Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(3):314-323. doi:10.7150/jca.13332 This issue Cite
Review
Survivin and Tumorigenesis: Molecular Mechanisms and Therapeutic Strategies
Xun Chen1,*, Ning Duan1,*, Caiguo Zhang2, ![]() , Wentao Zhang1,
, Wentao Zhang1, ![]()
1. Hong-Hui Hospital, Xi'an Jiaotong University, College of Medicine, Xi'an, Shaanxi, China, 710054
2. Department of Biochemistry and Molecular Genetics, University of Colorado School of Medicine, Aurora, CO, USA, 80045
* These authors contribute equally to this work.
Received 2015-7-24; Accepted 2015-11-25; Published 2016-1-10
Abstract
Survivin is the smallest member of the inhibitor of apoptosis protein family, which has key roles in regulating cell division and inhibiting apoptosis by blocking caspase activation. Survivin is highly expressed in most human cancers, such as lung, pancreatic and breast cancers, relative to normal tissues. Aberrant survivin expression is associated with tumor cell proliferation, progression, angiogenesis, therapeutic resistance, and poor prognosis. Studies on the underlying molecular mechanisms indicate that survivin is involved in the regulation of cytokinesis and cell cycle progression, as well as participates in a variety of signaling pathways such as the p53, Wnt, hypoxia, transforming growth factor, and Notch signaling pathways. In this review, recent progress in understanding the molecular basis of survivin is discussed. Therapeutic strategies targeting survivin in preclinical studies are also briefly summarized.
Keywords: survivin, apoptosis, cell cycle, cancer, therapy
Introduction: overview of inhibitor of apoptosis proteins and cancer
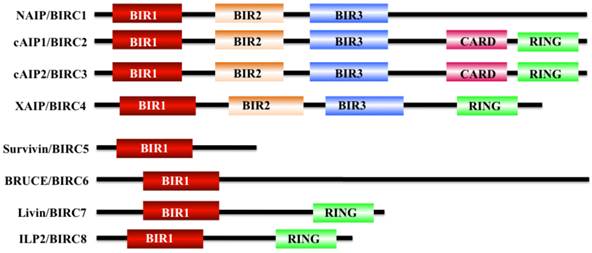
Apoptosis is a physiological cell death process that contributes to the development and maintenance of healthy cells and tissues [1-3]. Dysregulation of apoptosis pathways leads to a number of diseases such as cancer, autoimmune and immunodeficiency diseases, and neurodegenerative disorders [1]. Organisms have evolved two signaling pathways, namely, intrinsic and extrinsic pathways, to trigger apoptosis through the activation of caspases [4, 5]. At present, two types of apoptotic caspases including initiator caspases (caspase-2, 8, 9, and 10) and effector caspases (caspase-3, 6, and 7), have been widely identified in mammals [6]. Of them, initiator caspases mainly process and activate pro-forms of effector caspases in response to different signals [7]. Effector caspases, also known as death effector caspases, cleave protein substrates in the cell to trigger the apoptotic process [7]. Interestingly, the apoptosis process can be blocked by a family proteins termed as inhibitor of apoptosis proteins (IAPs) through direct binding to caspases [8]. All of these IAP family proteins contain a domain of approximately 70 amino acids known as baculoviral IAP repeat (BIR) [8]. The mammalian genome encodes eight IAP family members including BIRC1 (also known as neuronal apoptosis inhibitory protein, NAIP), BIRC2 (also known as cellular inhibitor of apoptosis protein 1, cIAP1), BIRC3 (also known as cIAP2), BIRC4 (also known as X-linked inhibitor of apoptosis, XIAP), BIRC5 (also known as survivin), BIRC6 (also known as BRUCE), BIRC7 (also known as livin), and BIRC8 (also known as IAP-like Protein 2, ILP2) (Figure 1) [9, 10]. BIRC1, 2, 3, 4, 7, and 8 have been shown to bind to caspase-3, 7, and 9, thereby inhibiting activation of these caspases; BIRC5 and BIRC6 directly interact with caspase-3 and 7, and they also regulate cytokinin to inhibit apoptosis [10, 11]. BIRC5/survivin, the focus of this review, only contains one BIR domain for the suppression of apoptosis, and it is structurally organized as a stable dimer (Figure 1) [11, 12].
