Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(8):1002-1009. doi:10.7150/jca.14645 This issue Cite
Research Paper
Glypican-1 as a Biomarker for Prostate Cancer: Isolation and Characterization
Quach Truong, Irene O. Justiniano, Aline L. Nocon, Julie T. Soon, Sandra Wissmueller, Douglas H. Campbell, Bradley J. Walsh ![]()
Minomic International Ltd, Suite 2, Ground Floor, 75 Talavera Rd, Macquarie Park, NSW 2113, Australia
Received 2015-12-10; Accepted 2016-3-22; Published 2016-5-21
Abstract
Prostate cancer is the most frequently diagnosed male visceral cancer and the second leading cause of cancer death in the United States. Standard tests such as prostate-specific antigen (PSA) measurement have poor specificity (33%) resulting in a high number of false positive reports. Consequently there is a need for new biomarkers to address this problem. The MIL-38 antibody was first described nearly thirty years ago, however, until now, the identification of the target antigen remained elusive. By a series of molecular techniques and mass spectrometry, the MIL-38 antigen was identified to be the highly glycosylated proteoglycan Glypican-1 (GPC-1). This protein is present in two forms; a membrane bound core protein of 55-60 kDa and secreted soluble forms of 40 kDa and 52 kDa. GPC-1 identification was confirmed by immuno-precipitation, western blots and ELISA. An ELISA platform is currently being developed to assess the levels of GPC-1 in normal, benign prostatic hyperplasia (BPH) and prostate cancer patients to determine whether secreted GPC-1 may represent a clinically relevant biomarker for prostate cancer diagnosis.
Keywords: Prostate Cancer, Glypican 1, Proteoglycan, Theranostic
Introduction
Many potential prostate cancer biomarkers have been identified to assist in the diagnosis and prognosis of the disease1. As prostate cancer is the most frequently diagnosed male visceral cancer and the second leading cause of cancer death in the United States it is unsurprising that there has been such a broad effort. The American Cancer Society estimates that in 2015 about 220 800 new cases of prostate will be diagnosed and 27 540 men will die of the disease.
Digital rectal exam (DRE) and measurement of PSA in the blood of patients are the two major prostate cancer detection tests currently used by medical professionals. Unfortunately, neither of these tests are entirely conclusive and both can result in false negatives (leaving real cancers undetected), and false positives (signalling cancer where there is none). In fact, the poor specificity (33%) exhibited by the PSA test has led to controversy about how and when it should be used2,3. Consequently, a number of biomarkers have been developed as companion diagnostics to improve the PSA test4,5. Multianalyte tests such as the PHI test (measuring three different PSA markers: PSA, free PSA and p2PSA)6 and the OPKO 4K score (measuring four prostate-specific kallikreins)7 have improved the sensitivity of the PSA test but specificity still remains poor. As one of the most powerful tools against cancer is early accurate detection, the search continues for biomarkers that will help to address this issue.
Cell surface markers of disease have potential to be theranostics, that is, they have both diagnostic and therapeutic applications. There is likelihood that such surface molecules will be shed into the body fluids, thus allowing their measurement in a non-invasive fashion. In situ antibodies or aptamers to cell surface molecules can be used to target the location of tumors using medical imaging and to deliver toxic payloads to the tumor in the form of radio-immunotherapy or antibody drug conjugates8,9.
The MIL-38 monoclonal antibody, previously known as BLCA-38, was originally raised to a primary bladder cancer tumor10,11. MIL-38 showed specific binding to prostate cancer cell lines and prostate cancers, with no reactivity to normal prostate or other normal tissues tested12. The MIL-38 antigen therefore represents a potential theranostic marker for prostate cancer. This paper describes for the first time the identity of the MIL-38 antigen as the cell surface antigen Glypican-1.
Materials and Methods
DU-145 and C3 cell lines
The prostate cancer cell line DU-145 cells were purchased from ATCC. Antigen negative C3 cells12 were kindly provided by Professor Pamela Russell (Australian Prostate Cancer Research Centre, Institute of Health and Biomedical Innovation, Queensland University of Technology, Australia). MIL-38 hybridomas were provided by Professor Pamela Russell and also obtained from the ATCC. Cells were cultured according to standard tissue culture techniques at 37°C with 5% CO2. DU-145 cells were grown in RPMI-1640 medium supplemented with 10% Newborn Calf Serum (NCS) and C3 cells in RPMI-1640 supplemented with 20% Foetal Bovine Serum (FBS) in T175 flasks (Greiner Bio-one, Frickenhausen, Germany). To harvest conditioned media, cells were grown in RPMI in the absence of NCS for 24 to 48 h. For scale up and harvest, cells were briefly washed with PBS and detached using 2mM EDTA in PBS at 37°C for 15 min. Cells were resuspended in PBS and kept on ice until required.
