Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(7):872-882. doi:10.7150/jca.13855 This issue Cite
Research Paper
Role of Four Different Kinds of Polyethylenimines (PEIs) in Preparation of Polymeric Lipid Nanoparticles and Their Anticancer Activity Study
Kongtong Yu1, Jinlong Zhao1, Changhui Yu1, Fengying Sun1, Yan Liu1, Yang Zhang1, Robert J. Lee1,2, Lesheng Teng1,3 ![]() , Youxin Li1,3
, Youxin Li1,3 ![]()
1. School of Life Sciences, Jilin University, Changchun, Jilin Province, 130012, China.
2. College of Pharmacy, The Ohio State University, Columbus, OH 43210, USA.
3. State Key Laboratory of Long-acting and Targeting Drug Delivery System, Yantai 264000, China.
Received 2015-9-15; Accepted 2016-3-22; Published 2016-4-28
Abstract
A series of polyethylenimines-coated poly(d,l-lactide-co-glycolide)/lipid nanoparticles (PPLs) were fabricated for delivering paclitaxel via a simple nano-precipitation method. Four kinds of polyethylenimines (PEIs) (800 Da-, 2000 Da- and 25 kDa-branched PEIs, and 25 kDa-linear PEI) were selected as a polymeric coating for the nanoparticles. The PPLs were evaluated for their cytotoxic effects towards tumor cells. The nanoparticles were uniform spheres with particle sizes ranging from 135.8 to 535.9 nm and zeta potentials between 13.5 and 45.4 mV. The content of lipids and PEIs were optimized at lipids content from 0 to 40% and PEI content from 2.5% to 10%, respectively. At 20% lipid content and 5% PEI content, the formulation was found to be optimal. In vitro experiments showed that 25 kDa-branched PEI coated PLGA/lipid nanoparticles (25k-bPPLs) had higher cytotoxicity than other PPLs in several cancer cell lines. Meanwhile, 25k-bPPLs maintained high cellular delivery efficiency without excessive toxicity, which was confirmed by confocal microscopy and flow cytometry analyses. Furthermore, 25k-bPPLs displayed excellent colloidal stability in pH 7.4 PBS. In conclusion, 25k-bPPLs are promising drug delivery vehicles for cancer therapeutics.
Keywords: Drug delivery, Polyethylenimine, Polymeric lipid nanoparticles.
Introduction
Nanoparticles can potentially play a key role in delivering anticancer drugs. Nanoparticles can escape from the vasculature around the leaky tumor endothelial tissue and thus accumulate in tumors by an effect called “Enhanced Permeability and Retention (EPR)”. They are promising drug delivery vehicles because of their ability to prolong circulation time and increase tumor accumulation 1,2. They can incorporate hydrophilic and lipophilic drugs, increasing the stability of loaded drug and enabling controlled release 3,4.
Currently available nanoparticles are mostly lipid- or polymer- based 5,6. Biodegradable polymer nanoparticles display enhanced therapeutic performance of anticancer drugs due to good size control, high encapsulation efficiency and sustained release behavior 7,8. Nanoparticles prepared from polyesters like poly(lactide-co-glycolic acid) (PLGA), is widely studied due to their biodegradability, biocompatibility, storage stability and controlled release.
However, polymer nanoparticles are prone-to-aggregate and have limited cellular uptake 9. These problems require the establishment of a novel stable and targeted delivery system. Recent studies have suggested that a combination of polymers and lipids for drug delivery could improve stability, enhance cellular uptake and alter biodistribution 10-12. Polymeric lipid nanoparticles combine the advantages of polymers and lipids and avoid their disadvantages in physicochemical and biological properties 4. It is hypothesized that lipids release the loaded drug into cytosol via membrane fusion or destabilization 13. Egg phosphatidylcholine (Egg PC), a neutral lipid capable of mixing with polymers and forming solid nanoparticles based on hydrophobic interaction, has been used as components in solid nanoparticles to protect its surfaces, reduce cytotoxicity and increase cellular uptake 14.
Polyethylenimine (PEI), a cationic polymer, can be synthesized in different molecular weights and configurations, including linear or branched structures. Different PEIs have varying degrees of toxic effects on cells 15. PEIs have been shown to be efficacious vectors in gene delivery. Their high transfection efficiency is believed to originate from the so-called “proton sponge effect” 16. Based on their high surface charge, PEIs are promising candidates for delivery of negatively charged nucleic acids 17,18. In general, the transfection capability and cytotoxicity increases with the increase of molecular weight 4. Linear PEI shows lower cytotoxicity and higher efficiency than branched PEI for gene transfection 19,20. The transfection capability and cytotoxicity effect depend on the molecular weight and structure of PEIs 21. Only a few researches have systematically investigated the role of various PEIs in fabrication of polymeric lipid nanoparticles for chemotherapy drugs delivery. PEI coating may enhance the permeability of PLGA which has opposite charge.
Paclitaxel (PTX) has been used as a chemotherapy drug for the treatment of a variety of cancers, including breast, ovarian as well as lung cancers. It acts by binding to microtubules and inhibiting the depolymerization of microtubule, leading to cell death 22. However, its utility has been limited due to poor aqueous solubility and undesired side effects 23,24. Recently, various targeted drug vehicles have been developed for improving solubility and reducing non-targeted toxicity.
In the present study, we entrapped PTX in PEI-coated polymer-lipid nanoparticles (PPLs) by a nano-precipitation method. Herein, we evaluated four different kinds of PEIs for nanoparticles coatings and studied the in vitro cytotoxicity of the nanoparticles. We systematically optimized formulation by varying components and the type of PEI. Overall PPLs displayed good biocompatibility and enhanced cellular uptake efficacy. Our results indicated that the combination of cationic PEIs with PLGA/lipid matrix opens new possibilities for drug delivery in cancer cells.