Structures of IAP proteins in mammals. The mammalian IAP proteins consist of eight members, including NAIP/BIRC1, cAIP1/BIRC2, cAIP2/BIRC3, XAIP/BIRC4, survivin/BIRC5, BRUCE/BIRC6, livin/BIRC7, and ILP2/BIRC8 [10]. The main domains of these proteins are indicated in different colors.
A number of studies have indicated IAP genes are highly expressed in different cancer cells and primary tumor biopsy samples [13-18]. Among these IAPs, survivin exhibits the most dramatic overexpression in many tumors and fetal tissues [19], but it is present at low levels or is completely absent in healthy cells and tissues. The National Cancer Institute's (NCI) cancer drug-screening program found that survivin is expressed in all 60 human tumor lines, with the highest levels in breast and lung cancer cells and the lowest levels in renal cancer [20, 21]. This aberrant expression of survivin in tumor cells is regulated by numerous factors including microRNAs (miRNAs) [9] and receptor tyrosine kinases (RTKs), as well as in its downstream signaling cascades, such as phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) [22, 23], mitogen-activated protein kinase kinase/mitogen-activated protein kinase (MEK/MAPK) [24], mammalian target of rapamycin (mTOR) [25], and signal transducer and activator of transcription 3 (STAT3) pathways [26]. In addition, survivin is also involved in tumorigenesis through diverse mechanisms, including interaction with caspase-3 and 7 [27], inhibition of Bax- and Fas-induced apoptosis [28], regulation of cytokinesis and cell cycle progression [29], and participation in a variety of signaling pathways such as the p53 [30], Wnt [31], hypoxia [32], transforming growth factor (TGF), and Notch signaling pathways [33-36].
Regulation of survivin expression
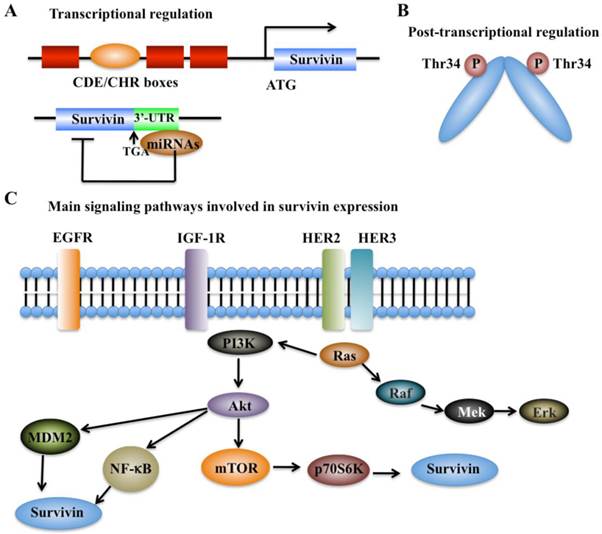
The expression of survivin can be regulated at the transcriptional and post-translational levels with precise mechanisms (Figure 2). Moreover, survivin expression is also identified to associate with aberrant activation of a number of RTKs, such as the epidermal growth factor receptor (EGFR) [37], epidermal growth factor receptor 2 (erbB2, also known as Her2) [38], insulin-like growth factor-1 (IGF-1) [39], and various cell survival signaling cascades, including PI3K/Akt [22, 23], MEK/MAPK [24], mTOR [25], STAT3, and hypoxia-inducible factor-1 (HIF-1) [26, 40].
Regulation of survivin expression. (A) Transcriptional mechanisms that control survivin expression involve CDE/CHR G1 repressor elements in the survivin promoter and binding of miRNAs to 3'-UTR region of the survivin gene [41]. (B) Post-transcriptional mechanisms that control survivin expression involve increased protein stability by phosphorylation at Thr34 [41]. (C) Survivin expression is associated with aberrant activation of several RTKs, such as EGFR, HER2 and HER3, and IGF-1. These kinases can trigger various cell survival signaling cascades such as PI3K/Akt-mTOR, thereby regulating survivin expression.
