Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(9):1114-1124. doi:10.7150/jca.14120 This issue Cite
Research Paper
Vasculogenic Mimicry in Prostate Cancer: The Roles of EphA2 and PI3K
Hua Wang*, Hao Lin*, Jincheng Pan*, Chengqiang Mo, Faming Zhang, Bin Huang, Zongren Wang, Xu Chen, Jintao Zhuang, Daohu Wang ![]() , Shaopeng Qiu
, Shaopeng Qiu ![]()
Department of Urology, The First Affiliated Hospital, Sun Yat-Sen University, Guangzhou, China
*Hua Wang, Hao Lin and Jincheng Pan are Co-First authors.
Received 2015-10-14; Accepted 2016-3-24; Published 2016-6-5
Abstract
BACKGROUND. Aggressive tumor cells can form perfusable networks that mimic normal vasculature and enhance tumor growth and metastasis. A number of molecular players have been implicated in such vasculogenic mimicry, among them the receptor tyrosine kinase EphA2, which is aberrantly expressed in aggressive tumors. Here we study the role and regulation of EphA2 in vasculogenic mimicry in prostate cancer where this phenomenon is still poorly understood.
METHODS. Vasculogenic mimicry was characterized by tubules whose cellular lining was negative for the endothelial cell marker CD34 but positive for periodic acid-Schiff staining, and/or contained red blood cells. Vasculogenic mimicry was assessed in 92 clinical samples of prostate cancer and analyzed in more detail in three prostate cancer cell lines kept in three-dimensional culture. Tissue samples and cell lines were also assessed for total and phosphorylated levels of EphA2 and its potential regulator, Phosphoinositide 3-Kinase (PI3K). In addition, the role of EphA2 in vasculogenic mimicry and in cell migration and invasion were investigated by manipulating the levels of EphA2 through specific siRNAs. Furthermore, the role of PI3K in vasculogenic mimicry and in regulating EphA2 was tested by application of an inhibitor, LY294002.
RESULTS. Immunohistochemistry of prostate cancers showed a significant correlation between vasculogenic mimicry and high expression levels of EphA2, high Gleason scores, advanced TNM stage, and the presence of lymph node and distant metastases. Likewise, two prostate cancer cell lines (PC3 and DU-145) formed vasculogenic networks on Matrigel and expressed high EphA2 levels, while one line (LNCaP) showed no vasculogenic networks and lower EphA2 levels. Specific silencing of EphA2 in PC3 and DU-145 cells decreased vasculogenic mimicry as well as cell migration and invasion. Furthermore, high expression levels of PI3K and EphA2 phosphorylation at Ser897 significantly correlated with the presence of vasculogenic mimicry and in vitro inhibition of PI3K by LY294002 disrupted vasculogenic mimicry, potentially through a reduction of EphA2 phosphorylation at Ser897.
CONCLUSIONS. The expression levels of PI3K and EphA2 are positively correlated with vasculogenic mimicry both in vivo and in vitro. Moreover, phosphorylation levels of EphA2 regulated by PI3K are also significantly associated with vasculogenic mimicry in vivo. Based on its functional implication in vasculogenic mimicry in vitro, EphA2 signaling may be a potential therapeutic target in advanced prostate cancer.
Keywords: Vasculogenic mimicry, Prostate cancer, EphA2, PI3K.
Introduction
Prostate cancer (PCa) is the most commonly diagnosed tumor in males and is second only to lung cancer as a leading cause of cancer-related deaths in western countries [1]. Treatment options for localized PCa are radical surgery and radiation, and for tumors that are not confined to the prostate, androgen deprivation therapy (ADT). However, within an average of 18 months, most patients with metastatic disease ultimately progress to ADT-resistant PCa, for which no effective therapies exist [2, 3]. Hence, new therapeutic approaches to advanced PCa are urgently required.
It is well known that aggressive tumor growth depends on a variety of distinct mechanisms. For instance, to ensure a sufficient nutrient supply, tumors release angiogenic factors that promote neo-vascularization, suggesting that inhibition of angiogenesis might be a powerful therapeutic strategy in controlling excessive tumor growth. Nevertheless, this approach, though widely applied in many tumors, often failed to provide satisfactory results and was largely unsuccessful when applied to advanced PCa [4, 5]. This suggested that tumors have alternative strategies for self-supply with nutrients. One of these strategies is called vasculogenic (or vascular) mimicry (VM) and was originally discovered in melanoma. It has since been observed in many tumors including hepatocellular carcinoma, gastric adenocarcinoma, gall bladder carcinoma, ovarian cancer, and PCa [6-10]. VM is characterized by the ability of aggressive tumor cells to directly form a blood vessel-like network with connections to normal or leaky blood vessels. In addition, VM could possibly also function as a route for metastatic dissemination of tumor cells [11]. As VM is promoted by intratumoral hypoxia, the above mentioned anti-angiogenic therapy can lead to an undesirable increase in VM [12, 13]. Hence, treatment strategies aimed at cutting off blood supply to advanced PCa should target both endothelium-dependent vessels and VM. Therefore, it is important to analyze the underlining mechanisms of PCa VM from its initiation to its final form in order to design rational therapeutic strategies for advanced PCa.
EphA2 (which denotes erythropoietin-producing hepatocellular A2 protein, also referred to as epithelial cell kinase, ECK) is a receptor tyrosine kinase that is expressed in the majority of epithelial cells [14] and detected in various normal tissues including lung, intestine, kidney, and ovary [15]. EphA2 is overexpressed in several types of cancers and may modulate tumor cell motility, invasion and metastasis [16]. Recently, EphA2 has been implicated in VM formation by melanoma cells [17]. In head and neck cancer, EphA2 has been shown to promote VM through the regulation of epithelial-to-mesenchymal transition (EMT) [18]. Wang et al. found that EphA2 was required for VM formation by interacting with vascular endothelium growth factor-A (VEGF-A) in ovarian cancer [19]. Furthermore, in cellular extension of epithelial kidney cells (MDCK cells), the first step of tubule formation is initiated by a PI3K/AKT-dependent phosphorylation of Ser897 of EphA2 [20]. However, whether PI3K and EphA2 also play specific roles in PCa VM has not been tested so far.