MIL-38 production
Frozen hybridoma cell stocks were thawed followed by resuspension in RPMI-1640 medium (Life Technologies, Carlsbad, California, US) and allowed to grow at 37°C with 5% CO2 for 72 h. Cells were expanded, split and scaled up in a sequential process. At each step, cells were resuspended in fresh medium and incubated at 37°C with 5% CO2. After scale up, cells were transferred to sterile serum-free medium and grown till the start of death phase. The supernatant was harvested to collect the MIL-38 antibody, filter sterilized and stored at -80°C until required. Antibody was purified using Pierce Protein G Agarose (Thermofisher Scientific, Waltham Massachusetts, US) according to the manufacturer's recommendations.
Antibodies and proteins used
MIL-38 antibody was produced in-house as described above. Rabbit anti-GPC1 antibody was purchased from Abcam (Cat no. PN000-06211, Cambridge, UK). A recombinant GPC-1 protein produced in murine NS0 cells was purchased from R&D Systems (Cat no. 4519-GP, Minneapolis, Minnesota, US).
Flow cytometry
Detached cells were counted and aliquoted to a concentration of 2 x 105 cells per sample. Primary antibody (MIL-38) and secondary antibodies were prepared in Wash Solution (5% FBS in PBS). Cells were incubated with MIL-38 (2μg in 50 μL) at 4°C for 45 min followed by three washes with Wash Solution. Goat anti-mouse Alexa Fluor® 488 (Life Technologies: 2μg in 50 μL) secondary antibody incubations were performed at 4°C for 30 min in the dark followed by three washes in Wash Solution. The cells were stored as a pellet on ice until flow cytometric analysis. Samples were run on a BD LSR Fortessa X-20 (Becton, Dickinson and Company, Franklin Lakes, New Jersey, US). All parameters were set to log scale at the following voltages: FSC: 115. SSC: 88, Alexa Fluor 488: 280.
Protein Extraction, SDS PAGE and Western Blot Analysis
Membrane proteins were extracted from DU-145 and C3 cells using the ProteoExtract Native Membrane Protein Extraction Kit (MPEK: Merck Millipore, Darmstadt, Germany) according to manufacturer's instructions. Extracted proteins were mixed with non-reducing SDS sample buffer unless specified and loaded onto 4-15% precast Criterion TGX (Tris-Glycine) gels (Bio-Rad, Hercules, California, US). Gels were run for 10 min at 80V and additional 50 min at 200V in Tris-Glycine running buffer. Proteins were transferred to nitrocellulose membranes (Bio-Rad) using the Transblot Turbo system (Bio-Rad). Transfer was at 2.5A, 25V maximum for 10 min.
For visualisation, all incubations were performed at room temperature. Gels were fixed with staining/destaining solution (10% Ethanol, 7% acetic acid) for 2 h followed by overnight incubation with Sypro®Ruby (Thermofisher Scientific) in the dark. Gels were rinsed and washed with destaining solution for a minimum of 2 h. Imaging was performed with a Pharos X Scanner (Bio-Rad). If required, gels were post stained with Bio-SafeTM Coomassie (Bio-Rad).
Western blot was performed by blocking the membranes with 5% skim milk in PBS-0.1% Tween-20 for 2h at room temperature. MIL-38 or rabbit anti-GPC-1 antibodies (1µg/ml in 5% skim milk - PBS- 0.1% Tween) were applied and incubated overnight at 4ºC. The primary antibody was omitted for negative controls. After washing (3 x 10 min PBS-0.1% Tween) membranes were incubated with either 1:2,000 sheep-anti-mouse HRP-labelled secondary antibody (GE Healthcare Life Sciences, Cleveland, Ohio, US) or 1:2,000 goat anti-rabbit HRP labelled secondary antibody (Abcam) in 5% skim milk - PBS-0.1% Tween for 1 h at room temperature. After washing (3 x 10 min PBS-0.1%Tween) antigen was detected using ECL detection kit (Bio-Rad) and imaged with a LAS4000 mini imager (GE Healthcare Life Sciences).
Protein expression levels were quantified by using Image J (National Institutes of Health, Bethesda, Maryland). Comparative protein bands were individually selected and the area measured for each band.