Transcriptional regulation
Cell cycle-dependent genes usually contain unique elements such as the cell cycle-dependent element (CDE) and the cell cycle genes homology region (CHR) to control gene transcription [41]. Interestingly, the survivin genome sequence contains a CDE/CHR element within its promoter region, suggesting that survivin may be a cell cycle-regulated gene (Figure 2A) [42].
In recent years, a variety of miRNAs have also been identified to regulate survivin expression via binding to the 3'-untranslated region (UTR) of survivin mRNA, thereby resulting in alteration of survivin protein translation or leading to its mRNA degradation (Figure 2A) [9]. Multiple survivin-targeting miRNAs, such as miR-16, miR-34a, miR-143, miR-150, miR-203, miR-218, miR-320a, miR-494, miR542-3p, and miR-708, have been identified [9]. The underlying molecular mechanisms of miR-34a and miR-203 have been extensively studied. miR-34a has been shown to reduce survivin expression through various mechanisms in different types of cancer [9, 43]. miR-34a regulation of survivin includes direct regulation and indirect regulation. For direct regulation, the 3'-UTR of survivin contains a unique sequence (position 1940-1946) for miR-34a binding [9]. For indirect regulation, elevated expression of miR-34a in cancer cells can repress the upstream activators or transcriptional factors of survivin, thereby leading to decreased survivin expression [9]. miR-203 is also able to directly target survivin mRNA, which significantly contributes to prostate cancer progression and metastasis [9].
Post-translational regulation
Protein modifications such as phosphorylation and polyubiquitination are able to affect survivin levels. Mitotic phosphorylation of survivin at Thr34 by cell division cycle protein 2 (CDC2, also known as cyclin-dependent kinase 1, CDK1), can promote its stability at metaphase (Figure 2B) [42, 44]. In addition, the ubiquitin-proteasome pathway has been identified to regulate survivin degradation in a cell cycle-dependent manner, and the BIR domain of survivin is essential for maintaining its stability at G2/M phase [42, 45].
Receptor tyrosine kinase-associated regulation
In breast cancer, the increased expression of survivin shows association with chemotherapeutic resistance, poor diagnosis and prognosis [37]. Based on immunohistochemical staining in invasive breast cancer specimens, co-expression of epidermal growth factor receptor (EGFR) members (e.g., EGFR and erbB2) is able to regulate survivin protein level in clinical breast cancer samples (Figure 2C) [37], thereby resulting in increased resistance against etoposide-induced apoptosis [37]. Activation of EGFR can increase survivin protein levels, and this process is dependent on the PI3K pathway but not on the MAPK pathway [37, 46]. Survivin protein levels and apoptosis resistance dramatically decrease when cells are treated with herceptin, a monoclonal antibody against erbB2 [37]. Moreover, in prostate cancer cells, stimulation with IGF-1 results in increased survivin expression through a mechanism dependent on the mTOR pathway [39]. Further studies indicate that forced expression of p70S6K1, an mTOR target gene, led to increased survivin expression [39]. Thus, the IGF-1/mTOR signaling pathway regulates survivin expression via rapid changes in mRNA translation to control prostate cancer cell growth and survival [39].
The PI3K/Akt signaling-dependent transcription has been identified to implicate in the expression of a number of cancer-related genes such as survivin and vascular endothelial growth factor (VEGF), a critical regulator of tumor angiogenesis [47]. Recently, it was shown that a positive feedback cycle exists between survivin expression and PI3K/Akt pathway, by which survivin can trigger VEGF-induced tumor angiogenesis [48]. In pancreatic β-cells, EGF can regulate survivin protein stability and prolong survivin protein half-life through the Raf-1/MEK pathway [49, 50]. In acute myelogenous leukemia cells, survivin expression is predominantly regulated through the MEK/MAPK pathway [50, 51]. In primary effusion lymphoma, activation of STAT3 leads to malignant progression through a mechanism involving elevated survivin expression; in contrast, inhibition of STAT3 signaling induces apoptosis due to the decrease of survivin expression [52].
Molecular mechanisms of survivin in tumorigenesis
Survivin is involved in tumorigenesis through diverse mechanisms, including inhibition of apoptosis pathways (Figure 3), regulation of cytokinesis and cell cycle progression, and participation in a variety of pathways such as the p53, Wnt, hypoxia, TGF, and Notch signaling pathways (Figure 4).