Phosphoinositide 3-Kinase (PI3K) is a heterodimeric protein, consisting of a catalytic subunit (p110α/β/γ/δ) and a regulatory subunit (p85α/β) [21]. In melanoma, EphA2-mediated cell invasion was shown to be AKT-dependent and could readily be abrogated through PI3K and AKT inhibition [22]. Miao et al. suggested that EphA2 stimulation coupled with activation of the PI3K/AKT pathway could promote invasion and migration of PCa cells [23]. These findings implied a close relationship between EphA2 and PI3K. Indeed, PI3K mediates melanoma VM [24]. Therefore, we hypothesized that PI3K may be part of the EphA2 signaling pathway and so regulate PCa VM.
In the present study, we demonstrate that both PI3K and EphA2 are important players in PCa VM. Hence, manipulations of EphA2 signaling may become a therapeutic strategy to reduce VM in advanced PCa.
Materials and Methods
Patient Samples and Immunohistochemistry (IHC)
IHC was performed on paraffin-embedded samples obtained from 92 patients with PCa whose tumors were removed by radical prostatectomy at the First Affiliated Hospital of Sun Yat-sen University between 2009 and 2013. All patients were diagnosed with adenocarcinoma. Their median age at the time of surgery was 68 (range: 54-78 y). The study was approved by the Medical Ethics Committee of Sun Yat-sen University and informed consent was obtained from each patient to use their tissues for the purpose of research. IHC was conducted as previously described [25], using polyclonal rabbit IgG anti-EphA2 antibody (1:200, Santa Cruz), polyclonal rabbit IgG anti-PI3K p110 (alpha) antibody (1:100, Proteintech), p-EphA2S897 (1:100, Cell Signaling Technology) and goat anti-rabbit IgG-HRP as secondary antibody (1:5000, Good-Science). The evaluation of the expression of EphA2, PI3K and p-EphA2S897 were performed as previously reported [26-28], whereby high level expression of EphA2 was defined as a staining index ≥ 3 and low level expression as a staining index <3. High level expression of PI3K and p-EphA2S897 were defined as a staining index ≥ 2 and low level expression as a staining index <2.
CD34/periodic acid Schiff Double Staining
CD34-PAS double staining was used for detection of VM in paraffin-embedded sections. First, CD34 staining (brown) was performed using a mouse monoclonal antibody (1:50, Zhong Shan Goldenbridge, China) to identify endothelial cells. The sections were then incubated with periodic acid-Schiff (PAS) (purple-magenta) to label the basement membrane of tubular structures. The assay was conducted as previously described [13].
Cell Culture
The human PCa cell lines, PC3 and LNCaP, were purchased from the ATCC. DU-145 cells were a kind gift from Prof. Franky L. Chan (Faculty of Medicine, The Chinese University of Hong Kong). The following media, each supplemented with 1% penicillin/streptomycin (Gibco) and 10% fetal bovine serum (Gibco) were used: for PC3 cells, F-12K medium (Gibco); for DU-145 cells, DMEM medium (Gibco); and for LNCaP cells, RPMI 1640 medium (Gibco). All cells were incubated at 5% CO2, 37oC.
Three-dimensional Culture
96-well plates were pre-coated with growth factor-reduced Matrigel (50µl/well, BD Biosciences, Bedford, MA) which was allowed to polymerize for 2h at 37oC. Then, 4×104 cells in 100 µl were added to each well and incubated at 37oC for 4 hours. The number of tube-like structures in 3 random fields was counted, and the mean values of the 3 fields were used for analysis.
RNA Purification and RT-qPCR Analysis
Total RNA was extracted by an E.Z.N.A® HP Total RNA Kit (OMEGA, USA). One µg of total RNA was used for generating first-strand cDNA by Revert Aid First Strand cDNA Synthesis Kit (Thermo Scientific, USA). RT-qPCR was performed by using SYBR® Premix Ex Taq™ (TaKaRa, Japan) according to the manufacturer's protocol. The primers used had the following sequences: EphA2 forward: 5'-CCGCAACATCCTCGTCAAC-3', reverse: 5'-ACAATGCCAAAGCTCCACACGTC-3'; PI3K forward: 5'-CGTGGAGGCATTGTTCTGAT-3', reverse: 5'-GGAGCCTGGAAGAGCCC-3'; GAPDH forward: 5'-CGACCACTTTGTCAAGCTCA-3', reverse: 5'-AGGGGAGATTCAGTGTGGTG-3'.
RNA Interference
Transfection was carried out using Lipofectamine 2000™ transfection reagent following the manufacturer's instructions. siRNAs against EphA2 were obtained from RiboBio, with the following target sequences: siEphA2#1: GGGACCTGATGCAGAACAT; siEphA2#2: GCAAGAAGGGAGACTCCAA; siEphA2#3: ACAAGAAGATCACCCTGAA. Transfections were performed according to the manufacturer's instructions. The expression of EphA2 was confirmed by RT-qPCR and western blot.
Pharmacological inhibition of PI3K
PC3 and DU-145 cells were serum-starved overnight before experiments. Cells were harvested and re-suspended in complete medium with various concentrations of the PI3K inhibitor, LY294002 (MedChemExpress, USA) dissolved in DMSO and used at concentrations between 0μM-80μM. Control cells were treated with DMSO alone. Forty thousand cells per well were then plated in 96-well plates pre-coated with Matrigel and incubated for 4h.
