Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(9):1044-1048. doi:10.7150/jca.14815 This issue Cite
Research Paper
Genomic Profiling of Metastatic Gastroenteropancreatic Neuroendocrine Tumor (GEP-NET) Patients in the Personalized-Medicine Era
Seung Tae Kim, Su Jin Lee, Se Hoon Park, Joon Oh Park, Ho Yeong Lim, Won Ki Kang, Jeeyun Lee ![]() , Young Suk Park
, Young Suk Park ![]()
Division of Hematology-Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea
Received 2015-12-27; Accepted 2016-4-26; Published 2016-5-25
Abstract
Background: We have conducted molecular profiling through a high-throughput molecular test as part of our clinical practice for patients with advanced gastrointestinal (GI) cancer or rare cancers including gastroenteropancreatic neuroendocrine tumors (GEP-NETs). Herein, we report on the molecular characterization of 14 metastatic GEP-NET patients.
Methods: We conducted the Ion AmpliSeq Cancer Hotspot Panel v2 (detecting 2,855 oncogenic mutations in 50 commonly mutated genes) and nCounter Copy Number Variation Assay, which was designed with 21 genes based on available targeted agents, as a high throughput genomic platform in 14 patients with metastatic GEP-NETs.
Results: Among the 14 GEP-NET patients analyzed in this study, 8 patients had grade III neuroendocrine carcinoma (NEC) and 6 had grade I/II NET. Primary sites included pancreas (n=3), small intestine and ascending colon (n=3), distal colon and rectum (n=5), and unknown primary origin (n=3). The most common metastatic site was the liver. Of 14 GEP-NET patients available for mutational profiling, 7 (50.0%) patients had one or more aberrations detected. Common aberrations were as follows: SMARCB1 mutation (n=2), TP53 mutation (n=2), STK11 mutation (n=1), RET mutation (n=1), and BRAF mutation (n=1). Gene amplification by nCounter was detected in only 1 patient, showing CCNE1 amplification, and this patient also had a TP53 mutation.
Conclusions: This high throughput genomic test may be useful to identify new drug targets in metastatic GEP-NET patients. Currently, we plan to conduct further genomic analysis to develop predictive and prognostic biomarkers in a larger number of GEP-NET patients.
Keywords: molecular profiling, Gastroenteropancreatic neuroendocrine tumor (GEP-NET)
Introduction
Neuroendocrine tumors (NETs) are composed of a heterogeneous group of malignancies derived from neuroendocrine cell compartments, with roles in both the endocrine and the nervous system. The majority of NETs are gastroenteropancreatic (GEP) in origin, arising in the foregut, midgut, or hindgut [1]. While NETs are very rare [2], recent studies on NETs based on the Surveillance, Epidemiology, and End Results (SEER) cancer registry and European studies demonstrated that the rate incidence of NETs is increasing [2]. Moreover, a Korean study showed a remarkable increase in the incidence of GEP-NET during the last decade [3]. Although GEP-NETs are being increasingly diagnosed, a concomitant improvement in outcomes has not been noted.
The management of patients with GEP-NETs is individualized, based mainly on tumor biology and the presence of distal metastases. In patients with inoperable advanced disease, there have been some therapeutic options such as octreotide for hormonal control, cytotoxic agents (doxorubicin, streptozocin, capecitabine, dacarbazine and temozolamide), and peptide receptor radionuclide therapy [4]. Recently, the use of two molecularly targeted agents, sunitinib and everolimus, has been approved but only for advanced pancreatic NETs [5, 6]. However, most GEP-NET patients with distant metastases experience resistance to these treatments. In these cases, alternative effective treatment options are very limited. Thus, new drug therapies and potential molecular markers for novel therapies are needed in GEP-NET patients with distant metastases.
Personalized medicine is defined as the use of an individual patient's molecular information to inform diagnosis, prognosis, treatment, and prevention of cancer and has become a primary focus of many studies in oncology [7]. Indeed, the identification of genomic alterations associated with responses to molecularly targeted agents, such as tyrosine kinase inhibitors, has changed the paradigm of cancer treatment into precision medicine by identification of multiple actionable targets across cancer types, especially with the emergence of advanced genomic techniques [8, 9]. Thus, it is necessary to improve our understanding of the heterogeneity of a disease at the genomic and molecular levels. However, there is little data available on the integrated comprehensive molecular profiling for GEP-NETs.