Materials and methods
Materials
Paclitaxel (PTX) was obtained from Huiang Biopharma Co., Ltd. (Guilin, China). Poly(d,l-lactide-co-glycolide) (PLGA, Mw 5-10 kDa, 50:50) was purchased from Boehringer Ingelheim Pharm GmbH & Co. KG (Ingelheim, Germany). Egg phosphatidylcholine (Egg PC) was obtained from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). Linear polyethylenimine (lPEI, Mw 25 kDa), branched polyethylenimines (bPEIs, Mw 800 Da, 2000 Da, 25 kDa), poloxamer 188 (F68), acetone, methanol, acetonitrile and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). All of the solvents and reagents were of analytical grade. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), 9-(2-carboxyphenyl)-3,6-bis(diethylamino)xanthylium chloride (Rhodamine B) and 4',6-diamidino-2-phenylindole (DAPI) were obtained from Yuanye Biotech Co., Ltd. (Shanghai, China). All cell culture media and supplements were obtained from Thermo Fisher Scientific (Waltham, MA, USA). All experiments used ultrapure water.
Cell cultures
The A549, HepG2 and MCF7 cancer cells were obtained from American Type Culture Collection (ATCC). Cells were continuously cultured as a monolayer in RPMI 1640 medium that was supplemented with 100 μg/mL of streptomycin, 100 units/mL of penicillin, and 10% fetal bovine serum (FBS) at 37 °C.
Preparation of nanoparticles
The preparation of PPLs was conducted using a nano-precipitation technique with slight modifications 25,26. Briefly, PLGA, Egg PC and PTX were dissolved together in an acetone-methanol solvent (3: 2, v/v) as the organic phase. PEI was ultrasonically dissolved in an aqueous F68 solution (1%, w/v) and served as the aqueous phase. The organic phase was then added dropwise into the aqueous phase with an oil-water ratio of 1: 15 (v/v) and homogenized at 22,000 rpm for 30 s at the same time (T25, IKA, Germany). The suspension was stirred at room temperature overnight to evaporate the organic solvent. Larger aggregates were removed by centrifugation at 5,000 rpm for 10 min (Allegra 64R, Beckman Coulter, USA). Then supernatant containing PPLs was collected through centrifugation at 15,000 rpm for 15 min. The obtained PPLs were washed twice with Tween-80 solution (0.1%, w/v) in order to remove unencapsulated PTX or free polymers and then were lyophilized at -40 °C for 24 h using a BenchTop Freeze Dryer (AdVantage 2.0, SP Scientific, USA). The lyophilized PPLs were stored at 4 °C until use. For PLGA/lipid nanoparticles (PLs) and PLGA nanoparticles (Ps), PEI was not added into aqueous phase. For Ps, only PLGA and PTX were dissolved in organic phase.
Various formulation parameters such as the percentage contents of PLGA and Egg PC, the kinds and percentage contents of PEIs were optimized on the basis of their effects on particle size, z-potential and drug entrapment efficiency of PPLs.
Size and z-potential measurements
The particle size, polydispersity and z-potential of nanoparticles were determined by dynamic light scattering (DLS) using a Zetasizer Nano ZS90 (Malvern, England) at 25 °C. The lyophilized nanoparticles were ultrasonically dispersed in ultrapure water and diluted to nanoparticles concentration of 0.25 mg/mL.
Morphology examination
The surface morphology of nanoparticles was visualized using a scanning electron microscopy (SEM, JSM-6700F, JEOL, Japan) with an acceleration voltage of 3 kV. The lyophilized nanoparticles were ultrasonically dispersed in ultrapure water before they were introduced onto a silicon wafer and were dried to evaporate the surface liquid. The nanoparticles were sputter-coated with platinum before detection.
Stability study
To investigate the stability, the lyophilized nanoparticles were resuspended in PBS (0.01 M, pH 7.4) and kept at 25 °C. The particle size and polydispersity were analyzed on days 0, 3, 7 and 14. The colloidal stability of the nanoparticles was evaluated based on particle size and polydispersity.
Drug loading determination
The quantity of PTX in the nanoparticles was measured by high performance liquid chromatography (HPLC, Waters, USA) on a system equipped with an UV-visible detector, an autosampler and a binary pump. The analysis was performed at 228 nm using an Extend-C18 column (4.6 mm × 250 mm). Briefly, 1 mg of lyophilized nanoparticles was redissolved in 2 mL of acetonitrile and sonicated for 10 min. Subsequently, 8 mL of mobile phase was added for HPLC analysis. The encapsulation efficiency (EE) and drug loading (DL) were calculated using the following Eqs.:
DL = (Amount of PTX actually present nanoparticles/ Amount of nanoparticles) × 100% (1)
EE = (Amount of PTX actually present nanoparticles/ Amount of PTX actually used) × 100% (2)
Drug release study
The PTX release experiment was performed using a dialysis bag method. PBS (0.01 M, pH 7.4) containing 1% Tween-80 (v/v) was used as dissolution medium to increase the solubility of PTX. Dialysis bags (8 kDa molecular cut off) were soaked in ultrapure water for 24 h before use. One mL of nanoparticles suspension was poured into the dialysis bag with two ends clamped. These dialysis bags were immersed in glass bottles and 50 mL of dissolution medium was added. These glass bottles were then placed into a water bath shaker (SY-2230, Crystal, USA) that was shaking at 120 rpm at 37 °C. At selected sampling times, 5 mL of the release medium was removed and replaced with same volume of fresh medium. The release medium was analyzed by HPLC for its PTX content.
Differential scanning calorimetry (DSC) analysis
Individual components, PTX free and lyophilized nanoparticles were analyzed on a differential scanning calorimeter (DSC 1, Mettler Toledo, Switzerland). In brief, 8 ~ 10 mg of dried sample was transferred into an aluminum pan (40 µL), and an empty standard aluminum pan (40 µL) was used as a reference. DSC analysis was performed by heating the sample from 0 °C to 300 °C at a heating rate of 10 °C/min while flushing with nitrogen at 50 mL/min. The thermograms of the samples were recorded.