Two dimensional PAGE
Membrane protein lysates (180 μg) were desalted using 7M Urea, 2M thiourea in 3,000 MWCO spin columns (Sartorius AG, Göttingen, Germany) until conductivity was 210 μS. Four percent CHAPS (w/v) and bromophenol blue was added. Samples were spun at 20 000 x g to remove precipitates and used to rehydrate pI 3-10 IPG strips (Bio-Rad) overnight. Isoelectric focusing (IEF) was performed at 500V for 1 h (step and hold), 100V for 1 h (gradient), 8 000V for 3 h (gradient) and 8 000V for a further 3 h (step and hold). IPG strips were then equilibrated twice with non-reducing solution (2% SDS, 6M Urea, 20% glycerol, 0.375M Tris-HCl, pH 8.8). Strips were run on precast 4-15% TGX gels (Bio-Rad) at 50V for 10 min followed by 200V for 1 h.
Immuno-precipitation
Dynabeads (Life Technologies, Cat no. 14311D) were conjugated with MIL-38 (5μg per mg of beads) at 37°C overnight as per manufacturer's instructions. The conjugated beads were then washed and incubated with membrane protein extracts at 4°C overnight. Prior to gel running or mass spectrometry, captured antigen was eluted using a low pH buffer (0.2M glycine, pH 3.5, 0.5% Triton-X 100) for 5 min followed by neutralization (1M Tris-HCl, pH 9.0).
Immuno-precipitations were performed as above with a commercial rabbit anti-GPC-1 antibody. This antibody was used for all confirmatory and comparative analysis.
Antigen selection and Mass Spectrometry
Antigen was immuno-precipitated from DU-145 extracts, eluted with low pH buffer, then run on two parallel gels. One gel was processed using the western blot procedure and the other run and stained by Sypro®Ruby and Coomassie Blue. The western blots were used as a reference to confirm recognition of MIL-38 to the antigen. Bands were cut blind from stained gels for mass spectrometry using molecular weight markers as a guide. Gel bands were destained, reduced (25 mM DTT) and alkylated (55 mM iodoacetamide (IAA) in 25 mM ammonium bicarbonate), dried, then digested with trypsin in ammonium bicarbonate (pH 8) overnight. Peptides were extracted with acetonitrile (ACN):water:formic acid (50:45:5) then vacuum concentrated.
For gel free analysis, immuno-precipitated antigen was reduced for 30 min at 65°C in 10 mM DTT in 7M urea, 2M thiourea in water. Proteins were alkylated with 55 mM IAA in 100 mM ammonium bicarbonate for 20 min in the dark. Liquid samples were diluted 1:10 with 25 mM ammonium bicarbonate, followed by overnight TPCK trypsin (500 ng) digestion at 37°C. Peptides were extracted with ACN:water:formic acid (50:45:5) then vacuum concentrated.
For mass spectrometry, sample (10 μL) was injected onto a peptide trap column (Michrome peptide Captrap) for pre-concentration and desalted with 0.1% formic acid, 2% acetonitrile, at 5 μL/min for 10 minutes. The peptide trap column was then switched into line with the analytical column. Peptides were eluted from the column using a linear gradient from H2O:ACN (98:2; + 0.1% formic acid) to H2O:ACN (10:90; + 0.1% formic acid) at 1 µL/min over a 120 min period. The eluent was subject to positive ion nanoflow electrospray MS analysis in an information dependent acquisition mode (IDA). In the IDA mode a TOFMS survey scan was acquired (m/z 350-1200, 0.5 sec), with the ten largest multiply charged ions (counts >150) in the survey scan sequentially subjected to MS/MS analysis. MS/MS spectra were accumulated for 100 milli-seconds (m/z 100-1500) with rolling collision energy. Peak lists generated were then searched using Mascot (Matrix Science, Boston, Massachusetts, USA) against Homo sapiens proteins to identify the antigen.
Results
Flow cytometric analysis of MIL-38 binding
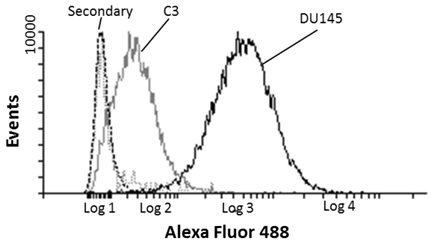
Prostate cancer DU-145 and C3 negative control cells were indirectly labelled with MIL-38 and analysed by FLOW cytometry. MIL-38 recognised a surface antigen of DU-145 cells with a two log increase over C3 cells (Figure 1). Secondary antibody controls only were used as the baseline. These results were consistent with those reported previously12.