We have conducted molecular profiling through high-throughput molecular tests as part of a clinical practice for patients with advanced gastrointestinal (GI) cancer, or rare cancers including (GEP-NETs). Herein, we evaluated the molecular characteristics for 14 metastatic GEP-NET patients and identified new drug targets.
Methods
Patients
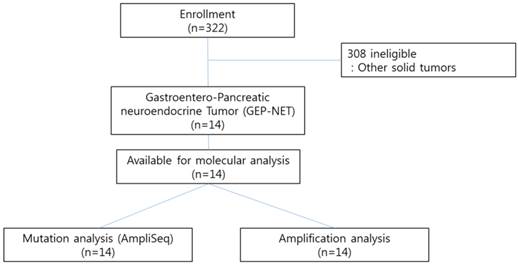
We conducted the Ion AmpliSeq Cancer Hotspot Panel v2 (detecting 2,855 oncogenic mutations in 50 commonly mutated genes) and nCounter Copy Number Variation Assay (includes 21 genes based on available targeted agents) as high throughput genomic platforms as part of our clinical practice for 322 patients with advanced gastrointestinal (GI) cancer or rare cancers including gastroenteropancreatic neuroendocrine tumors (GEP-NETs) at Samsung Medical Center between November 2013 and August 2014. Patients with pathologically confirmed cancer and who had either archived or fresh tissues were candidates for genomic analysis. All patients provided written informed consent before molecular profiling. Of the 322 patients examined, 14 GEP-NET patients with distant metastases were included in this study (Figure 1). The definition of GEP-NET in this study was NET arising in the gastrointestinal, pancreatic (GEP), and hepatobiliary (HB) systems. The confirmation for NET was based on the 2010 WHO classification. The following clinicopathological characteristics of all fourteen patients were collected: age, gender, Eastern Cooperative Oncology Group performance status (ECOG PS), primary site, site of metastasis, and liver metastasis.
CONSORT diagram
Ion AmpliSeq cancer panel v2
We used the Ion AmpliSeq Cancer Panel v2 (Ion Torrent) to detect frequent somatic mutations that were selected based on a literature review. This panel examines 2855 mutations in 50 commonly mutated oncogenes and tumor suppressor genes (Supplementary Table 1). First, 10 ng of DNA from each of 322 FFPE tumor samples (14 GEP-NETS and 308 other solid tumors) (Figure 1) was used for single-tube, multiplex PCR amplification using the Ion AmpliSeqCancer Primer Pool and the Ion AmpliSeqKit reagents (Life Technologies). Treatment of the resulting amplicons with FuPa Reagent partially digested the primers and phosphorylated the amplicons. The phosphorylated amplicons were ligated to Ion Adapters and purified. For barcoded library preparation, we substituted barcoded adapters from the Ion Xpress™ Barcode Adapters 1-96 Kit for the non-barcoded adapter mix supplied in the Ion AmpliSeq™ Library Kit. The ligated DNA underwent nick-translation and amplification to complete the linkage between adapters and amplicons and to generate sufficient material for downstream template preparation. Two rounds of Agencourt® AMPure® XP Reagent binding at 0.6 and 1.2 bead-to-sample volume ratios removed input DNA and unincorporated primers from the amplicons. The final library molecules were 125-300 bp in size. We then transferred the libraries to the Ion OneTouch™ System for automated template preparation. Sequencing was performed on the Ion PGM™ sequencer according to the manufacturer's instructions. We used IonTorrent Software for automated data analysis.
nCounter Copy Number Variation CodeSets
For detection of CNV, nCounter Copy Number Variation CodeSets were used with 300 ng of purified genomic DNA extracted from 2-3 sections of 4-μm-thick FFPE representative tumor blocks using a QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany). DNA was fragmented via AluI digestion and denatured at 95°C. Fragmented DNA was hybridized with the codeset of 86 genes in the nCounter Cancer CN Assay Kit (Nanostring Technologies) for 18 hours at 65°C and processed according to the manufacturer's instructions. The nCounter Digital Analyzer counted and tabulated the signals of reporter probes.