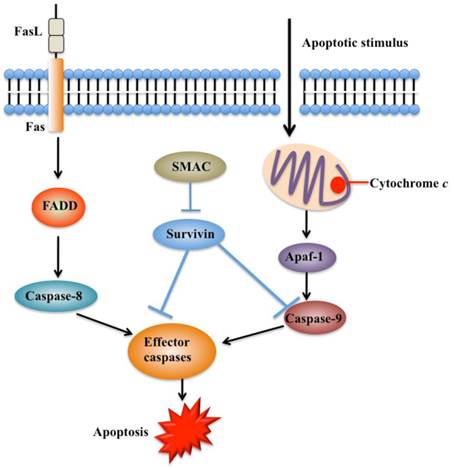
Functions of survivin in apoptosis pathways. Apoptosis can be initiated either by the extrinsic pathway that acts through caspase-8 or by intrinsic pathway that acts through caspase-9. These pathways converge to activate the effector caspases, which eventually leads to apoptosis. Survivin prevents processing of initiator caspase-9 and effector caspases, thereby leading to apoptosis inhibition. Second mitochondria-derived activator of caspase (SMAC) can relieve the caspase-inhibitory function of survivin. FADD, FAS-associated death domain [41, 54].
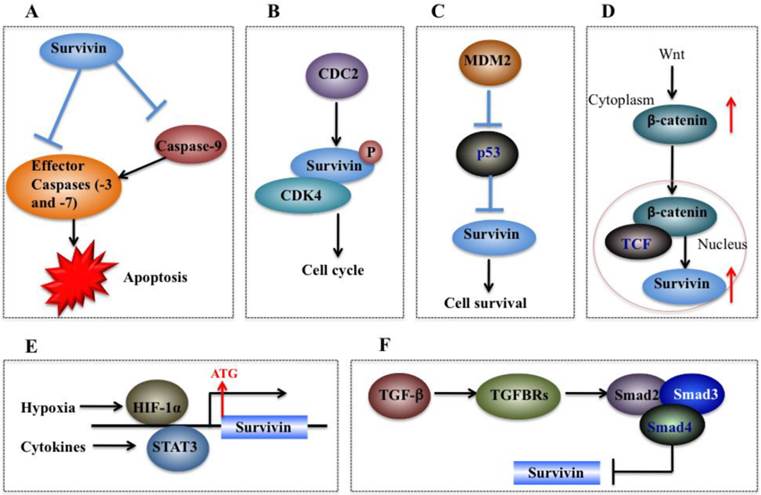
Molecular mechanisms of survivin in tumorigenesis. (A) Survivin binds and suppresses effector caspases (caspase-3 and 7) and caspase-9, thereby resulting in decreased apoptosis in cancer cells. (B) CDC2 can directly phosphorylate survivin. Survivin also interacts with CDK4, which results in nuclear translocation, thereby leading to S phase shift. (C) Wild-type p53 represses survivin expression at the transcriptional level. (D) Activation of the Wnt signaling pathway leads to accumulation of β-catenin in the cytoplasm, which then translocates to the nucleus to form the β-catenin/TCF enhancer factor transcriptional machinery and upregulate survivin. (E) Both HIF-1α and STAT3 can directly bind to the survivin promoter and function as transcriptional activators of the survivin gene. (F) TGF-β is a negative regulator of survivin. The TGF signaling pathway transcriptionally downregulates survivin expression through a mechanism dependent on Smads 2, 3, and 4.