Cytotoxicity analysis
The cytotoxicity of free drug and drug loaded nanoparticles was evaluated using MTT assay. Briefly, A549, HepG2 and MCF7 cells were cultured at 96-well plates (Thermo Fisher Scientific, China) at a density of 1 × 104 cells/well and cultured overnight. Subsequently, the medium was replaced with fresh medium containing free drug dissolved in DMSO and different formulations of drug loaded nanoparticles (at a drug concentration of 2 µM, at the nanoparticles concentration of 26.39, 20.7, 20.83, 22.33 and 22.61 µg/mL, respectively). The cells were further incubated for 4 h and the culture medium was substituted by fresh complete medium following by incubation for 24 h, 48 h and 72 h, respectively. MTT solution was added to each well to achieve a concentration of 1 mg/mL. After a further 4 h of incubation, the culture medium was discarded, followed by adding 150 µL of DMSO to dissolve the formazan crystals. The cell viability was quantified by testing the absorbance of the formazan with a microplate reader (Multiskan Spectrum, Thermo, USA) at a wavelength of 490 nm. The relative cell viability was defined by dividing the absorbance of the drug treated wells by the absorbance of the untreated controls. Each test was performed in eight replicates. The lyophilized nanoparticles were sterilizated with ultraviolet light prior to use.
Confocal laser scanning microscopy (CLSM) analysis
Rhodamine B, a lipophilic dye, was used to track the nanoparticles. Rhodamine B-labeled nanoparticles were prepared by the method described above except replacing PTX with Rhodamine B. For cellular uptake experiments, A549 and MCF7 cells (2 × 104) were seeded into glass-bottom Petri dish (NEST, China) and cultured in complete medium to reach 70% confluency. The cells were then incubated with different formulations of Rhodamine B-labeled nanoparticles suspended in serum-free medium at the nanoparticles concentration of 0.021 mg/mL for 2 h. The medium was discarded and cells were washed three times with cold PBS (0.01 M, pH 7.4). The cells were fixed with 4% paraformaldehyde for 15 min and the nuclei were stained with DAPI for 5 min. Then, they were washed three times with PBS solution to remove free dye and debris. This suspension was kept in the glass-bottom Petri dish and cells were imaged using a confocal laser scanning microscope (LSM710, Carl Zeiss, Germany) equipped with an argon laser and an Rhodamine B filter (Ex: 540 nm, Em: 625 nm).
Cellular uptake analysis
The cellular uptake of the nanoparticles was also quantified by flow cytometry. Briefly, 2 × 105 of A549 and MCF7 cells were placed in 6-well plates (Thermo Fisher Scientific, China) and cultivated overnight. The cells were incubated with a fixed concentration (0.021 mg/mL) of different formulations of Rhodamine B-labeled nanoparticles for 3 h. The cells were washed, trypsinized and resuspended in PBS. After filtering through a 35 µm nylon mesh, the cells were analyzed using a flow cytometer (EPICS XL/XL-MCL, Beckman Coulter, USA) under the excitation wavelength (540 nm) and emission wavelength (625 nm), respectively. At least 15000 events were acquired and analyzed per group.
Statistical analysis
All statistical analyses were performed using Student's t-test with p < 0.05 as indicating statistical significance. All results are presented as the average ± standard deviation.
Results
Preparation and characterization of PLs
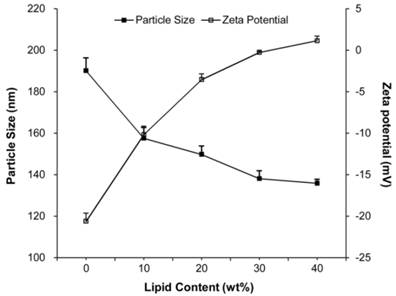
Effects of lipid content on the particle size, z-potential and EE of PLs were investigated to obtain optimal lipid-to-PLGA ratio and the results were shown in Table 1 and Fig. 2. With the increase of lipid content, the particle size and EE decreased simultaneously while the zeta potential increased. The formulation with smaller particle size and higher EE was chosen for subsequent experiments. The optimal lipid-to-PLGA ratio was found to be 3: 1 with a particle size of 149.8 ± 4.08 nm, a zeta potential of -0.6 ± 1.0 mV and an EE of 64.7 ± 1.35%.
Characteristics of PLs of various PLGA and Egg PC amounts used in the preparation process. N=3.
| Lot | PLGA (wt, %) | Egg PC (wt, %) | Size (nm) | PDI | ZP (mV) | EE (%) |
|---|---|---|---|---|---|---|
| 1 | 80 | 0 | 190.1 ± 6.21 | 0.297 ± 0.028 | -12.7 ± 1.8 | 70.5 ± 2.31 |
| 2 | 70 | 10 | 157.6 ± 5.15 | 0.264 ± 0.048 | -9.3 ± 3.2 | 65.9 ± 1.07 |
| 3 | 60 | 20 | 149.8 ± 4.08 | 0.254 ± 0.033 | -0.6 ± 1.0 | 64.7 ± 1.35 |
| 4 | 50 | 30 | 138.1 ± 3.80 | 0.286 ± 0.026 | -0.4 ± 1.7 | 62.9 ± 2.24 |
| 5 | 40 | 40 | 135.8 ± 1.98 | 0.108 ± 0.041 | 1.8 ± 0.5 | 60.5 ± 1.36 |
Abbreviations: PDI, polydispersity index; ZP, zeta potential; EE, encapsulation efficiency.
Effects of the lipid content on the particle size and zeta potential of PLs.