Analytical methods
We excluded all synonymous changes after an automated mutation-calling algorithm was used to detect supposed mutations. We used cutoff values of greater than 6% variant frequency and more than X100 coverage to detect true mutational changes in accordance with previous studies and our own experience. We filtered out single-nucleotide polymorphisms after a manual review of each polymorphism in the Catalogue of Somatic Mutations in Cancer (COSMIC, http://cancer.sanger.ac.uk/cancergenome/projects/cosmic). Descriptive statistics were reported as proportions and medians.
Ethics statement
The Ethics Committee at Samsung Medical Center approved the study in accordance with the Declaration of Helsinki. All individuals gave written informed consent for participation in the study.
Results
Patient characteristics
Among the 322 patients with refractory GI and rare cancers analyzed by molecular profiling using a high-throughput molecular test between November 2013 and August 2014, a total of 14 patients with a diagnosis of NET arising from the digestive system were found. Baseline characteristics of these 14 patients are listed in Table 1. The median age of the patients was 50.5 years (range, 34-72) and the male to female ratio was 1.00. All patients had an ECOF performance status of 1. According to WHO classification, 8 patients had grade III neuroendocrine carcinoma (NEC) and 6 had grade I/II NET. The most common primary site was the distal colon and rectum (N=5), followed by the small intestine and ascending colon (N=3), pancreas (N=3), and unknown primary origin (N=3). Liver (50.0%) was the predominant metastatic site, followed by the lymph node (28.6%), bone (7.2%), and spleen (7.2%). The most commonly used regimen as first line therapy was etoposide plus cisplatin (EP) (N=8, 57.1%), followed by everolimus (n=3, 21.4%), somatostatin analogues (n=2, 14.3%), and pazopanib (n=1, 7.2%).
Baseline characteristics
| Variables | N (%) | |
|---|---|---|
| Gender | ||
| Male | 7 | 50% |
| Female | 7 | 50% |
| Age, years | ||
| Median, range | 50.5 | 34-72 |
| ECOG performance status | ||
| 0-1 | 14 | 100% |
| WHO grade | ||
| Grade I/II neuroendocine tumor (NET) | 6 | 42.9 |
| Grade III neuroendocrine carcinoma (NEC) | 8 | 57.1 |
| Primary site | ||
| Pancreas | 3 | 21.4% |
| Small intestine and ascending colon | 3 | 21.4% |
| Distal colon and rectum | 5 | 35.7% |
| Unknown primary sites | 3 | 21.4% |
| Metastatic sites | ||
| Liver | 7 | 50.0% |
| Lymph nodes | 4 | 28.6% |
| Bone | 1 | 7.2% |
| Spleen | 1 | 7.2% |
| 1st line therapies | ||
| Etoposide/Cisplatin | 8 | 57.1% |
| Everolimus | 3 | 21.4% |
| Octreotide | 2 | 14.3% |
| Pazopanib | 1 | 7.2% |
Mutational profiling
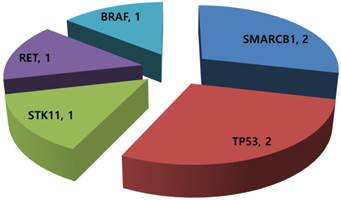
Of the 14 GEP-NET patients available for mutational profiling, 7 (50.0%) patients had one or more aberrations detected. Any patients did not have multiple genetic aberrations. Three patients were grade I/II NET and four were grade III neuroendocrine carcinoma (NEC). Common aberrations were as follows: SMARCB1 mutation (n=2), TP53 mutation (n=2), STK11 mutation (n=1), RET mutation (n=1), and BRAF mutation (n=1) (Figure 2). The two patients with a TP53 mutation were grade III NEC.
Mutational profiling in 14 metastatic GEP-NETs patients.
nCounter CNV analysis
Gene amplification by nCounter was detected in only one patient, who showed CCNE1 amplification (Table 2) and simultaneously had a TP53 mutation. This patient was grade III NEC with unknown primary origin.