Western Blot Analysis
Cell lysates were prepared using total protein extract kit (Keygen, China) according to the supplied protocol. Protein concentrations were measured by Pierce BCA Protein Assay Kit (Thermo, USA), following the manufacturer's protocol. The same amounts of protein (30μg) were resolved on 10% SDS-PAGE (Keygen, China) and transferred to PVDF membranes (Pierce Biotechnology, Inc., Rockford, IL, USA). Membranes were blotted overnight at 4oC with primary antibody. Rabbit antibodies against EphA2 (1:500, Santa Cruze), AKT (1:1000, Cell Signaling Technology), p-AKTS473 (1:1000, Cell Signaling Technology), p-EphA2S897 (1:1000, Cell Signaling Technology), GAPDH (1:5000, Cwbiotech, China), and goat anti-rabbit IgG-HRP (1:5000, Good-Science) were used for detecting the specific bands.
Wound-healing Assay
Forty-eight hours after transfection, cells were seeded in six-well plates at a density of 5×105/well and incubated for 24 h. Following the application of a scratch, the assay was performed as previously described [29]. The scratched area was photographed at 0h and 6h, or 12h after scratching, and cellular repopulation analyzed by Image pro-plus software.
Invasion Assay
Cellular invasion was assayed in transwell migration assays as previously described [30]. Cells that had invaded the empty space were counted microscopically at a magnification of 400×.
Statistical Analysis
Data were analyzed using the software package SPSS13.0 (SPSS Inc., Chicago, IL) and are shown as mean ± SEM. Unpaired two-tailed student's t-test was used to compare the differences between groups. The associations between VM, EphA2, PI3K and clinico-pathological parameters were assessed using the chi-square (χ2) test or Fisher's exact test. The correlation between VM and EphA2, PI3K and p-EphA2S897 expressions were evaluated by Spearman's correlation. Significance was defined as p<0.05.
Results
Presence of VM in PCa tissues correlates with high expression levels of EphA2
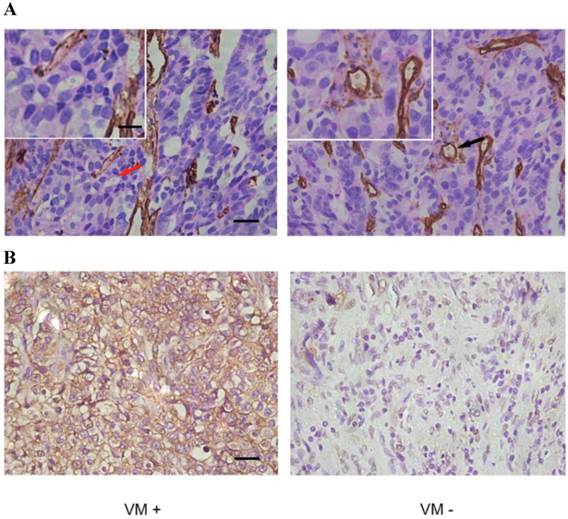
The presence of VM was validated by the presence of PAS-positive loops and/or contained red blood cells that were negative for the endothelial cell marker CD34 (Fig. 1A). As shown in Table 1, 25 tissue samples from 92 cases of PCa (27.2%) were VM positive. In addition, there was a significant association between the presence of VM and the Gleason score, the TNM stage, and lymph node and distant metastases. According to subgroup analysis, the occurrence of VM was 10.3% (3/29), 33.3% (12/36) and 37.0% (10/27) in Gleason score subgroups of ≤6, =7, and ≥8. Furthermore, VM seemed to be more prevalent in the T3 stage than the T2 stage (48.5% vs. 15.3%) but there was no association between VM and a patient's age at surgery.
IHC of paraffin-embedded prostate specimens. (A) The panel on the left shows VM, indicated by a red arrow (magnified in inset), which are tubule-like structures that contain luminal erythrocytes and are delimited by cells that are negative for CD34 staining (brown). The panel on the right shows endothelium-dependent vessels (one of which is indicated by a black arrow and magnified in the inset) that are positive for CD34 staining. Note that red blood cells can be seen in the lumens of both VM and endothelium-dependent vessels. (B) EphA2 in PCa shows mainly cytoplasmic staining. Different expression levels of EphA2 in VM-positive and VM-negative tumors from the two distinct PCa patient groups. (Original magnification ×400; bar: 10 µm; for insets, 2.5µm).
Relationship between VM and EphA2 expression and clinico-pathological factors in PCa a: GS ≤ 6 vs. GS = 7; b: GS ≤ 6 vs. GS ≥ 8; c: GS =7 vs. GS ≥ 8; d: T2 vs. T3. Patients (n=92).
| Variables | N | VM | p value | EphA2 | p value | ||
|---|---|---|---|---|---|---|---|
| positive | negative | High expression (staining index≥3) | Low expression (staining index<3) | ||||
| Age, years | |||||||
| ≥68 | 48 | 11 | 37 | 0.338 | 24 | 24 | 0.828 |
| <68 | 44 | 14 | 30 | 23 | 21 | ||
| Gleason score | |||||||
| ≤6 | 29 | 3 | 26 | 0.029a | 8 | 21 | 0.024a |
| =7 | 36 | 12 | 24 | 0.018b | 20 | 16 | 0.001b |
| ≥8 | 27 | 10 | 17 | 0.760c | 19 | 8 | 0.231c |
| T classification | |||||||
| T2a | 28 | 3 | 25 | 0.001d | 13 | 15 | 0.025d |
| T2b | 31 | 6 | 25 | 12 | 19 | ||
| T3a | 9 | 4 | 5 | 7 | 2 | ||
| T3b | 24 | 12 | 12 | 15 | 9 | ||
| Lymph node metastasis | |||||||
| Yes | 12 | 8 | 4 | 0.001 | 10 | 2 | 0.017 |
| No | 80 | 17 | 63 | 37 | 43 | ||
| Distant metastasis | |||||||
| Yes | 6 | 4 | 2 | 0.025 | 6 | 0 | 0.026 |
| No | 86 | 21 | 65 | 41 | 45 | ||
As shown in Fig 1B, high levels of expression of EphA2 (staining index≥3) was present in 47 of the 92 cases (51.0%) and in 18 of the 25 VM-positive samples (72.0%) but in only 29 of the 67 VM-negative samples (43%). EphA2 staining was mainly cytoplasmic (Fig. 1B). Statistically, there was a significant correlation between high expression of EphA2 and presence of VM (r=0.256 p = 0.014) (Table 2). Moreover, expression of EphA2 was positively correlated with Gleason score, TNM stage, lymph node and distant metastases (Table 1). The subgroup analysis showed the highest EphA2 expression in Gleason score subgroup of ≥ 8 (70.4%), and a higher likelihood of high-level EphA2 expression in T3 compared to T2 stage (66.7% vs. 42.4%). Taken together, the results show a positive association of VM and EphA2 in aggressive PCa.