Preparation and characterization of PPLs
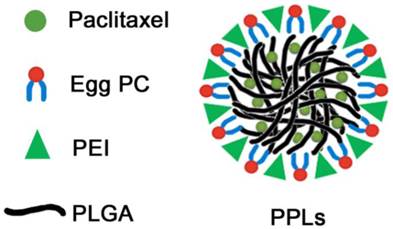
The schematic illustration of the structure of PPLs was shown in Fig. 1. We fabricated a series of PPLs with PEI contents of 2.5%, 5% and 10% to achieve different surface charges (Table 2). With increase in the PEI content, the particle size and surface charges increased. Considering both particle size and surface potential, PPLs coated with 5% PEI were chosen for the subsequent experiments. At the same PEI content (5%), the 25k-bPPLs possessed the smallest particle size following by the 2000-bPPLs, 800-bPPLs and 25k-lPPLs, at 249.9 ± 4.76 nm, 273.5 ± 5.94 nm, 286.7 ± 2.99 nm and 451.8 ± 12.5 nm, respectively. So the particle size depended on the molecular weight and structure of PEI. Table 2 showed that PPLs were more efficient in drug encapsulation than PLs.
Schematic illustration of the structure of the PPLs.
Characteristics of PPLs of various PEI types and amounts used in the preparation process. N=3.
| Lot | Mw of PEI (Da) | PEI structure | PEI (wt, %) | Size (nm) | PDI | ZP (mV) | EE (%) |
|---|---|---|---|---|---|---|---|
| 6 | 800 | Branched | 2.5 | 267.6 ± 8.20 | 0.293 ± 0.072 | 31.2 ± 1.8 | 82.0 ± 1.23 |
| 7 | 800 | Branched | 5 | 286.7 ± 2.99 | 0.238 ± 0.064 | 39.1 ± 2.1 | 82.5 ± 2.21 |
| 8 | 800 | Branched | 10 | 325.5 ± 6.19 | 0.307 ± 0.091 | 45.4 ± 1.5 | 88.5 ± 1.70 |
| 9 | 2000 | Branched | 2.5 | 236.6 ± 6.29 | 0.303 ± 0.046 | 31.6 ± 3.4 | 77.0 ± 1.36 |
| 10 | 2000 | Branched | 5 | 273.5 ± 5.94 | 0.159 ± 0.070 | 38.7 ± 3.2 | 82.0 ± 0.78 |
| 11 | 2000 | Branched | 10 | 304.7 ± 2.69 | 0.271 ± 0.076 | 44.0 ± 1.4 | 88.0 ± 1.01 |
| 12 | 25000 | Branched | 2.5 | 209.1 ± 5.35 | 0.160 ± 0.041 | 29.2 ± 4.2 | 74.5 ± 0.52 |
| 13 | 25000 | Branched | 5 | 249.9 ± 4.76 | 0.255 ± 0.045 | 35.3 ± 2.3 | 76.5 ± 1.61 |
| 14 | 25000 | Branched | 10 | 297.7 ± 5.71 | 0.217 ± 0.039 | 41.4 ± 2.6 | 85.0 ± 1.36 |
| 15 | 25000 | Linear | 2.5 | 355.8 ± 8.30 | 0.288 ± 0.095 | 13.5 ± 1.3 | 71.5 ± 2.45 |
| 16 | 25000 | Linear | 5 | 451.8 ± 12.5 | 0.497 ± 0.066 | 19.0 ± 1.2 | 75.5 ± 0.25 |
| 17 | 25000 | Linear | 10 | 535.9 ± 13.5 | 0.447 ± 0.096 | 23.2 ± 2.9 | 83.5 ± 0.61 |
Abbreviations: Mw, molecular weight; PDI, polydispersity index; ZP, zeta potential; EE, encapsulation efficiency.
Morphology
SEM images showed that Ps and PPLs were spherical with a smooth morphology and a narrow size distribution (Fig. 3D and 3F). By contrast, the surface of the PLs was relatively uneven (Fig. 3E). Some particle agglomerates were observed in Ps and PLs (Fig. 3A and 3B). However, the dispersion of the PPLs was better than that of Ps and PLs (Fig. 3C). Besides, an absence of crystalline structure of PTX in SEM images indicated that no unencapsulated PTX crystals formed during the preparation.
SEM images of the Ps (A, D), PLs (B, E) and PPLs (C, F). N=3. Note that PPLs used was 25k-bPPLs.

DSC
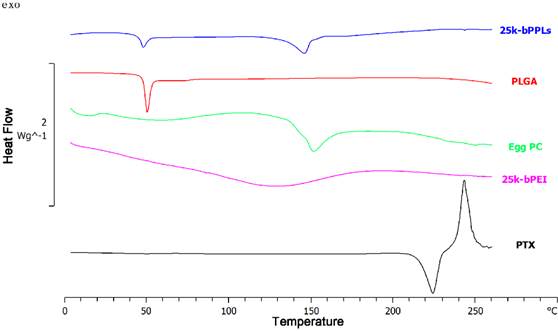
DSC study was performed to understand the blending behavior and crystallinity of PTX during the formation. Fig. 4 showed DSC curves of free PTX, PLGA, Egg PC, 25k-bPEI and lyophilized 25k-bPPLs. At 47 °C and 146 °C, two endotherms corresponded to the melting peak of the PLGA and the fusion peak of lipid and PEI, respectively. The free PTX displayed a sharp endothermic peak at 224 °C, indicating that it was crystalline. For PPLs, the absence of the PTX endothermic melting peak showed that the PTX incorporated in PPLs was not in a crystalline state but rather in an amorphous form.
DSC thermograms of paclitaxel, PLGA, Egg PC, 25k-bPEI and 25k-bPPLs, respectively.