Survivin and apoptosis pathways
Apoptosis is an important process in both carcinogenesis and cancer therapy. Inhibition of apoptosis has been widely reported in a number of cancers such as breast, liver and lung cancers [53-55]. Currently, there are two known apoptotic mechanisms in mammals: the intrinsic and the extrinsic apoptotic pathway [51]. Both of these pathways are controlled by a number of caspase proteins, such as initiator caspases (caspase-8 and 9) and executioner caspases (caspase-3, 6, and 7) [55]. Briefly, apoptosis is initiated by cytochrome c, which is able to bind to cardiolipin in the inner mitochondrial membrane and activates caspase-9 (Figure 3) [56]. Caspase-9 further activates caspase-3 and caspase-7, which are responsible for destroying cells (Figure 3) [57]. Overexpression of survivin is associated with inhibition of cell death initiated via the extrinsic or intrinsic apoptotic pathways (Figure 4A) [58]. Interestingly, reports on the interaction between survivin and caspases are contradictory. A few studies indicate that survivin binds and suppresses caspase-3, 7, and 9, whereas others failed to demonstrate a direct effect on these proteases [59]. Active caspase-3 and 7 co-immunoprecipitated with survivin, but their inactive pro-forms did not [60, 61]. The interaction disrupts the caspase cascade and cleavage mediated by caspases, thereby resulting in decreased apoptosis [28]. In a similar manner, survivin inhibits cytochrome c- and caspase-8-induced DEVD (Asp-Glu-Val-Asp)-cleavage activity [28]. An in vitro study indicates that phosphorylation of survivin at Thr34 by CDC2 is required for the interaction of survivin with caspase-3, 7, and 9 [62]. Further studies found that a mutation of survivin (T34A) can induce the release of cytochrome c from the mitochondria, leading to apoptosis [63].
Survivin and the cell cycle
The cell cycle is very important for cell development, and cell cycle dysfunction may eventually lead to malignant tumorigenesis [35]. As mentioned previously, sequence alignment indicates that survivin contains a CDE/CHR element, implying survivin may be involved in cell cycle process [59]. Experimental studies have consistently indicated that survivin expression is regulated by the cell cycle, and it peaks in the G2/M phase and rapidly declines in the G1 phase [45, 59]. Survivin is dynamically localized at different regions of chromosomes during the cell cycle. During mitosis, survivin localizes to the mitotic spindle, where it interacts with tubulin and plays an important role in regulating mitosis [64]. Survivin accumulates in the centromeres in G2 phase, then it starts to diffuse to the chromosome arms and is abundant at the inner centromeres in prophase and metaphase of mitosis [65]. In anaphase, survivin relocalizes to the central spindle because it fails to associate with the centromeres [66]. During cytokinesis, survivin is concentrated at the midbodies [66]. The cell cycle-specific regulation of survivin implies that it may play important roles in cancer cell proliferation [59]. Moreover, survivin is able to be phosphorylated by CDK1 (Figure 4B), a key kinase for cell cycle regulation [59]. Based on results from co-immunoprecipitation studies, CDK1 can interact with survivin during mitosis [59]. In addition, overexpression of survivin in human hepatoma cells can accelerate S phase shift through a mechanism involving interaction with CDK4, counteract G1 arrest, and result in phosphorylation of retinoblastoma protein (Rb), a tumor suppressor protein [67].
Survivin and p53
It is clear that the tumor suppressor p53 can block cell cycle progression and/or induce apoptosis through specific transactivation of a number of target genes such as p21Cip1, the growth arrest and DNA damage-inducible 45 (GADD45), cyclin G, and Bcl-2-associated X protein (Bax) [68-70]. Given that aberrant expression of survivin and disruption of wild-type p53 are commonly associated with tumorigenesis [71], there exists a high possibility that survivin is functionally linked with p53. A variety of studies have indicated that wild-type p53, but not mutated p53, can repress survivin expression at the transcriptional level [72] and that survivin loss of function partially mediates the p53-dependent apoptotic pathway [73]. Two putative p53-binding sites have been identified within the survivin promoter [74]. However, functional analyses suggest that neither site can repress survivin expression [74]. Further analyses via chromatin deacetylation of the survivin promoter suggest that modification of chromatin may contribute to survivin expression silencing by p53 [74, 75].
On the other hand, survivin can also regulate p53 expression. Overexpression of survivin in human lung cancer cells blocked p53-dependent apoptosis through a dose-dependent manner [73, 75], suggesting that survivin regulates (at least in part) the p53-dependent apoptotic pathway. In addition, survivin regulates p53 expression and modifies p53 degradation through the caspase-3/mouse double minute 2 homolog (MDM2) complex (Figure 4C) [30]. In adriamycin-treated MCF7 human breast cancer cells, aberrant expression of survivin decreased both p53 and MDM2 mRNA levels [30]. Further studies have demonstrated that inhibition of MDM2 cleavage mediated by survivin leads to increased degradation of p53, and thus eventually results in decreased p53 protein level in cells overexpressing survivin [30].