Relationship between VM and expression of EphA2 and analysis by chi-square (χ2) test.
| VM | EphA2 | p value | |
|---|---|---|---|
| High expression | Low expression | ||
| Positive (25) | 18 | 7 | 0.014 |
| Negative (67) | 29 | 38 |
VM formation and EphA2 expression in PCa cell lines
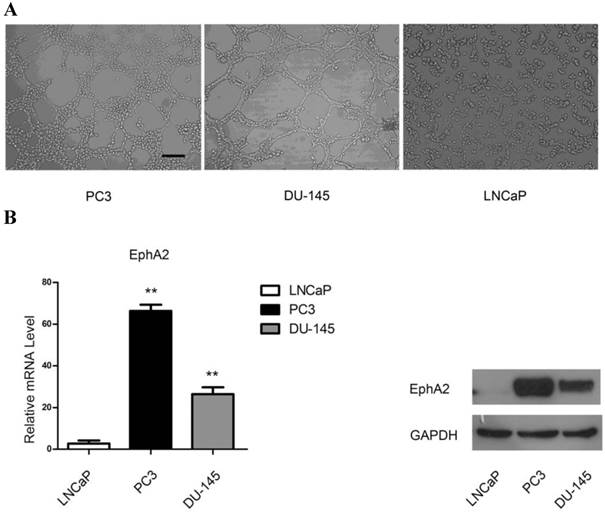
In order to elucidate the ability of PCa cells to form VM-associated tubes, we used three PCa cell lines, LNCaP, PC3 and DU-145, in a three-dimensional culture system. As shown in Fig. 2A, both PC3 and DU-145 cells, but not LNCaP cells, formed tubular structures after 4h of culture on Matrigel. Interestingly, both mRNA and protein levels of EphA2 were much higher in PC3 and DU-145 compared to LNCaP cells (Fig. 2B). Hence, it is conceivable that EphA2 plays a role in PCa VM.
(A) PCa cell lines were cultured on Matrigel. PC3 and DU-145 (4×105cell/ml) were able to form vascular channels 4 hours after seeding, whereas LNCaP (3×105cell/ml) failed to form patterned networks even when cultured for up to 48 hours. (Original magnification ×100, bar: 40µm). (B) Expression of EphA2 in PCa cell lines. Both mRNA and protein levels of EphA2 in PC3 and DU-145 were significantly higher than in LNCaP. (*, P<0.05, **, P <0.001).
Down-regulation of EphA2 impairs VM formation by PC3 and DU-145 cells
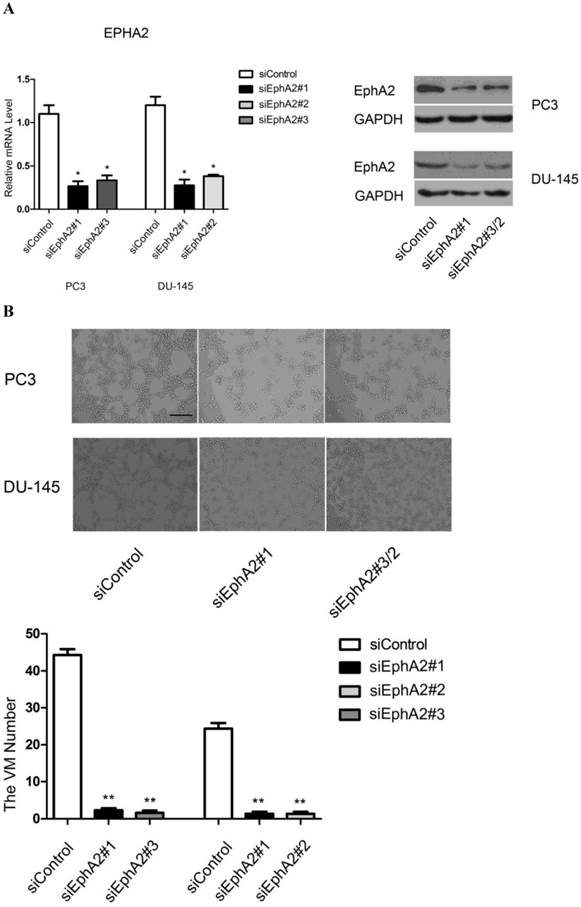
To confirm that EphA2 is required for VM formation in vitro, we used three distinct siRNAs to inhibit EphA2 expression. After evaluating the efficiency of each siRNA, we chose siEphA2#1 and siEphA2#3 for PC3 and siEphA2#1 and siEphA2#2 for DU-145 (Fig. 3A). As shown in Fig. 3B, down-regulation of EphA2 in either PC3 or DU-145 cells markedly reduced the formation of tubular structures. This suggests that EphA2 levels not only correlate with the presence of VM, but that EphA2 is also functionally important for VM in PCa.