Stability
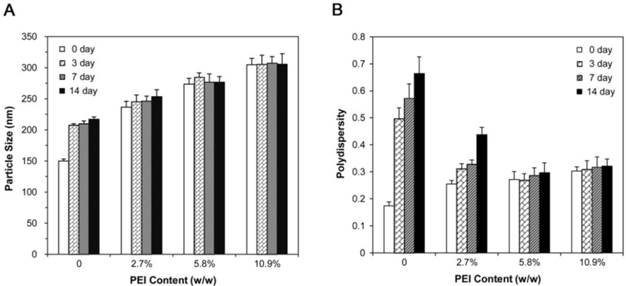
To study the stability of PPLs, we chose PLs and 25k-bPPLs (2.5%, 5% and 10% in PEI content) and evaluated the effects of the cationic layer on the physical stability of nanoparticles. The nanoparticles were incubated in PBS (0.01 M, pH 7.4) and the particle size and polydispersity were measured at predetermined time intervals (Fig. 5). After two weeks of incubation, a significant increase of 50-100 nm in the particle size was observed for the PLs. During this period, the polydispersity of PLs also increased. However, no significant changes were observed in the 25k-bPPLs (5% and 10% in PEI content) in particle size and polydispersity because of their positively-charged surfaces (35.3 and 41.4 mV). Meanwhile, 25k-bPPLs (2.5% in PEI content) emerged with a slight increase in particle size and polydispersity. The result illustrated that the PPLs (5% in PEI content), which had a surface potential above 35 mV, could maintain good stability.
Stability of the PLs and 25k-bPPLs (2.5%, 5% and 10% of PEI content). Particle size (A) and polydispersity (B). N=3.
In vitro drug release
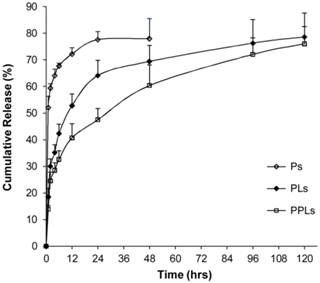
The cumulative release of drug was plotted as a function of time (Fig. 6). There was an initial burst release of drug in the first 12 h, during which 72.2%, 52.8% and 40.7% of drug were released from Ps, PLs and 25k-bPPLs, respectively. In the following hours, drug was released from PLs and PPLs in a sustained manner at a slower rate. The drug release of PPLs was significantly slower than that of PLs. At 120 h, 78.6% and 76.1% of PTX had been released from the PLs and PPLs, respectively.
In vitro drug releases from the Ps, PLs and 25k-bPPLs. N=3.
Cytotoxicity results
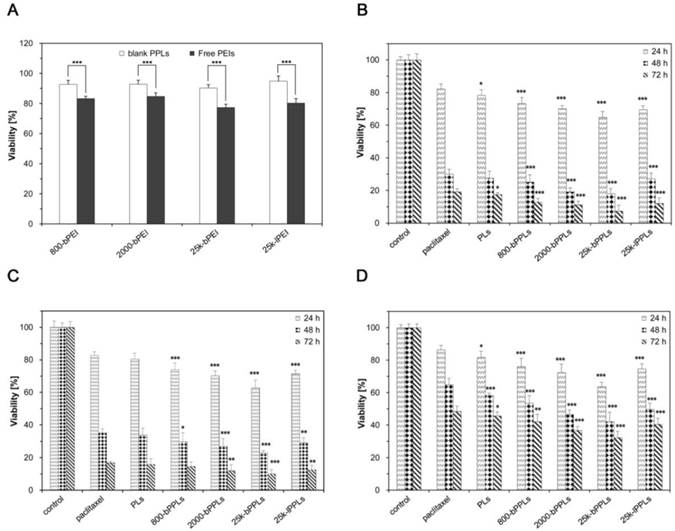
In vitro cytotoxicity of the free PTX, free PEIs, blank PPLs, PLs and four kinds of drug loaded PPLs was measured by MTT assay. The cytotoxicity of free PEIs and blank PPLs were evaluated using A549 cell lines after 24 h of incubation. As shown in Fig. 7A, the blank PPLs did not exhibit notable toxic effects compared with free PEIs at an identical PEI concentration (10 µg/mL). Fig. 9B, C and D illustrated the cytotoxic analysis of three different cell lines (A549, HepG2 and MCF7 cells, respectively) incubated with free PTX, PLs and four kinds of drug-loaded PPLs, aiming at a general assessment. There was no apparent difference in trend in cytotoxicity observed among three different cell lines. For A549 cells, treatment with 800-bPPLs, 2000-bPPLs, 25k-bPPLs and 25k-lPPLs at a PTX concentration of 2 µM resulted in cell viabilities of 12.9 ± 2.25%, 11.3 ± 2.07%, 7.4 ± 3.64% and 12.1 ± 3.28% after 72 h of incubation, respectively, which were lower than that of free PTX (19.2 ± 1.76%) and PLs (17.5 ± 1.06%). 25k-bPPLs treatment resulted in the lowest cell viability among four kinds of PPLs (P<0.001). Fig. 9 also showed that the cell inhibition was time-dependent.
In vitro cytotoxicity. Effects of the blank PPLs and free PEIs on A549 cells (A); effects of free paclitaxel, drug-loaded PLs and four kinds of drug-loaded PPLs on A549 cells (B), HepG2 cells (C) and MCF7 cells (D). N=8. The reference was set as the cell viability of free paclitaxel group at the same incubation time.
Flow cytometry results of A549 cells (A) and MCF7 cells (B) after 3 h of incubation with PLs and four kinds of PPLs. N=3.