Tissue molecular alterations
| No. of patients | |
|---|---|
| Molecular analysis ordered | 14 |
| Adequate tissue avalable | 14 |
| No. of mutation | |
| 0 | 7 |
| 1 | 7 |
| No. of patients with mutation | 7 |
| No. of amplification | 1 |
| No. of patients with amplification | 1 |
Discussion
We analyzed a heterogeneous collection of metastatic GEP-NETs refractory to standard systemic therapies for comprehensive molecular profiling. Molecular profiling with a combination of cancer panel screening and a nanostring 21-gene CNV assay had a success rate of 100%. We were able to apply the molecular profiling from all 14 GEP-NET patients to a comprehensive molecular test. Our analysis showed that 7 (50.0%) of the 14 patients had one or more aberrations detected. The most common aberration was a SMARCB1 mutation (n=2), followed by a TP53 mutation (n=2), STK11 mutation (n=1), RET mutation (n=1), and BRAF mutation (n=1). Gene amplification by nCounter was detected in only 1 patient, who showed CCNE1 amplification and also had a TP53 mutation. Currently, genetic alterations including mutation and amplification from this analysis might be considered as novel molecular targets for newly developed targeted agents. Our findings provide useful information and serve as a background for future clinical trials of genomic-based therapy in GEP-NETs.
Through the clinical and molecular genetic studies of family members of patients with GEP NETs, the underlying alterations in those families have been characterized. In contrast, less is known about the genetic mechanism of sporadic GEP NETs, although genes involved in their familial counterparts also play a role in the molecular pathology of sporadic tumors [10]. Maitra A et al attempted to analyze malignant pancreatic NETs using a genome-wide expression microarray [11]. That study revealed a greater than three-fold increase in the expression levels of 66 transcripts, including IGFBP3, and underexpression of 119 transcripts, including p21, O6-MGMT, and JunD. More recently, Duerr et al conducted DNA microarray analysis and hierarchical clustering of 19 pancreatic NETs [12]. The most highly upregulated genes in the malignant group of pancreatic NET were FEV, adenylate cyclase 2 (ADCY2), nuclear receptor subfamily 4, group 4, member 2 (NR4A2), and growth arrest and DNA-damaged inducible beta (GADD45b). However, these molecular genetic studies, including our analysis, used different molecular-analysis platforms and applied them to different patient populations. Thus, the molecular genetic profiles produced from these studies were inconsistent [11, 12]. We conducted a comprehensive integrated genomic analysis including identification of mutations and CNVs. Our profiling focused on identifying new drug targets in GEP-NETs to use the developed or developing targeted agents.
Our analysis detected some genetic alterations in GEP-NETs, as follows: SMARCB1 mutation (n=2), TP53 mutation (n=2), STK11 mutation (n=1), RET mutation (n=1), BRAF mutation (n=1), and CCNE1 amplification (n=1). These genetic alterations have molecularly matched targeted agents [13]. Those molecularly matched agents are as follows: Pazopanib for the SMARCB1 mutation, AZD1775 for TP53, everolimus for STK11, imatinib for the RET mutation, vemutafenib for the BRAF mutation, and cyclin dependent kinase (CDK) inhibitors for CCNE1 amplification. Currently, there are very limited therapeutic options for treatment of metastatic GEP-NETs. Matched therapies based on molecular profiling could provide new therapeutic opportunities in metastatic GEP-NET patients who are lacking in therapeutic options.
Our study has some limitations including a small sample size and heterogeneous patient population. This study couldn't present the definitive key for treatment-strategies for GEP-NETs. However, this analysis identified novel molecular therapeutic targets such as TP53 mutation, and BRAF mutation in metastatic GEP-NETs. Molecular targeted agents could be considered to GEP-NETs patients with these genetic aberrations. Thus, this data may provide useful information and serve as a background for future clinical trials with new novel targeted agents for GEP-NET. We are now performing prospective molecular profiling with comprehensive genomic analysis for supporting matched targeted agents to refractory solid cancer patients.