Analysis of VM formation after specific knockdown of EphA2 in PC3 and DU-145 cells (original magnification ×100, bar: 40µm). (A) Messenger RNA and protein levels of EphA2 were significantly decreased 48 hours after transfection. (B) siRNA-mediated down-regulation of EphA2 in PC3 and DU-145 cells significantly impaired VM formation on Matrigel in both PC3 and DU-145 cells.
Suppression of EphA2 reduces migration and invasion of PC3 and DU-145 cells
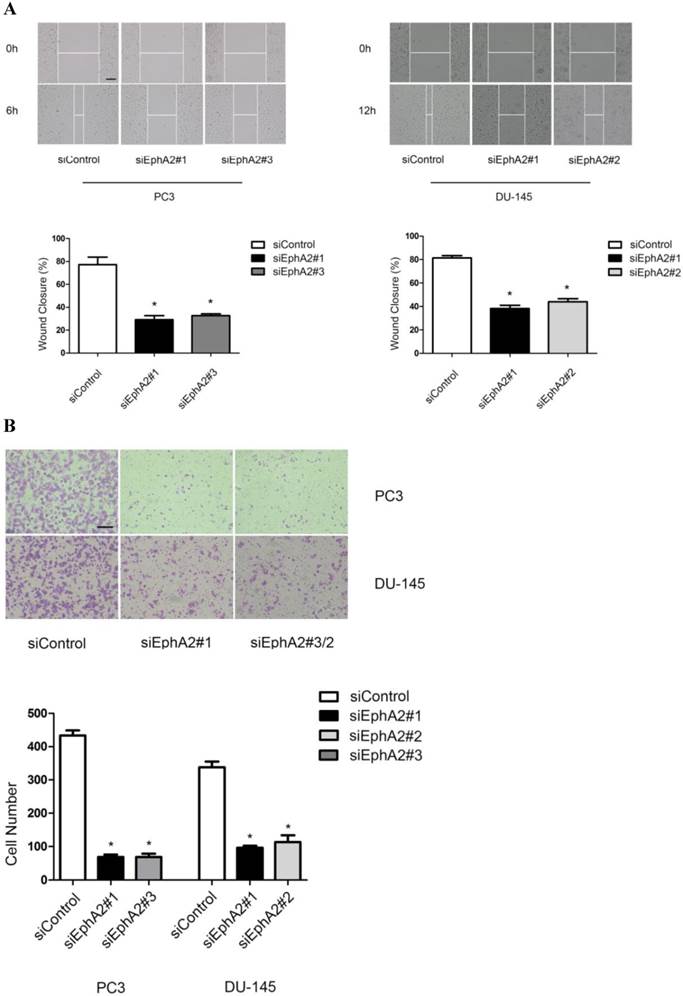
In addition to VM formation, aggressive PCa cells also show enhanced migration and invasion compared to LNCaP cells. Hence, we also tested whether reduction of EphA2 expression would have an effect on these two parameters in wound-healing and transwell assays. As shown in Fig. 4A and B, both the migratory and invasive capacity of PC3 and DU-145 cells were markedly reduced after down-regulation of EphA2, suggesting that EphA2 regulates additional parameters of cell behavior besides influencing VM.
Effects of siEphA2 on cell migration and invasion in PC3 and DU-145 cells. (A) Wound-healing assay: cell migration was significantly inhibited in response to silencing of EphA2 at 6 hours (PC3) and 12 hours (DU-145). (B) Transwell invasion assay: cell numbers on the lower side of the membrane, reflecting transmembrane invasion, were significantly reduced after EphA2 silencing by EphA2 siRNA compared with control siRNA. (Original magnification ×100, bar: 20µm).
PI3K is required for VM formation and potentially regulates EphA2 phosphorylation
We first used the specific PI3K inhibitor, LY294002, to investigate the role of PI3K in VM formation. As shown in Fig. 5A, the level of p-AKTS473, resulting from phosphorylation by PI3K, decreased in a dose-dependent manner after application of the drug, indicating that the drug was active. Concomitantly, application of LY294002 also reduced the appearance of tubular structures in a dose-dependent manner (Fig. 5B).
Effects of PI3K inhibition. (A) Protein levels of p-AKTS473 and (B) the number of tubular structures formed by PC3 and DU-145 cells were decreased in a dose-dependent manner after treatment with different concentrations of LY294002 (0-80µM). (Original magnification ×100, bar: 40µm). (C) Inhibition of PI3K dose-dependently decreased the levels of p-EphA2S897 in vitro.
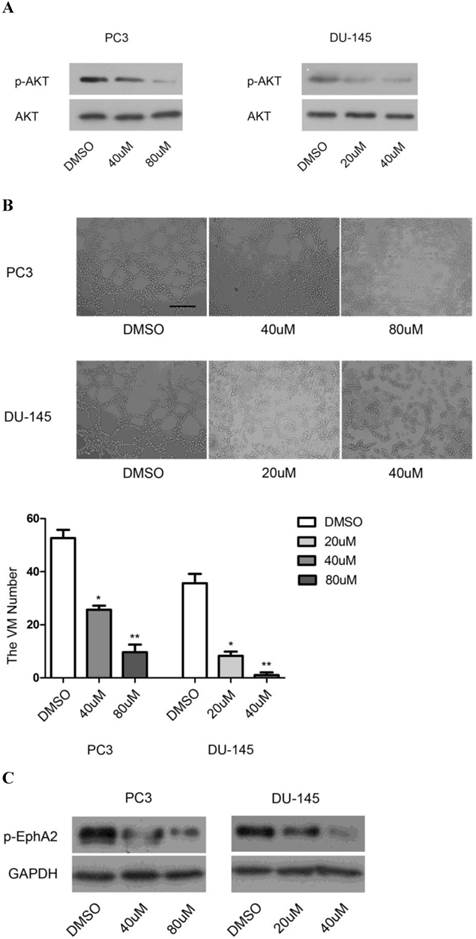
Given the previous demonstration that PI3K/AKT-dependent phosphorylation of Ser897 of EphA2 may be involved in the initiation of cellular tubule formation [20], we tested whether inhibition of PI3K would also affect the levels of p-EphA2S897. Indeed, in both PC3 and DU-145 cells, application of LY294002 resulted in a dose-dependent reduction in p-EphA2S897 (Fig. 5C). These results suggest that PI3K and EphA2 are linked in a common pathway to regulate VM formation. Nevertheless, it was conceivable that EphA2 was not only a target of PI3K activity but also one of its regulators. However, we found that downregulation of EphA2 had no effect on PI3K expression as determined by RT-PCR for PI3K RNA (data not shown). The results suggest that PI3K is required for VM in PCa and that it might exert its function through the regulation of EphA2 signaling.