Flow cytometric analysis of MIL-38 binding to the surface antigen on DU-145 and C3 cells. The secondary antibody only control (DU-145 (dash) and C3 (dotted) cells) represents the baseline of the analysis. DU-145 cells show strong staining and C3 cells represent a negative population sample.
Characterization of the membrane associated MIL-38 antigen
Western blots of DU-145 membrane extracts indicated that MIL-38 recognized a protein band of between 55-60 kDa under non-reduced conditions (Figure 2A). Increasing the concentration of dithiothreitol (DTT) in the sample buffer resulted in decreased binding of MIL-38 to the antigen. Reactivity was lost if DTT concentration was greater than 0.8mM. These results indicate that the MIL-38 epitope is reduction sensitive and the epitope is most likely a structural determinant.
A. Western Blot of DU-145 membrane protein extracts treated with increasing concentrations of DTT. DU-145 (Lane 1) and C3 (Lane 2) are positive and negative controls respectively. B. Western Blot of immuno-precipitated of DU-145 membrane protein extracts (Lane A). The solid arrow indicates the immuno-precipitated antigen and the outlined arrow indicates the MIL-38 antibody. Antigen is not immuno-precipitated in Lanes (B) representing protein extracts with unconjugated beads or Lane (C) which represents antibody conjugated beads with no protein extracts. C. Western blot of membrane protein extracts from prostate cancer and normal prostate samples. The prostate cancer sample shows strong reactivity with MIL-38 at thirty times more expression than normal prostate samples.
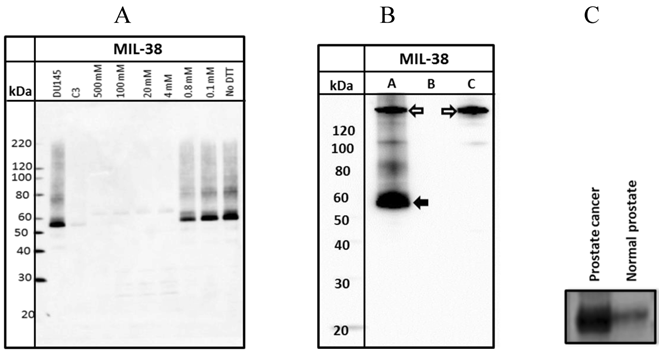
MIL-38 immuno-precipitations of DU-145 membrane extracts followed by MIL-38 western blotting also detected the 55-60 kDa antigen (Figure 2B). In the absence of either conjugated beads or protein lysate, no antigen was captured (negative controls). No antigen was detected in C3 protein lysates and C3 immuno-precipitated precipitated extracts (results not shown).
To determine if the MIL-38 antigen could be detected in membrane protein extracts from normal prostate and prostate cancer, one sample of each was obtained from Novus Biologicals (Littleton, Colorado, USA). Equivalent amounts of protein were western blotted using MIL-38 antibody (Figure 2C). The prostate cancer extract demonstrated thirty times more protein expression of the MIL-38 antigen than the normal prostate sample.
MIL-38 can detect antigen secreted into cell culture supernatant
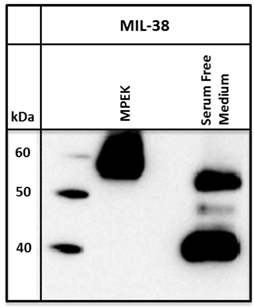
Conditioned serum-free media from DU-145 cells was harvested and immuno-precipitated with MIL-38 conjugated beads. A standard immuno-precipitation from DU-145 membrane protein extracts was run for comparison. For the serum free medium, bands of approximately 40 and 50-52 kDa were observed on the Western blot, compared to the 55-60 kDa band isolated from DU-145 membrane protein extracts (Figure 3).
Western blot of MIL-38 antigen secreted into conditioned serum free medium by DU-145 cells. Two proteins of sizes 40kDa and 52kDa were identified. DU-145 membrane protein extracts (MPEK) immuno-precipitates were run as controls and show the 55-60kDa antigen.
Identification of the MIL-38 antigen by mass spectrometry
To identify the MIL-38 antigen by mass spectrometry, DU-145 immuno-precipitates were processed in two ways. The immuno-precipitates were eluted ('gel free') and subjected to mass spectrometry. Alternatively, the immuno-precipitates were run on two identical gels; a western blot was performed using one gel and this was used to approximate the location of the band corresponding to the antigen. A region of the gel pertaining to the antigen was excised from the second gel and then subjected to mass spectrometry. Band excision was performed for the 40kDa and 52 kDa secreted antigen. Control samples were analyzed in similar manners.