Survivin and Wnt/β-catenin
Wnt/®-catenin signaling is implicated in the development of multiple cancers such as breast, colorectal, lung, prostate cancers and melanoma [76]. During tumorigenesis, accumulation of β-catenin in the cytoplasm is recognized as the hallmark of the Wnt signaling pathway [77]. Subsequently, Wnt signaling induces β-catenin translocation to the nucleus to form the β-catenin/T-cell factor (TCF) transcriptional activator which up-regulates a number of target genes such as survivin, c-Myc and VEGF [78]. The elevated expression of survivin then protects cells from apoptosis and enhances cell proliferation (Figure 4D) [9]. In breast cancer cells, it was found that herceptin, a widely used agent for erbB2-targeting therapy, induced degradation of β-catenin, disrupting the β-catenin/TCF complex and thus eventually leading to repression of survivin expression [38].
Survivin and hypoxia signaling
Hypoxic conditions are very common in most tumors [79]. Experimental studies and clinical trials have shown that hypoxia can contribute to multiple biological processes such as tumor angiogenesis, invasiveness, metastasis, and therapeutic resistance [79]. In response to hypoxia, cells undergo gene expression changes initiated by the HIF-1 transcription factor, which is a dimeric protein complex consisting of HIF-1α and HIF-1β [80]. Generally, the expression of HIF-1 increases under hypoxic conditions, which can activate transcription of numerous genes critical for cellular function under hypoxic conditions [81]. HIF-1α regulates survivin expression in a variety of cancer types such as breast, lung, and pancreatic cancer cells [40, 82]. For example, down-regulation of HIF-1α by RNA interference significantly decreased survivin levels in pancreatic and breast cancer cells [83]. Further studies indicate that HIF-1α directly binds to the promoter of survivin to activate expression (Figure 4E) [83]. In malignant lymphoma cells, NF-κB has been identified to mediate aberrant activation of HIF-1 through a mechanism dependent on survivin, which is one of the downstream targets of the NF-κB/HIF-1 signaling [84].
Survivin and STAT3
STAT3, an oncogene, is persistently activated by cytokines or growth factors in multiple cancer types, and it plays important roles in tumor cell proliferation, differentiation and survival [85]. STAT3 can translocate to the nucleus and regulate transcription of specific target genes such as survivin [86]. In primary lymphoma cells, inhibition of STAT3 signaling has been found to trigger apoptosis through a mechanism involving transcriptional repression of survivin [52]. Further studies found that STAT3 directly binds to the survivin promoter and regulates its expression (Figure 4E) [52].
The survivin protein sequence has a lysine-rich region at the C-terminal, which forms a basic platform for protein-protein interactions and modifications at the post-translational level [49, 87]. Interestingly, survivin shares structural similarities with STAT3, and, following lysine acetylation of survivin, it binds to the N-terminal domain of STAT3 [49, 87], thereby inhibiting STAT3 transactivation [49, 87].
Survivin and the Notch signaling pathway
Notch signaling is critical for cell development, differentiation and survival, as well as tissue morphogenesis [88]. The dysregulation of Notch signaling is implicated in a number of human cancers such as breast, prostate, liver, ovarian, renal, lung and colon cancers [88]. Notch can act as an oncogene in hematological malignancies and solid tumors through multiple mechanisms involved in influencing tumor initiation, regulating the critical events in tumor progression (such as angiogenesis, chemotherapeutic resistance and metastasis), and maintenance of cancer stem cells [88, 89].
Growing evidence also indicates that Notch signaling can regulate survivin expression. For example, Notch-1 signaling activated by hypoxia regulated survivin expression in human non-small cell lung cancer (NSCLC) cells [90]. Further studies have shown that Notch-1 signaling activation is associated with elevated HIF-1α, which functions as a co-activator through its interaction with an activated form of Notch-1 (N1ICD, Notch 1 intracellular domain), facilitating the association of RBP-Jκ (recombination signal binding protein for immunoglobulin kappa J region) to the survivin promoter, ultimately leading to the transcriptional activation of survivin [90]. In addition, up-regulation of survivin by Notch-1 signaling has also been found in estrogen receptor (ER)-negative breast cancer cells, which results in inhibition of apoptosis and acceleration of mitotic transitions [91].