VM correlates with high expression levels of PI3K in PCa
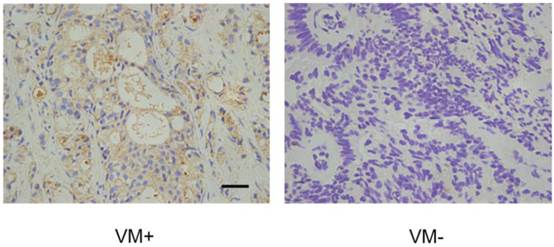
The above suggestion that EphA2 and PI3K are linked in a common pathway to regulate VM formation prompted us to test whether PI3K levels, similar to those of EphA2, were also positively correlated with the presence of VM in PCa tissue. Immunohistochemical assays showed that 15 of the 25 VM positive tumors (60.0 %) showed high PI3K levels, while only 24 of the 67 VM-negative tumors (35.8%) showed high PI3K levels (Fig. 6). As shown in Table 3, this result indicate a significant correlation between high expression levels of PI3K and the presence of VM (r = 0.218, p = 0.037). Nevertheless, there is no significant correlation between high expression levels of EphA2 and high expression levels of PI3K (r = 0.047) (Table 4).
IHC analysis of the PI3K catalytic subunit PI3K p110 (alpha) in PCa specimens. Cytoplasmic staining of PI3K p110 (alpha) was found in prostate cancer tissues. Note different expression levels of PI3K in VM-positive and VM-negative PCa specimens. (Original magnification ×400, bar: 10µm).
Relationship between VM and expression of PI3K and analysis by chi-square (χ2) test.
| VM | PI3K | p value | |
|---|---|---|---|
| High expression | Low expression | ||
| Positive (25) | 15 | 10 | 0.037 |
| Negative (67) | 24 | 43 |
Relationship between expression of EphA2 and PI3K and analysis by Spearman's correlation test.
| EphA2 | PI3K | r value | |
|---|---|---|---|
| High expression | Low expression | ||
| High expression | 21 | 26 | 0.047 |
| Low expression | 18 | 27 |
High levels of p-EphA2S897 correlate with VM in PCa
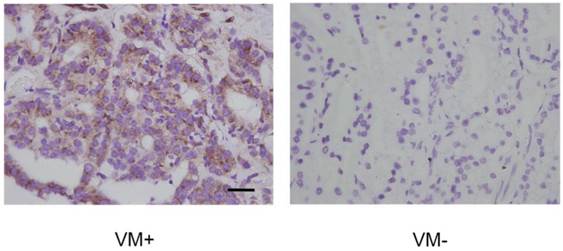
As we speculated that PI3K was required for VM in PCa and that it might exert its function through regulating phosphorylation of EphA2, we tested whether p-EphA2 S897 levels were elevated in VM in PCa. IHC data showed that high p-EphA2S897 levels could be detected in 16 of the 25 (64.0%) samples in the VM-positive group while 24 of the 67 (35.8%) samples were high in the VM-negative group (Fig. 7). As shown in Table 5, this observation suggests a significant correlation between high expression levels of p-EphA2S897 and the existence of VM (r = 0.253, p = 0.015).
Levels of EphA2 phosphorylation at Ser897 in PCa specimens assessed by IHC. P-EphA2S897 was predominantly localized in the cytoplasm. Note the different expression levels of p-EphA2S897 in VM-positive and VM-negative PCa specimens. (Original magnification ×400, bar: 10µm).
Relationship between VM and expression of p-EphA2S897 and analysis by chi-square (χ2) test.
| VM | p-EphA2S897 | p value | |
|---|---|---|---|
| High expression | Low expression | ||
| Positive (25) | 16 | 9 | 0.015 |
| Negative (67) | 24 | 43 |
Discussion
Here we analyzed both in vivo and in vitro, the formation of VM in PCa and its correlation with high levels of both EphA2 and PI3K. Reducing EphA2 levels or inhibiting PI3K activity in vitro led to a reduction in VM formation in at least two PCa lines. Because the activity of EphA2 can be regulated by a PI3K/AKT-dependent phosphorylation at S897 [23] and we found p-EphA2S897 levels to be positively correlated with VM, PI3K and EphA2, this may well be linked in a common pathway, which affects VM formation in PCa.