Analysis of the mass spectrometry data using Mascot identified the MIL-38 antigen as Glypican-1 (GPC-1; Swissprot P35053). Samples were either excised from gels (n=4) or directly eluted (n=2) prior to analysis. The highest peptide score of 4278 represents 18 distinct peptide sequences covering 46% of the GPC-1 sequence (Figure 4). GPC-1 was not detected in any of the control samples (MIL-38 conjugated Dynabeads alone or DU-145 extracts incubated with unconjugated Dynabeads).
Mass spectrometric identification of the MIL-38 antigen (GPC-1). Identified amino acids are in bold (78% coverage). Amino acids identified in the secreted form are highlighted in grey.

The MIL-38 antigens from the 40 kDa and 52 kDa bands were also identified as GPC-1. The 40 kDa band (Table 1) identified 48 peptides with sequence coverage of 18%. The 52 kDa band (Table 1) identified 39 peptides with sequence coverage of 24%. These results suggest that a MIL-38-reactive form of GPC-1 can be released into cell culture supernatant from the DU145 prostate cancer cell line.
Identification of the MIL-38 antigen (GPC-1) by various characterization experiments. Scoring is based on the number of peptides isolated and the frequency using the MASCOT protein database
| Score | Number of peptides | Protein Coverage (%) | |
|---|---|---|---|
| MIL-38 Immuno-precipitation | 4278 | 166 | 46 |
| Secreted 52 kDa | 1364 | 39 | 24 |
| Secreted 40 kDa | 1531 | 48 | 18 |
Confirmation of the MIL-38 antigen as GPC-1
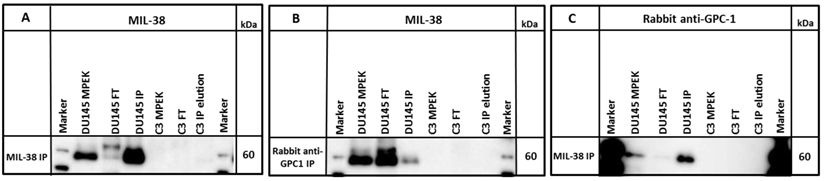
A commercial rabbit anti-GPC-1 was used to probe MIL-38 immuno-precipitates and DU-145 membrane extracts. The antibody detected an antigen of ~60 kDa in both DU-145 membrane extracts and also in the MIL-38 immuno-precipitates. Similarly, rabbit anti-GPC-1 immuno-precipitates probed with MIL-38 identified the same size 60 kDa antigen (Figure 5) and extracts showed almost complete depletion of the 60 kDa antigen following immuno-depletion with either antibody.
Western blot of immuno-precipitates (IP) of MIL-38 or rabbit anti-GPC-1 conjugated beads with DU145 or C3 protein lysates (MPEK). DU145 protein lysates were immuno-precipitated by MIL-38 (A, C) and rabbit anti-GPC-1 (B) and probed by MIL-38 (A, B) or rabbit anti-GPC-1 (C). C3 protein lysates were not immuno-precipitated by either antibody. FT (flow through) is defined as the proteins in the supernatant not captured by the conjugated beads.
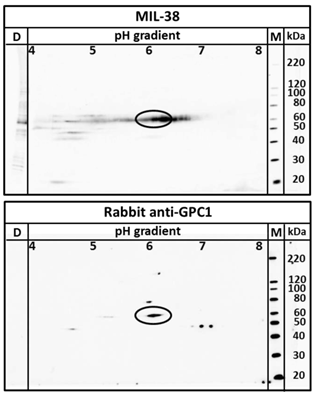
Two dimensional electrophoresis was used to confirm MIL-38 and rabbit anti-GPC-1 recognised the same antigen in DU-145 protein extracts. As shown in Figure 6, MIL-38 antibody and the rabbit anti-GPC-1 antibodies showed overlapping reactivity detecting a protein with 60 kDa molecular weight and isoelectric points ranging from 5 to 7.
Two dimensional western blot using MIL-38 and rabbit anti-GPC-1 on DU-145 membrane protein extracts. The antigen identified by both antibodies overlap at 60kDa at isoelectric point 6 (indicated by ellipse).