Survivin and TGF-β signaling
TGF-β and its signaling effectors such as Smad2 (mothers against decapentaplegic homolog 2) and Smad3 are considered as key factors in controlling tumor cell proliferation and differentiation [92]. TGF-β is a tumor suppressor due to its ability to inhibit cell growth through induction of cell apoptosis [93]. In recent years, TGF-β has been identified as an important negative regulator of survivin [94]. Generally, TGF-β signaling pathway represses survivin expression at the transcriptional level through a mechanism dependent on Smad2 and 3 and survivin CDE/CHR elements (Figure 4F), which are essential for TGF-β repression [94]. Further studies indicate that the recruitment of the Rb/E2F4 repressive complex to the CDE/CHR elements of the survivin promoter by TGF-β leads to hypophosphorylation of Rb through a Smad3-dependent mechanism [95].
Therapeutic strategies
In recent years, considerable efforts have been made to validate survivin as a preferential target and an important prognostic marker in cancer therapy. Collectively, four types of strategies based on targeting survivin to enhance tumor cell response to apoptosis and inhibit tumor growth have been developed. These promising approaches are: (1) inhibitors of survivin transcription, including antisense oligonucleotides, ribozymes and siRNAs; (2) inhibitors of survivin at the post-translational level; (3) survivin-based vaccines; and (4) gene therapy approaches with survivin suppressor mutants (Figure 5).
Different strategies targeting survivin in cancer therapy. (A) Antisense oligonucleotides, ribozymes, and siRNAs can target survivin mRNA and suppress its expression [10, 41]. (B) Small molecule antagonists can inhibit survivin phosphorylation at Thr34 residue (CDK inhibitors) or counteract survivin-Hsp90 interaction (Hsp90 inhibitors), thereby leading to dysfunction of survivin [97]. (C) Survivin-based vaccines or epitopes have strong immunodominant and immunoprevalent T-cell reactions against survivin, thereby resulting in survivin decrease after treatment.
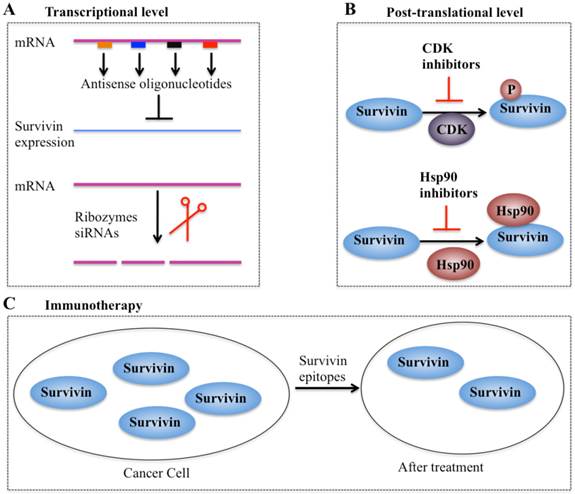
Inhibitors of survivin transcription
Antisense oligonucleotides (AOs) of survivin are designed to specifically target survivin mRNA to suppress its expression [10]. A variety of survivin antisense oligonucleotides have been designed and successfully delivered to cells as chemically synthesized molecules or through vectors, which significantly inhibit survivin expression (Figure 5A) and eventually trigger apoptosis and decrease cell proliferation in different cancer types such as lung and head and neck cancer cells [10]. Importantly, the first antisense oligonucleotide drug, LY2181308, has entered phase II clinical trials in patients with advanced cancers [97-99]. Ribozymes are a class of small RNA molecules that are capable of cleaving target RNA through endonucleolytic activity [10, 99]. Several ribozymes such as CUA110 (RZ7) and GUC294 (RZ1) have been successfully transfected into human melanoma cell lines, resulting in to decreased survivin protein levels and increased caspase-9-dependent apoptosis [99], indicating their potential anticancer activities. However, none of the ribozymes have entered clinical trials because of their limitations (e.g., easy degradation and aberrant cell trafficking). In addition, siRNAs, a class of short (20-25 base pairs in length), double-stranded RNAs that inhibit gene expression from complementary nucleotide sequences, are also utilized in preclinical studies. siRNAs are considered to be more effective than other antisense strategies, and preclinical studies indicate that cancer cells transfected with survivin-specific siRNAs have increased apoptosis and reduced cell proliferation and growth [10, 99].
