl ester in the resistant hepatocyte model involves modifications of cytochrome P450. Toxicol Sci. 2008;104:100-6
30. Wu CS, Chen MF, Lee IL, Tung SY. Predictive role of nuclear factor-kappaB activity in gastric cancer: a promising adjuvant approach with caffeic acid phenethyl ester. J Clin Gastroenterol. 2007;41:894-900
31. Wang D, Xiang DB, He YJ, Li ZP, Wu XH, Mou JH. et al. Effect of caffeic acid phenethyl ester on proliferation and apoptosis of colorectal cancer cells in vitro. World J Gastroenterol. 2005;11:4008-12
32. He YJ, Liu BH, Xiang DB, Qiao ZY, Fu T, He YH. Inhibitory effect of caffeic acid phenethyl ester on the growth of SW480 colorectal tumor cells involves beta-catenin associated signaling pathway down-regulation. World J Gastroenterol. 2006;12:4981-5
33. Lee YJ, Kuo HC, Chu CY, Wang CJ, Lin WC, Tseng TH. Involvement of tumor suppressor protein p53 and p38 MAPK in caffeic acid phenethyl ester-induced apoptosis of C6 glioma cells. Biochem Pharmacol. 2003;66:2281-9
34. McEleny K, Coffey R, Morrissey C, Fitzpatrick JM, Watson RW. Caffeic acid phenethyl ester-induced PC-3 cell apoptosis is caspase-dependent and mediated through the loss of inhibitors of apoptosis proteins. BJU International. 2004;94:402-6
35. Lee KW, Chun KS, Lee JS, Kang KS, Surh YJ, Lee HJ. Inhibition of cyclooxygenase-2 expression and restoration of gap junction intercellular communication in H-ras-transformed rat liver epithelial cells by caffeic acid phenethyl ester. Ann N Y Acad Sci. 2004;1030:501-7
36. Jaiswal AK, Venugopal R, Mucha J, Carothers AM, Grunberger D. Caffeic acid phenethyl ester stimulates human antioxidant response element-mediated expression of the NAD(P)H:quinone oxidoreductase (NQO1) gene. Cancer Res. 1997;57:440-6
37. Weyant MJ, Carothers AM, Bertagnolli ME, Bertagnolli MM. Colon cancer chemopreventive drugs modulate integrin-mediated signaling pathways. Clin Cancer Res. 2000;6:949-56
38. Na HK, Wilson MR, Kang KS, Chang CC, Grunberger D, Trosko JE. Restoration of gap junctional intercellular communication by caffeic acid phenethyl ester (CAPE) in a ras-transformed rat liver epithelial cell line. Cancer Lett. 2000;157:31-8
39. Orsolic N, Sver L, Terzic S, Tadic Z, Basic I. Inhibitory effect of water-soluble derivative of propolis and its polyphenolic compounds on tumor growth and metastasizing ability: a possible mode of antitumor action. Nutrition Cancer. 2003;47:156-63
40. Carrasco-Legleu CE, Sanchez-Perez Y, Marquez-Rosado L, Fattel-Fazenda S, Arce-Popoca E, Hernandez-Garcia S. et al. A single dose of caffeic acid phenethyl ester prevents initiation in a medium-term rat hepatocarcinogenesis model. World J Gastroenterol. 2006;12:6779-85
41. Chiang EP, Tsai SY, Kuo YH, Pai MH, Chiu HL, Rodriguez RL. et al. Caffeic acid derivatives inhibit the growth of colon cancer: involvement of the PI3-K/Akt and AMPK signaling pathways. PLoS One. 2014;9:e99631
42. Terao K, Nakata D, Fukumi H, Schmid G, Arima H, Hirayama F. et al. Enhancement of oral bioavailability of coenzyme Q10 by complexation with γ-cyclodextrin in healthy adults. Nutrition Res. 2006;26:503-8
43. Miyoshi N, Wakao Y, Tomono S, Tatemichi M, Yano T, Ohshima H. The enhancement of the oral bioavailability of γ-tocotrienol in mice by γ-cyclodextrin inclsion. J Nutritional Biochem. 2011;22:1121-6
44. Uchida R, Iwamoto K, Nagayama S, Miyajima A, Okamoto H, Ikuta N. et al. Effect of g-cyclodextrin inclusion complex on the absorption of R-α-lipoic acid in rats. Int J Molecular Sci. 2015;16:10105-20
45. Wadhwa R, Takano S, Kaur K, Deocaris CC, Pereira-Smith OM, Reddel RR. et al. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. International J Cancer. 2006;118:2973-80
46. Chang JT, Nevins JR. GATHER: a systems approach to interpreting genomic signatures. Bioinformatics. 2006;22:2926-33
47. Draghici S, Khatri P, Tarca AL, Amin K, Done A, Voichita C. et al. A systems biology approach for pathway level analysis. Genome Res. 2007;17:1537-45
48. Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27-30
49. Guo Z, Mohanty U, Noehre J, Sawyer TK, Sherman W, Krilov G. Probing the alpha-helical structural stability of stapled p53 peptides: molecular dynamics simulations and analysis. Chem Biology Drug Design. 2010;75:348-59
50. Pikkemaat MG, Linssen AB, Berendsen HJ, Janssen DB. Molecular dynamics simulations as a tool for improving protein stability. Protein Eng. 2002;15:185-92
51. Schneider G, Bohm HJ. Virtual screening and fast automated docking methods. Drug Discovery Today. 2002;7:64-70
52. Du J, Sun H, Xi L, Li J, Yang Y, Liu H. et al. Molecular modeling study of checkpoint kinase 1 inhibitors by multiple docking strategies and prime/MM-GBSA calculation. J Comput Chem. 2011;32:2800-09
53. Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT. et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739-49
54. Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G. et al. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Information Modeling. 2012;52:3099-105
55. Ryu J, Kaul Z, Yoon AR, Liu Y, Yaguchi T, Na Y. et al. Identification and functional characterization of nuclear mortalin in human carcinogenesis. J Biol Chem. 2014;289:24832-44
56. Yang J, Kerwin SM, Bowman PD, Stavchansky S. Stability of caffeic acid phenethyl amide (CAPA) in rat plasma. Biomed Chromatography. 2012;26:594-8
57. Celli N, Dragani LK, Murzilli S, Pagliani T, Poggi A. In vitro and in vivo stability of caffeic acid phenethyl ester, a bioactive compound of propolis. J Agri Food Chem. 2007;55:3398-407
58. Choi D, Han J, Lee Y, Choi J, Han S, Hong S. et al. Caffeic acid phenethyl ester is a potent inhibitor of HIF prolyl hydroxylase: structural analysis and pharmacological implication. J Nutritional Biochem. 2010;21:809-17
59. Kaul SC, Aida S, Yaguchi T, Kaur K, Taira K, Wadhwa R. Activation of wild type p53 function by its mortalin-binding cytoplasmically localizing carboxy-terminus peptides. J Biol Chem. 2005;280:39373-9
60. Lu WJ, Lee NP, Kaul SC, Lan F, Poon RT, Wadhwa R. et al. Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Different. 2011;18:1046-56
61. Lu WJ, Lee NP, Kaul SC, Lan F, Poon RT, Wadhwa R. et al. Induction of mutant p53-dependent apoptosis in human hepatocellular carcinoma by targeting stress protein mortalin. Intl J Cancer. 2011;129:1806-14
62. Na Y, Kaul SC, Ryu J, Lee J, Ahn HM, Kaul Z. et al. Stress chaperone mortalin contributes to epithelial-mesenchymal transition and cancer metastasis. Cancer Res. 2016;76:2754-65
63. Uekaji Y, Jo A, Urano A, Terao K. Application of g-cyclodextrin in nanomedicinal foods and cosmetics. In: (ed.) Bagchi D, Bagchi M, Moriyama H, Shahidi F. Bio-Nanotechnology: A Revolution in Food, Biomedical and Health Sciences. U. K, Wiley & Sons. 2013:179-211
64. Nomura M, Kaji A, Ma W, Miyamoto K, Dong Z. Suppression of cell transformation and induction of apoptosis by caffeic acid phenethyl ester. Mol Carcinogenesis. 2001;31:83-9
65. Song YS, Park EH, Hur GM, Ryu YS, Lee YS, Lee JY. et al. Caffeic acid phenethyl ester inhibits nitric oxide synthase gene expression and enzyme activity. Cancer Lett. 2002;175:53-61
66. Lin HP, Lin CY, Huo C, Hsiao PH, Su LC, Jiang SS. et al. Caffeic acid phenethyl ester induced cell cycle arrest and growth inhibition in androgen-independent prostate cancer cells via regulation of Skp2, p53, p21Cip1 and p27Kip1. Oncotarget. 2015;30:6684-707
67. Watabe M, Hishikawa K, Takayanagi A, Shimizu N, Nakaki T. Caffeic acid phenethyl ester induces apoptosis by inhibition of NFkappaB and activation of Fas in human breast cancer MCF-7 cells. J Biol Chem. 2004;279:6017-26
68. Liao HF, Chen YY, Liu JJ, Hsu ML, Shieh HJ, Liao HJ. et al. Inhibitory effect of caffeic acid phenethyl ester on angiogenesis, tumor invasion, and metastasis. J Agri Food Chem. 2003;51:7907-12
69. Hwang HJ, Park HJ, Chung HJ, Min HY, Park EJ, Hong JY. et al. Inhibitory effects of caffeic acid phenethyl ester on cancer cell metastasis mediated by the down-regulation of matrix metalloproteinase expression in human HT1080 fibrosarcoma cells. J Nutritional Biochem. 2006;17:356-62
70. Nagaoka T, Banskota AH, Tezuka Y, Harimaya Y, Koizumi K, Saiki I. et al. Inhibitory effects of caffeic acid phenethyl ester analogues on experimental lung metastasis of murine colon 26-L5 carcinoma cells. Biol Pharma Bulletin. 2003;26:638-41
71. Shigeoka Y, Igishi T, Matsumoto S, Nakanishi H, Kodani M, Yasuda K. et al. Sulindac sulfide and caffeic acid phenethyl ester suppress the motility of lung adenocarcinoma cells promoted by transforming growth factor-beta through Akt inhibition. J Cancer Res Clinical Oncol. 2004;130:146-52
Author contact
![]() Corresponding author: Sunil C Kaul, National Institute of Advanced Industrial Science & Technology (AIST), Central 5-41, 1-1-1 Higashi, Tsukuba - 305 8565, Japan. Phone: +81 29 861 6713, Fax: +81 29 861 2900, E-mail: s-kaulgo.jp Or Keiji Terao, CycloChem Co., Ltd., 7-4-5 Minatojima-minamimachi, Chuo-ku, Kobe 650-0047, Japan. E-mail: keiji.teraocom.
Corresponding author: Sunil C Kaul, National Institute of Advanced Industrial Science & Technology (AIST), Central 5-41, 1-1-1 Higashi, Tsukuba - 305 8565, Japan. Phone: +81 29 861 6713, Fax: +81 29 861 2900, E-mail: s-kaulgo.jp Or Keiji Terao, CycloChem Co., Ltd., 7-4-5 Minatojima-minamimachi, Chuo-ku, Kobe 650-0047, Japan. E-mail: keiji.teraocom.