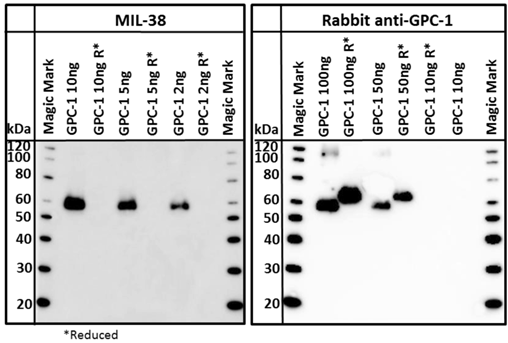
A commercially available recombinant GPC-1 protein was tested for reactivity with MIL-38 and rabbit anti-GPC-1 antibodies. This full-length GPC-1 was produced in murine NS0 cells. Strong reactivity of both MIL-38 and rabbit anti-GPC-1 antibodies with this protein was observed (Figure 7) under non-reducing conditions. Rabbit anti-GPC-1 was also able to react with the reduced form of the recombinant protein. The characterization and confirmatory results described all strongly suggest that the antigen is GPC-1.
Western blot of recombinant GPC-1. Both MIL-38 and rabbit anti-GPC-1 recognize the same non reduced forms of the protein. Rabbit anti-GPC-1 can also recognize the reduced form of the protein. R* indicates that the sample was processed under reducing conditions.
GPC-1 ELISA
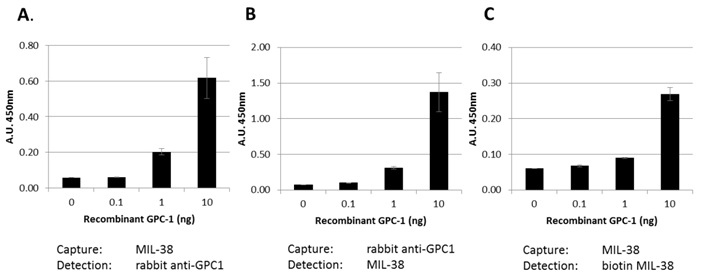
Our data indicates that a soluble form of GPC-1 can be detected in DU-145 conditioned medium. This suggests that the MIL-38 antibody may be useful in detecting soluble forms of GPC-1 in body fluids. Three MIL-38-based ELISA assay formats were used to test this (Figure 8). Recombinant GPC-1 was tested at 0, 0.1, 1 and 10 ng/ml concentrations using MIL-38 as a capture antibody and rabbit anti-GPC-1 as a detection antibody. Similar experiments were conducted using the rabbit anti-GPC-1 as the capture antibody and the MIL-38 as the detection antibody. A single antibody ELISA was also tested using the MIL-38 antibody as the capture antibody, and a biotinylated MIL-38 antibody as the detection antibody. All assay formats were able to detect recombinant GPC-1 indicating that MIL-38 is a suitable candidate for further ELISA development.
Bar graph showing various ELISA combinations of MIL-38 and rabbit anti-GPC-1. Recombinant GPC-1 was used as the analyte. All ELISA combinations show recombinant GPC-1 can be detected in a sandwich ELISA by MIL-38 as a capture antibody (A, C) and detector antibody (B, C).
Discussion
GPC-1 belongs to a six member family of heparan sulfate proteoglycans (HSPGs)13 linked to the cell surface by a glycosylphosphatidylinositol (GP1) anchor14,15. The size of the glypican core protein is 60-70 kDa16,17. The antigen target for MIL-38 was detected in two forms: a membrane bound cell surface form of 55-60 kDa and soluble secreted forms of 52 kDa and 40 kDa. This supports other findings which show cancer cells can shed GPC-1 into the tumor microenvironment18,19.
Another member of the family, Glypican-3 (GPC-3) is known to be processed in the Golgi apparatus to form an N-terminal 40 kDa and a C-terminal 30 kDa subunit which have been proposed as serological markers for early stage hepatocellular carcinoma20. When the protein is translocated to the cell surface, a second proteolytic event cleaves the GPI anchor to release the subunits. It is therefore reasonable to assume that GPC-1 could be similarly cleaved resulting in the observed 40 kDa and 52 kDa forms of GPC-1.
Studies have shown glypicans have the ability to regulate the activity of growth and survival factors in tissue, suggesting glypicans play a role in cancer progression21. Reduced GPC-1 expression in pancreatic cell lines (PANC-1) has been shown to inhibit tumor progression22 and reduced angiogenesis and metastasis23.
Of recent interest is the identification of GPC-1 on the surface of exosomes isolated from pancreatic cancer cell line PANC-124, breast cancer cell lines25 and prostate cancer cell lines26. Exosomes are secreted extracellular vesicles which contain the nucleic acids and protein information of the origin cell27,28 and are of interest regarding its role in immune response29, cell signalling30 cancer31 and tumor progression32-34. It can be suggested that GPC-1 is a specific marker for cancer exosomes and could be used to identify cancer in patients.