The rarity of GEP-NETs hinders clinicians from conducting definitive trials and from producing rigorous scientific data. Thus, coordination of trials among institutions and cooperative groups, both nationally and internationally, will be the key to improving treatment outcomes in GEP-NETs. Additionally, because of the heterogeneity of GEP-NETs, appropriate stratification using clinical and biological factors such as primary sites, WHO grade and KI67 will help to define more clearly outcomes of the research.
Conclusions
This high throughput genomic test may be useful to identify new drug targets in metastatic GEP-NET patients. Currently, we plan to conduct further genomic analysis to develop predictive and prognostic biomarkers in a larger number of GEP-NET patients.
Supplementary Material
Supplementary table 1.
Acknowledgements
This work was supported by funding from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI14C3418 and HI14C2750). Support was also provided by a grant from the Samsung Medical Center (SMX1161251).
Authors' contributions
All authors made substantial contributions to the conception and design of the study, and acquisition, analysis, and interpretation of the data. All authors were involved in drafting the manuscript (or revising it), and all read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Williams ED, Sandler M. The classification of carcinoid tum ours. Lancet. 1963;1:238-239
2. Yao JC, Hassan M, Phan A. et al. One hundred years after "carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063-3072
3. Gastrointestinal Pathology Study Group of Korean Society of P, Cho MY, Kim JM. et al. Current Trends of the Incidence and Pathological Diagnosis of Gastroenteropancreatic Neuroendocrine Tumors (GEP-NETs) in Korea 2000-2009: Multicenter Study. Cancer Res Treat. 2012;44:157-165
4. Rinke A, Muller HH, Schade-Brittinger C. et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27:4656-4663
5. Yao JC, Shah MH, Ito T. et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514-523
6. Raymond E, Dahan L, Raoul JL. et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501-513
7. Garraway LA, Verweij J, Ballman KV. Precision oncology: an overview. J Clin Oncol. 2013;31:1803-1805
8. Von Hoff DD, Stephenson JJ Jr, Rosen P. et al. Pilot study using molecular profiling of patients' tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol. 2010;28:4877-4883
9. Kruglyak KM, Lin E, Ong FS. Next-generation sequencing in precision oncology: challenges and opportunities. Expert Rev Mol Diagn. 2014;14:635-637
10. Starker LF, Carling T. Molecular genetics of gastroenteropancreatic neuroendocrine tumors. Curr Opin Oncol. 2009;21:29-33
11. Maitra A, Hansel DE, Argani P. et al. Global expression analysis of well-differentiated pancreatic endocrine neoplasms using oligonucleotide microarrays. Clin Cancer Res. 2003;9:5988-5995
12. Duerr EM, Mizukami Y, Ng A. et al. Defining molecular classifications and targets in gastroenteropancreatic neuroendocrine tumors through DNA microarray analysis. Endocr Relat Cancer. 2008;15:243-256
13. Le Tourneau C, Paoletti X, Servant N. et al. Randomised proof-of-concept phase II trial comparing targeted therapy based on tumour molecular profiling vs conventional therapy in patients with refractory cancer: results of the feasibility part of the SHIVA trial. Br J Cancer. 2014;111:17-24
Author contact
![]() Corresponding authors: Young Suk Park, M.D., Ph.D., Division of Hematology/Oncology, Department of Medicine Samsung Medical Center, Sungkyunkwan University School of Medicine, 50 Irwon-dong Gangnam-gu, Seoul 135-710 Korea. Tel.: +82-2-3410-3451, Fax: +82-2-3410-1754; Email: pys27hmoedu or Jeeyun Lee, MD, Division of Hematology/Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 135-710, Korea. Tel: +82-2-3410-1779; Fax: +82-2-3410-1754; E-mail: jyunleeedu
Corresponding authors: Young Suk Park, M.D., Ph.D., Division of Hematology/Oncology, Department of Medicine Samsung Medical Center, Sungkyunkwan University School of Medicine, 50 Irwon-dong Gangnam-gu, Seoul 135-710 Korea. Tel.: +82-2-3410-3451, Fax: +82-2-3410-1754; Email: pys27hmoedu or Jeeyun Lee, MD, Division of Hematology/Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 135-710, Korea. Tel: +82-2-3410-1779; Fax: +82-2-3410-1754; E-mail: jyunleeedu