CLSM results
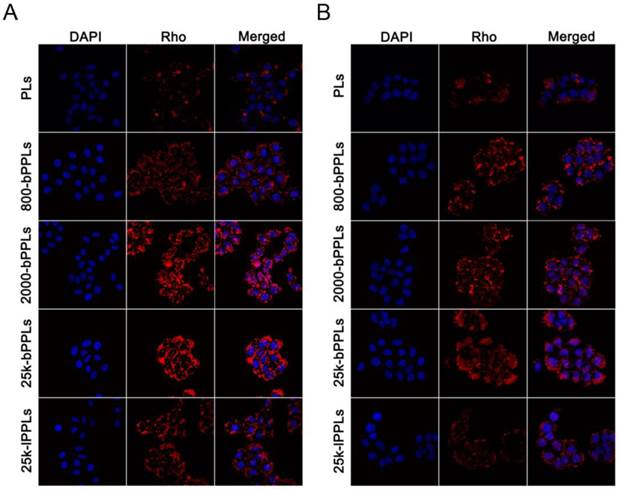
The PLs and four different PEI-coated PPLs were evaluated for their cellular uptake efficiency after 2 h of incubation in A549 and MCF7 cells using Rhodamine B as a marker. The intensity of incorporated dye was visualized using confocal microscopy. As shown in Fig. 8, red fluorescence was from Rhodamine B labeled PPLs and blue fluorescence was from nuclei stained with DAPI. As compared with the PLs, stronger red fluorescence of PPLs in the cytoplasmic regions was observed. The 25k-lPPLs showed much lower cellular uptake than 25k-bPPLs. 25k-bPPLs showed a stronger red fluorescence compared to other molecular weight of bPEI-coated PPLs. There was no significantly difference in trend of cellular uptake of nanoparticles between A549 and MCF7 cells, except that A549 cells consistently showed higher fluorescence uptake intensity than MCF7 cells.
CLSM images of A549 cells (A) and MCF7 cells (B) after 2 h of incubation with PLs and four kinds of PPLs. The red fluorescence represents the PPLs marked with Rhodamine B, and the blue fluorescence represents the cell nucleus stained by DAPI.
Cellular uptake results
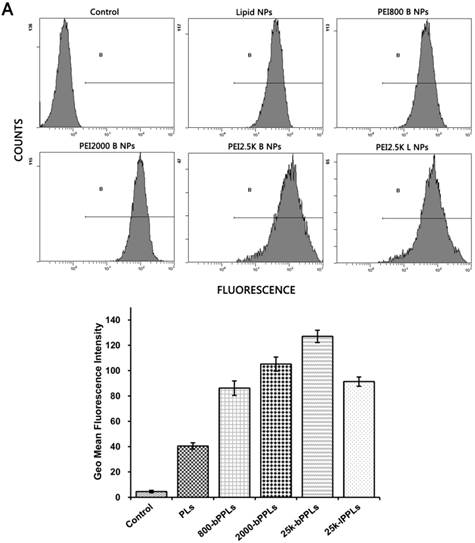
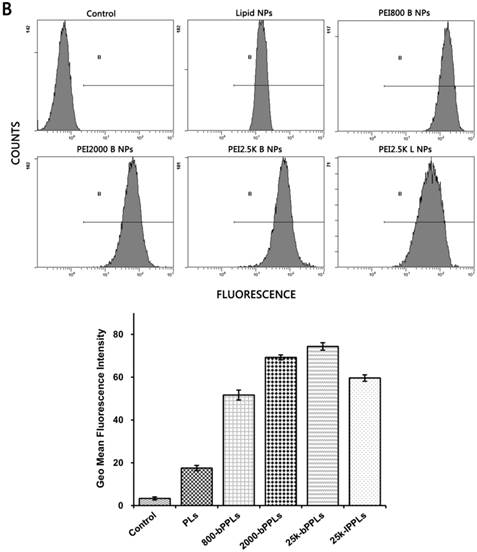
The internalization of PLs and four different PEI-coated PPLs was further quantified by flow cytometry. As shown in Fig. 9, similar results were obtained, which were consistent with CLSM results discussed above. In both A549 and MCF7 cells, the fluorescence intensity was higher in PPLs treated cells than those with PLs. In addition, cellular uptake of 25k-bPPLs in A549 and MCF7 cells was higher than that of other PPLs (P<0.05). In contrast, the mean intensity of cellular uptake of PLs and four different PEI-coated PPLs in A549 cells was higher than that in MCF7 cells. It is possible that the cellular uptake was depended on the cell surface potential and the resulting affinity for cationic vectors.
Discussion
Although PEI-functionalized nanoparticles are reported to be effective vectors in delivering negatively charged gene and chemotherapy drugs, not many attempts have been made to systematically investigate the role of PEI in fabrication of nanoparticles and in their anticancer activity. In this study, the PLs were firstly optimized to obtain smaller particle size and higher EE, which facilitates tumor uptake. With increase of lipid-to-PLGA ratio in PLs, particle size and EE decreased and zeta potential increased. The former might be attributed to the emulsification caused by lipid, leading to smaller particle size and lower EE 27,28. When PLGA content was low in the formulation, drug was not efficiently incorporated into PLs, suggesting a high affinity between PLGA and drug. The increasing of zeta potential was likely due to the surface neutral lipid covering up the negative charges of PLGA. The optimal lipid-to-PLGA ratio was 3: 1. Then we fabricated a series of PEI-coated polymeric lipid nanoparticles for PTX delivery. Four different kinds of PEIs (800 Da-, 2000 Da- and 25 kDa-bPEIs, and 25 kDa-lPEI) were selected to construct PPLs (defined as 800-bPPLs, 2000-bPPLs, 25k-bPPLs and 25k-lPPLs, respectively). PPLs were more efficient in drug encapsulation than PLs, suggesting that the cationic shell could prevent drug from rapid release. The weak repulsion of PEI to PTX kept the drug from release into the external phase, to a certain degree 29,30. The absence of the PTX in DSC spectrogram of PPLs also explained the interaction between polymer matrix and PTX 31. PEI coating was likely to promote the formation of high drug-loaded PPLs and held back the leakage of drug. The difference in morphology between PLs and PPLs might be attributed to the amphipathic nature of the lipids.