Our results are largely in accordance with previous studies in PCa. For instance, Liu et al. showed that high risk PCa had a higher incidence of VM [10], and we show here that presence of VM is more frequent in tumors with a higher Gleason score and higher TNM stage. Moreover, VM-positive cases more frequently displayed lymph node and distant metastases. This confirms that VM is a marker for poor prognosis [10] and suggests that therapeutic inhibition of VM may improve the course of the disease. In addition, EphA2 has also been shown to play important roles in regulating cell migration and invasion in PCa. J.Walker Daniels et al. reported that in comparison with non-invasive PCa cell lines, lines with metastatic capacity overexpress EphA2 [31]. Despite these extensive studies of EphA2 in PCa, however, no mechanistic details have so far emerged on its role in VM formation in PCa, while its role in VM formation in other tumors is better understood. For instance, EphA2 down regulation in head and neck cancer inhibits VM formation and decreases the expression of Twist and Vimentin, two molecules involved in EMT [18]. In ovarian cancer, matrix metalloproteinases (MMPs) are activated by EphA2, which in turn is up-regulated by VM-promoting VEGF-A [19].
To analyze the role of EphA2 in PCa VM in greater detail, we used three PCa cell lines. We found that the more aggressive lines, PC3 and DU-145, expressed higher levels of EphA2 and were able to form VM, while the less aggressive line LNCaP showed lower levels of EphA2 and failed to form VM under similar conditions. Most importantly, reduction in EphA2 expression using specific siRNAs reduced VM formation as visualized by the formation of tubular structures. Interestingly, Taddei et al. showed that down-regulation of EphA2 reduces the clonogenic potential and metastatic growth of PCa cells [32]. These observations are consistent with our findings and suggest that the EphA2-dependent capacity of aggressive PCa cells to form vascular-like structures is the mechanistic basis for the aggressive behavior of the associated tumors.
The discovery of a role for EphA2 in VM in PCa prompted us to search for possible regulators of EphA2. Such regulators could either affect EphA2 protein levels in general or more specifically its activity levels, for instance by phosphorylation. Although it is currently unknown what mechanisms lead to the differential levels of EphA2 in PCa in vivo or in distinct PCa cell lines, there is emerging evidence that EphA2 activity, and in turn VM formation, may be regulated by PI3K. For instance, in a murine choroidal melanoma model, curcumin inhibited VM formation through the suppression of the EphA2/PI3K/MMP pathway [33]. Here we found that pharmacological inhibition of PI3K in PCa cell lines led to reduced VM formation, suggesting a role for PI3K in PCa VM. Interestingly, PI3K inhibition also led to a dose-dependent reduction of Ser897 phosphorylated EphA2. Furthermore, both PI3K and p-EphA2S897 levels were positively correlated with VM in vivo. Hence, it is conceivable that PI3K may regulate VM formation in PCa by regulating EphA2 phosphorylation. The findings that PI3K and p-EphA2S897 activity are required for VM significantly extend the earlier observation by Miao et al. who showed that EphA2 signaling is part of the PI3K/AKT-regulated cell migration and invasion in PCa. It will be fascinating to explore the details of how PI3K/AKT and EphA2 affect migration, invasion and VM formation and thereby provide a potential mechanistic link between these different cellular behaviors.
Conclusions
In summary, our study demonstrates that the expression of EphA2 and PI3K are significantly correlated with VM formation in PCa in vivo. In vitro, EphA2 is essential for VM formation and may be regulated by PI3K through phosphorylation at Ser897. Hence, targeting EphA2 signaling may provide a potential therapeutic strategy to reduce VM in VM-positive, advanced PCa.
Abbreviations
VM: vasculogenic mimicry
PCa: prostate cancer
RTK: receptor tyrosine kinase
IHC: immunohistochemistry
ADT: androgen deprivation therapy
CRPC: castrate resistant prostate cancer
EphA2: EphA2 receptor tyrosine kinase
PI3K: Phosphoinositide 3-Kinase
WB: Western blot
EMT: epithelial-to-mesenchymal transition
VEGF: Vascular endothelial growth factor
Acknowledgements
We are grateful to Prof. Franky L. Chan (Faculty of Medicine, The Chinese University of Hong Kong) for providing DU-145 cells. This work was supported by grants from the National Natural Science Foundation of China (81272809), Science and Technology Planning Project of Guangdong (2012B031800351), the Guangdong province of integration of production and research projects (2012B090600021), Guangdong Natural Science Foundation (2014A030310034).
Conflict of Interest
The authors have declared that no competing interest exists.
References
1. Nelson WG, De Marzo AM, Isaacs WB. Prostate cancer. The New England journal of medicine. 2003;349:366-81
2. Bubendorf L, Schopfer A, Wagner U. et al. Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Human pathology. 2000;31:578-83
3. Gittes RF. Carcinoma of the prostate. The New England journal of medicine. 1991;324:236-45
4. Adesunloye BA, Karzai FH, Dahut WL. Angiogenesis inhibitors in the treatment of prostate cancer. Chemical immunology and allergy. 2014;99:197-215
5. Hwang C, Heath EI. Angiogenesis inhibitors in the treatment of prostate cancer. Journal of hematology & oncology. 2010;3:26
6. Li M, Gu Y, Zhang Z. et al. Vasculogenic mimicry: a new prognostic sign of gastric adenocarcinoma. Pathology oncology research: POR. 2010;16:259-66
7. Liu WB, Xu GL, Jia WD. et al. Prognostic significance and mechanisms of patterned matrix vasculogenic mimicry in hepatocellular carcinoma. Medical oncology. 2011;28(Suppl 1):S228-38