GPC-1 is an attractive prostate cancer biomarker as it is present on the cell surface and also shed by the cells. It's identification on exosomes from blood and urine allows an avenue for targeted personalized therapy as the genetic and proteomic mutations are specific to the cancer patient. Future studies are underway to develop a MIL-38 based GPC-1 ELISA to assess the levels of GPC-1 in normal, BPH and prostate cancer patients to determine whether secreted GPC-1 may represent a clinically relevant biomarker for prostate cancer diagnosis. Preliminary data presented at the 2015 American Urology Association supports GPC-1 as an unrelated candidate biomarker with improved specificity (79%) over PSA35, thereby reducing false positive reporting and the need for unnecessary biopsies. Furthermore, MIL-38 labelled with alpha emitting radioactive isotopes is able to suppress the growth of human prostate cancer subcutaneous xenografts in vivo36 and also as a targeting antibody for tissue specific gene therapy delivery37. The authors are currently studying the effects of GPC-1 modulation in prostate cancer using molecular techniques to examine growth and invasiveness in in vitro and in vivo models. Therefore, MIL-38 and its antigen target show great potential as a theranostic agent and biomarker for the early detection and treatment of prostate cancer.
Acknowledgements
The authors would like to thank Professor Pamela Russell for providing the C3 cell lines and BLCA-38 hybridoma and the Australian Proteome Analysis Facility for mass spectrometric analysis.
Conflict of Interest
The authors declare no conflicts of interest. The authors are employed by and are shareholders in Minomic International Ltd.
References
1. Velonas VM, Woo HH, Remedios CGd, Assinder SJ. Current status of biomarkers for prostate cancer. International Journal of molecular sciences. 2013;14(6):11034-11060
2. Barry MJ. Screening for prostate cancer—the controversy that refuses to die. New England Journal of medicine. 2009;360(13):1351-1354
3. McNaughton-Collins MF, Barry MJ. One man at a time—resolving the PSA controversy. New England Journal of medicine. 2011;365(21):1951-1953
4. Dijkstra S, Mulders P, Schalken J. Clinical use of novel urine and blood based prostate cancer biomarkers: a review. Clinical biochemistry. 2014;47(10):889-896
5. Bratt O, Lilja H. Serum markers in prostate cancer detection. Current opinion in urology. 2015;25(1):59-64
6. Ferro M, Bruzzese D, Perdona S, Marino A, Mazzarella C, Perruolo G. et al. Prostate Health Index (Phi) and Prostate Cancer Antigen 3 (PCA3) significantly improve prostate cancer detection at initial biopsy in a total PSA range of 2-10 ng/ml. PLoS one. 2013;8(7):e67687
7. Bryant RJ, Sjoberg DD, Vickers AJ, Robinson MC, Kumar R, Marsden L. et al. Predicting high-grade cancer at ten-core prostate biopsy using four kallikrein markers measured in blood in the ProtecT study. Journal of the National Cancer Institute. 2015;107(7):djv095
8. Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: targeted drug delivery for cancer. Current opinion in chemical biology. 2010;14(4):529-537
9. Senter PD. Potent antibody drug conjugates for cancer therapy. Current opinion in chemical biology. 2009;13(3):235-244
10. Russell PJ, Jelbart M, Wills E, Singh S, Wass J, Wotherspoon J. et al. Establishment and characterization of a new human bladder cancer cell line showing features of squamous and glandular differentiation. International Journal of cancer. 1988;41(1):74-82
11. Walker KZ, Russell PJ, Kingsley EA, Philips J, Raghavan D. Detection of malignant cells in voided urine from patients with bladder cancer, a novel monoclonal assay. The Journal of urology. 1989;142(6):1578-1583
12. Russell PJ, Ow KT, Tam PN, Juarez J, Kingsley EA, Qu CF. et al. Immunohistochemical characterisation of the monoclonal antibody BLCA-38 for the detection of prostate cancer. Cancer Immunology, Immunotherapy. 2004;53(11):995-1004
13. Perrimon N, Bernfield M. Specificities of heparan sulphate proteoglycans in developmental processes. Nature. 2000;404(6779):725-728
14. David G, Lories V, Decock B, Marynen P, Cassiman J-J, Van den Berghe H. Molecular cloning of a phosphatidylinositol-anchored membrane heparan sulfate proteoglycan from human lung fibroblasts. The Journal of cell biology. 1990;111(6):3165-3176