For PPLs, the hydrophobic polymer prevented itself from being exposed to the external aqueous solution leading to a dense structure 27. The stability of nanoparticles was closely correlated to their surface property. Due to the sticky nature of the materials, Ps and PLs produced particle agglomerates. The presence of PEIs on the surface of PPLs likely prevented the aggregation of the PPLs. PPLs showed higher stability than PLs due to its positively charged surfaces, which produced a repulsive force that prevented aggregation of PPLs 19. With increase of PEI ratio in PPLs, the particle size and z-potential increased at the same time. Therefore, the 5% of PEI content based 25k-bPPLs was chosen as the optimized formulation. For the in vitro release, there was an initial burst release of drug in the first 12 h, which might be due to the PTX located near the surface of the nanoparticles that could be helpful to suppress the growth of cancer cells in clinical setting. Subsequently, PPLs showed a significantly slower release than that of PLs, which might be attributed to the fact that the cationic layer prevented the external water permeating through the internal water channels and thus slowed down drug release 32. Generally, such a drug release profile of PTX is optimal for the delivery of anticancer drugs.
As expected, the blank PPLs showed lower cytotoxicity against free PEIs indicating good biocompatibility. The spatial distribution of PEIs in the surface of PPLs probably decreased or shielded partial positive charges and thus led to a decreased cytotoxic effect. PPLs showed more potency in inhibiting the growth of three cell lines: A549, HepG2 and MCF7, compared with PLs. PEIs in the surface of PPLs increased the cellular uptake of PTX and cytotoxicity, which might be attributed to the positive charge from PEIs binding with the negatively charged membrane 33,34. At equivalent molecular weight (25 kDa), lPPLs exhibited lower cellular uptake efficiency compared with bPPLs, which might be influenced by the linear structure of PEI, which led to larger particle size and stronger steric hindrance not conducive to endocytosis. Among branched PEIs, high molecular weight PEI based PPLs displayed higher cellular uptake efficiency compared to low molecular weight PEI based PPLs. Most importantly, due to the spatial structure and surface charge, 25k-bPPLs showed higher cytotoxicity against cancer cells than other PPLs. This result was consistent with the confocal and flow cytometry results on cellular uptake. Furthermore, cellular uptake of PLs and four different PEI-coated PPLs in A549 cells was higher than that in MCF7 cells, which might be due to a difference in cell surface potential 15,35. Taken together, the 25k-bPPLs had several advantages for drug delivery, including excellent stability and efficient cellular uptake due to the PEI cationic shell, high affinity and biocompatibility due to the lipid layer 36. In summary, 25k-bPPLs are promising as delivery vehicles and warrant further evaluation.
Acknowledgements
Supported by the Open Project Program of State Key Laboratory of Long-acting and Targeting Drug Delivery System, Yantai, 264000, China.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
1. Yang Z, Yu B, Zhu J, Huang X, Xie J, Xu S. et al. A microfluidic method to synthesize transferrin-lipid nanoparticles loaded with siRNA LOR-1284 for therapy of acute myeloid leukemia. Nanoscale. 2014;6:9742-9751
2. Wang X, Huang X, Yang Z, Gallego-Perez D, Ma J, Zhao X. et al. Targeted Delivery of Tumor Suppressor microRNA-1 by Transferrin- Conjugated Lipopolyplex Nanoparticles to Patient-Derived Glioblastoma Stem Cells. Curr Pharm Biotechno. 2014;15:839-846
3. Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery - a review of the state of the art. Eur J Pharm and Biopharm. 2000;50:161-177
4. Wong HL, Bendayan R, Rauth AM, Wu XY. Simultaneous delivery of doxorubicin and GG918 (Elacridar) by new Polymer-Lipid Hybrid Nanoparticles (PLN) for enhanced treatment of multidrug-resistant breast cancer. J Control Release. 2006;116:275-284
5. Liang H-F, Chen C-T, Chen S-C, Kulkarni AR, Chiu Y-L, Chen M-C. et al. Paclitaxel-loaded poly(γ-glutamic acid)-poly(lactide) nanoparticles as a targeted drug delivery system for the treatment of liver cancer. Biomaterials. 2006;27:2051-2059
6. Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. AdvDrug Deliver Rev. 2003;55:329-347
7. Cho K, Wang X, Nie S, Chen Z, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14:1310-1316
8. Feng S-S, Huang G. Effects of emulsifiers on the controlled release of paclitaxel (Taxol®) from nanospheres of biodegradable polymers. J Control Release. 2001;71:53-69
9. Feng S-S, Mu L, Chen B-H, Pack D. Polymeric nanospheres fabricated with natural emulsifiers for clinical administration of an anticancer drug paclitaxel (Taxol®). Mat Sci Eng: C. 2002;20:85-92
10. Freitas C, Müller RH. Effect of light and temperature on zeta potential and physical stability in solid lipid nanoparticle (SLN™) dispersions. Int J Pharm. 1998;168:221-229
11. Liu Y, Pan J, Feng S-S. Nanoparticles of lipid monolayer shell and biodegradable polymer core for controlled release of paclitaxel: Effects of surfactants on particles size, characteristics and in vitro performance. Int J Pharm. 2010;395:243-250