8. Sood AK, Fletcher MS, Coffin JE. et al. Functional role of matrix metalloproteinases in ovarian tumor cell plasticity. American journal of obstetrics and gynecology. 2004;190:899-909
9. Sun W, Shen ZY, Zhang H. et al. Overexpression of HIF-1alpha in primary gallbladder carcinoma and its relation to vasculogenic mimicry and unfavourable prognosis. Oncology reports. 2012;27:1990-2002
10. Liu R, Yang K, Meng C. et al. Vasculogenic mimicry is a marker of poor prognosis in prostate cancer. Cancer biology & therapy. 2012;13:527-33
11. Seftor RE, Hess AR, Seftor EA. et al. Tumor cell vasculogenic mimicry: from controversy to therapeutic promise. The American journal of pathology. 2012;181:1115-25
12. Qu B, Guo L, Ma J. et al. Antiangiogenesis therapy might have the unintended effect of promoting tumor metastasis by increasing an alternative circulatory system. Medical hypotheses. 2010;74:360-1
13. Xu Y, Li Q, Li XY. et al. Short-term anti-vascular endothelial growth factor treatment elicits vasculogenic mimicry formation of tumors to accelerate metastasis. Journal of experimental & clinical cancer research: CR. 2012;31:16
14. Lindberg RA, Hunter T. cDNA cloning and characterization of eck, an epithelial cell receptor protein-tyrosine kinase in the eph/elk family of protein kinases. Molecular and cellular biology. 1990;10:6316-24
15. Hess AR, Seftor EA, Gardner LM. et al. Molecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: role of epithelial cell kinase (Eck/EphA2). Cancer research. 2001;61:3250-5
16. Ireton RC, Chen J. EphA2 receptor tyrosine kinase as a promising target for cancer therapeutics. Current cancer drug targets. 2005;5:149-57
17. Hess AR, Seftor EA, Gruman LM. et al. VE-cadherin regulates EphA2 in aggressive melanoma cells through a novel signaling pathway: implications for vasculogenic mimicry. Cancer biology & therapy. 2006;5:228-33
18. Wang W, Lin P, Sun B. et al. Epithelial-mesenchymal transition regulated by EphA2 contributes to vasculogenic mimicry formation of head and neck squamous cell carcinoma. BioMed research international. 2014;2014:803914
19. Wang JY, Sun T, Zhao XL. et al. Functional significance of VEGF-a in human ovarian carcinoma: role in vasculogenic mimicry. Cancer biology & therapy. 2008;7:758-66
20. Harada K, Negishi M, Katoh H. HGF-induced serine 897 phosphorylation of EphA2 regulates epithelial morphogenesis of MDCK cells in 3D culture. Journal of cell science. 2015;128:1912-21
21. Shukla S, Maclennan GT, Hartman DJ. et al. Activation of PI3K-Akt signaling pathway promotes prostate cancer cell invasion. International journal of cancer Journal international du cancer. 2007;121:1424-32
22. Paraiso KH, Das Thakur M, Fang B. et al. Ligand-independent EPHA2 signaling drives the adoption of a targeted therapy-mediated metastatic melanoma phenotype. Cancer discovery. 2015;5:264-73
23. Miao H, Li DQ, Mukherjee A. et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer cell. 2009;16:9-20
24. Hess AR, Seftor EA, Seftor RE. et al. Phosphoinositide 3-kinase regulates membrane Type 1-matrix metalloproteinase (MMP) and MMP-2 activity during melanoma cell vasculogenic mimicry. Cancer research. 2003;63:4757-62
25. Zhuang J, Tu X, Cao K. et al. The expression and role of tyrosine kinase ETK/BMX in renal cell carcinoma. Journal of experimental & clinical cancer research: CR. 2014;33:25
26. Zeng G, Hu Z, Kinch MS. et al. High-level expression of EphA2 receptor tyrosine kinase in prostatic intraepithelial neoplasia. The American journal of pathology. 2003;163:2271-6
27. Shang L, Liu HJ, Hao JJ. et al. A panel of overexpressed proteins for prognosis in esophageal squamous cell carcinoma. PloS one. 2014;9:e111045
28. Huang J, Hu W, Bottsford-Miller J. et al. Cross-talk between EphA2 and BRaf/CRaf is a key determinant of response to Dasatinib. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:1846-55
29. Huang B, Cao K, Li X. et al. The expression and role of protein kinase C (PKC) epsilon in clear cell renal cell carcinoma. Journal of experimental & clinical cancer research: CR. 2011;30:88
30. Hart CA, Brown M, Bagley S. et al. Invasive characteristics of human prostatic epithelial cells: understanding the metastatic process. British journal of cancer. 2005;92:503-12
31. Walker-Daniels J, Coffman K, Azimi M. et al. Overexpression of the EphA2 tyrosine kinase in prostate cancer. The Prostate. 1999;41:275-80
32. Taddei ML, Parri M, Angelucci A. et al. EphA2 induces metastatic growth regulating amoeboid motility and clonogenic potential in prostate carcinoma cells. Molecular cancer research: MCR. 2011;9:149-60
33. Chen LX, He YJ, Zhao SZ. et al. Inhibition of tumor growth and vasculogenic mimicry by curcumin through down-regulation of the EphA2/PI3K/MMP pathway in a murine choroidal melanoma model. Cancer biology & therapy. 2011;11:229-35
Author contact
![]() Corresponding authors: Daohu Wang, M.D., Ph.D., Department of Urology, The First Affiliated Hospital, Sun Yat-Sen University, Guangzhou, China. E-mail: wangdaohsysu.edu.cn. Facsimile: +86 020 87333300, Phone: +86 13632302998 Or Shaopeng Qiu, M.D., Ph.D., Department of Urology, The First Affiliated Hospital, Sun Yat-Sen University, Guangzhou, China. E-mail: qiushpsysu.edu.cn. Facsimile: +86 020 87333300, Phone: +86 020 87755766-8227.
Corresponding authors: Daohu Wang, M.D., Ph.D., Department of Urology, The First Affiliated Hospital, Sun Yat-Sen University, Guangzhou, China. E-mail: wangdaohsysu.edu.cn. Facsimile: +86 020 87333300, Phone: +86 13632302998 Or Shaopeng Qiu, M.D., Ph.D., Department of Urology, The First Affiliated Hospital, Sun Yat-Sen University, Guangzhou, China. E-mail: qiushpsysu.edu.cn. Facsimile: +86 020 87333300, Phone: +86 020 87755766-8227.