15. Chen RL, Lander AD. Mechanisms underlying preferential assembly of heparan sulfate on glypican-1. Journal of biological chemistry. 2001;276(10):7507-7517
16. Iozzo RV, Sanderson RD. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. Journal of cellular and molecular medicine. 2011;15(5):1013-1031
17. Svensson G, Awad W, Håkansson M, Mani K, Logan DT. Crystal Structure of N-Glycosylated Human Glypican-1 Core Protein structure of Two Loops Evolutionarily Conserved in Vertebrate Glypican-1. Journal of biological chemistry. 2012;287(17):14040-14051
18. Matsuda K, Maruyama H, Guo F, Kleeff J, Itakura J, Matsumoto Y. et al. Glypican-1 is overexpressed in human breast cancer and modulates the mitogenic effects of multiple heparin-binding growth factors in breast cancer cells. Cancer research. 2001;61(14):5562-5569
19. Kleeff J, Ishiwata T, Kumbasar A, Friess H, Büchler MW, Lander AD. et al. The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. Journal of clinical investigation. 1998;102(9):1662
20. Hippo Y, Watanabe K, Watanabe A, Midorikawa Y, Yamamoto S, Ihara S. et al. Identification of soluble NH2-terminal fragment of glypican-3 as a serological marker for early-stage hepatocellular carcinoma. Cancer research. 2004;64(7):2418-2423
21. Filmus J, Selleck SB. Glypicans: proteoglycans with a surprise. Journal of clinical investigation. 2001;108(4):497-501
22. Kleeff J, Wildi S, Kumbasar A, Friess H, Lander AD, Korc M. Stable transfection of a glypican-1 antisense construct decreases tumorigenicity in PANC-1 pancreatic carcinoma cells. Pancreas. 1999;19(3):281-288
23. Aikawa T, Whipple CA, Lopez ME, Gunn J, Young A, Lander AD. et al. Glypican-1 modulates the angiogenic and metastatic potential of human and mouse cancer cells. The Journal of clinical investigation. 2008;118(1):89
24. Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J. et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523:177-182
25. Melo SA, Sugimoto H, O'Connell JT, Kato N, Villanueva A, Vidal A. et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer cell. 2014;26(5):707-721
26. Duijvesz D, Versluis CYL, van der Fels CA, Vredenbregt-van den Berg MS, Leivo J, Peltola MT. et al. Immuno-based detection of extracellular vesicles in urine as diagnostic marker for prostate cancer. International Journal of cancer. 2015;137(12):2869-2878
27. Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F. et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319(5867):1244-1247
28. Urbanelli L, Magini A, Buratta S, Brozzi A, Sagini K, Polchi A. et al. Signaling pathways in exosomes biogenesis, secretion and fate. Genes. 2013;4(2):152-170
29. Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nature reviews immunology. 2009;9(8):581-593
30. Bobrie A, Colombo M, Raposo G, Théry C. Exosome secretion: molecular mechanisms and roles in immune responses. Traffic. 2011;12(12):1659-1668
31. Taylor DD, Gercel-Taylor C. Exosomes/microvesicles: mediators of cancer-associated immunosuppressive microenvironments. Seminars in immunopathology. 2011;33(5):441-454
32. Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Curry WT. et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nature cell biology. 2008;10(12):1470-1476
33. Beckler MD, Higginbotham JN, Franklin JL, Ham A-J, Halvey PJ, Imasuen IE. et al. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Molecular & cellular proteomics. 2013;12(2):343-355
34. Janowska-Wieczorek A, Wysoczynski M, Kijowski J, Marquez-Curtis L, Machalinski B, Ratajczak J. et al. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. International journal of cancer. 2005;113(5):752-760
35. Shore N, Concepcion R, Saltzstein D, Paivanas T, Beebe-Dimmer J, Ruterbusch J. et al. Glypican-1 as a Biomarker for Prostate Cancer. The Journal of urology. 2015;193(4):e410-e411
36. Li Y, Song E, Abbas Rizvi SM, Power CA, Beretov J, Raja C. et al. Inhibition of micrometastatic prostate cancer cell spread in animal models by 213Bilabeled multiple targeted alpha radioimmunoconjugates. Clinical cancer research. 2009;15(3):865-875
37. Khatri A, Ho T, Lindholm L, Russell PJ. Promise of BLCA38 as a Targeting Antibody for Tissue-Specific Gene Delivery to Prostate Cancer. Austral-Asian Journal of cancer. 2010;9(3):195-203
Author contact
![]() Corresponding author: Dr Bradley J. Walsh, Email: brad.walshcom, Phone: +61 2 9850 4000
Corresponding author: Dr Bradley J. Walsh, Email: brad.walshcom, Phone: +61 2 9850 4000