12. Thevenot J, Troutier A-L, David L, Delair T, Ladaviere C. Steric stabilization of lipid/polymer particle assemblies by poly(ethylene glycol)-lipids. Biomacromolecules. 2007;8:3651-3660
13. Li Y, Wong HL, Shuhendler AJ, Rauth AM, Wu XY. Molecular interactions, internal structure and drug release kinetics of rationally developed polymer-lipid hybrid nanoparticles. J Control Release. 2008;128:60-70
14. Galletti P, Malferrari D, Samorì C, Sartor G, Tagliavini E. Effects of ionic liquids on membrane fusion and lipid aggregation of egg-PC liposomes. Colloid Surface B. 2015;125:142-150
15. Intra J, Salem AK. Characterization of the transgene expression generated by branched and linear polyethylenimine-plasmid DNA nanoparticles in vitro and after intraperitoneal injection in vivo. J Control Release. 2008;130:129-138
16. Lai W-F. In vivo nucleic acid delivery with PEI and its derivatives: current status and perspectives. Expert Rev Med Devic. 2011;8:173-185
17. Basarkar A, Devineni D, Palaniappan R, Singh J. Preparation, characterization, cytotoxicity and transfection efficiency of poly(dl-lactide-co-glycolide) and poly(dl-lactic acid) cationic nanoparticles for controlled delivery of plasmid DNA. Int J Pharm. 2007;343:247-254
18. Chumakova OV, Liopo AV, Andreev VG, Cicenaite I, Evers BM, Chakrabarty S. et al. Composition of PLGA and PEI/DNA nanoparticles improves ultrasound-mediated gene delivery in solid tumors in vivo. Cancer Lett. 2008;261:215-225
19. Ewe A, Schaper A, Barnert S, Schubert R, Temme A, Bakowsky U. et al. Storage stability of optimal liposome-polyethylenimine complexes (lipopolyplexes) for DNA or siRNA delivery. Acta Biomater. 2014;10:2663-2673
20. Kwok A, Hart SL. Comparative structural and functional studies of nanoparticle formulations for DNA and siRNA delivery. Nanomed-Nanotechnol. 2011;7:210-219
21. Wightman L, Kircheis R, Rossler V, Carotta S, Ruzicka R, Kursa M. et al. Different behavior of branched and linear polyethylenimine for gene delivery in vitro and in vivo. J Gene Med. 2001;3:362-372
22. Achim M, Tomuta I, Vlase L, Iuga C, Moldovan M, Leucuta SE. Paclitaxel-loaded poly(lactic-co-glycolic acid) microspheres: preparation and in vitro evaluation. J Drug Deliv Sci Tec. 2008;18:410-416
23. Harisa GI, Ibrahim MF, Alanazi F, Shazly GA. Engineering erythrocytes as a novel carrier for the targeted delivery of the anticancer drug paclitaxel. Saudi Pharm J. 2014;22:223-230
24. Tao Y, Han J, Dou H. Surface modification of paclitaxel-loaded polymeric nanoparticles: Evaluation of in vitro cellular behavior and in vivo pharmacokinetic. Polymer. 2012;53:5078-5086
25. Dinesh Kumar V, Verma PRP, Singh SK. Development and evaluation of biodegradable polymeric nanoparticles for the effective delivery of quercetin using a quality by design approach. LWT - Food Sci Technol. 2015;61:330-338
26. Govender T, Stolnik S, Garnett MC, Illum L, Davis SS. PLGA nanoparticles prepared by nanoprecipitation: drug loading and release studies of a water soluble drug. J Control Release. 1999;57:171-185
27. Ma T, Wang L, Yang T, Ma G, Wang S. Homogeneous PLGA-lipid nanoparticle as a promising oral vaccine delivery system for ovalbumin. Asian J Pharm Sci. 2014;9:129-136
28. Mu L, Feng SS. Fabrication, characterization and in vitro release of paclitaxel (Taxol®) loaded poly (lactic-co-glycolic acid) microspheres prepared by spray drying technique with lipid/cholesterol emulsifiers. J Control Release. 2001;76:239-254
29. Silva AC, González-Mira E, García ML, Egea MA, Fonseca J, Silva R. et al. Preparation, characterization and biocompatibility studies on risperidone-loaded solid lipid nanoparticles (SLN): High pressure homogenization versus ultrasound. Colloid Surface B. 2011;86:158-165
30. Yang X-Z, Dou S, Sun T-M, Mao C-Q, Wang H-X, Wang J. Systemic delivery of siRNA with cationic lipid assisted PEG-PLA nanoparticles for cancer therapy. J Control Release. 2011;156:203-211
31. Hao J, Wang F, Wang X, Zhang D, Bi Y, Gao Y. et al. Development and optimization of baicalin-loaded solid lipid nanoparticles prepared by coacervation method using central composite design. Eur J Pharm Sci. 2012;47:497-505
32. Zhao P, Wang H, Yu M, Liao Z, Wang X, Zhang F. et al. Paclitaxel loaded folic acid targeted nanoparticles of mixed lipid-shell and polymer-core: In vitro and in vivo evaluation. Eur J Pharm Biopharm. 2012;81:248-256
33. Bivas-Benita M, Romeijn S, Junginger HE, Borchard G. PLGA-PEI nanoparticles for gene delivery to pulmonary epithelium. Eur J Pharm Biopharm. 2004;58:1-6
34. Huang W, Lv M, Gao Z. Polyethylenimine grafted with diblock copolymers of polyethylene glycol and polycaprolactone as siRNA delivery vector. J Control Release. 2011;152(Supplement 1):e143-e145
35. Dehshahri A, Oskuee RK, Shier WT, Hatefi A, Ramezani M. Gene transfer efficiency of high primary amine content, hydrophobic, alkyl-oligoamine derivatives of polyethylenimine. Biomaterials. 2009;30:4187-4194
36. Shau MD, Shih MF, Lin CC, Chuang IC, Hung WC, Hennink WE. et al. A one-step process in preparation of cationic nanoparticles with poly(lactide-co-glycolide)-containing polyethylenimine gives efficient gene delivery. Eur J Pharm Sci. 2012;46:522-529
Author contact
![]() Corresponding authors: Lesheng Teng, Email: tengleshengedu.cn Or Youxin Li, Email: liyouxinedu.cn.Tel: +86-431-85155320 Fax: +86-431-85155320.
Corresponding authors: Lesheng Teng, Email: tengleshengedu.cn Or Youxin Li, Email: liyouxinedu.cn.Tel: +86-431-85155320 Fax: +86-431-85155320.