Inhibitors of survivin transcription at the post-translational level
Agents involved in survivin inhibition at the post-translational level include CDK inhibitors and Hsp90 inhibitors (Figure 5B) [10, 99]. A number of CDK inhibitors such as flavopiridol and purvalanol A have the ability to counteract mitotic phosphorylation of survivin at Thr34, thereby accelerating survivin degradation [10, 99]. Interestingly, administration of CDK inhibitors results in the decrease of survivin expression and induction of apoptosis in an in vivo study [10, 99].
Inhibitors of molecular chaperones have been used in preclinical studies because they can counteract the Hsp90-survivin interaction. Shepherdin is a cell-permeable antagonist of the Hsp90-survivin complex, and it also inhibits the Hsp90 function by competing with ATP binding [10, 99]. Moreover, shepherdin is able to destabilize the interactions between Hsp90 and its associated proteins such as Akt, telomerase and CDK6 [10, 99]. Functional analyses indicate that shepherdin induces apoptosis through mechanisms dependent on or independent of caspases in various cancer cells [10, 99]. Importantly, shepherdin is highly selective in its anticancer activity because treatment with shepherdin only had a weak toxicity on normal fibroblasts following increase in drug dose and no other serious side effects on normal cells [10, 99]. This suggests that shepherdin has significant potential for future therapeutic use in treating cancer.
Vaccines and immunotherapy
Immunotherapy for targeting survivin in tumor cells is based on the specific recognition of specific tumor-associated antigens by T lymphocytes (Figure 5C) [100, 101]. Earlier studies have revealed that a survivin-based vaccine had significant cytotoxic activity against liver cancer cells [10, 100, 101]. Thereafter, recent efforts have been made to develop survivin-based anticancer vaccines. These vaccines have been shown to result in tumor suppression in multiple cancer types such as lymphomas, neuroblastomas, pancreatic cancer, and lung cancer [10, 100], without inducing significant toxicity. Due to the promising capabilities survivin-based immunotherapeutic strategies, a variety of studies focused on identifying epitopes have developed powerful vaccines against survivin [10, 100], and some of these vaccines have already moved on to phase II clinical trials [10, 100].
Therapy based on gene editing
Gene therapy approaches that target survivin were one of the most promising strategies used to inhibit survivin, and two main approaches have been successfully developed [10, 97]. One is based on the activation of apoptosis in tumor cells transfected with survivin-containing vectors [94, 96]. The other approach involves the expression of cytotoxic genes driven by the promoter of survivin in tumor cells [10, 97]. Interestingly, in recent years, with the advancing development of “genome editing” techniques such as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), or clustered regularly interspaced short palindromic repeat (CRISPR/Cas-based) RNA-guided DNA endonucleases, researchers have proposed to use these approaches to target the survivin gene in different cancer cells [10]. These strategies would specifically trigger cell death in targeted cancer cells while having minimal or nontoxic effects on healthy cells.
Conclusion
Survivin is involved in tumorigenesis through complicated mechanisms. Survivin is considered to be a tumor marker due to aberrant survivin expression in tumor tissues and its causal role in cancer progression. Many studies have demonstrated that survivin is involved in multiple cancer survival-related signaling pathways and that survivin expression is highly correlated with tumor progression, therapeutic resistance and poor prognosis. Elucidation of the various mechanisms of survivin regulation has helped in the development of suitable therapeutic strategies in preclinical studies; some drugs based on these strategies are currently in phase I or phase II clinical trials. However, the fact that survivin is not an enzyme or a cell-surface molecule has led to limited therapeutic benefits. Thus, suitable survivin carrier molecules should be developed to overcome these limits. Moreover, strategies based on immunotherapy and gene therapy have exhibited significant advantages in targeting survivin with more efficiency and specificity for tumor cells, but less cytotoxicity against healthy tissues and cells. These strategies should receive more attention to develop promising agents that target survivin for cancer treatment.
Acknowledgements
We acknowledge all authors whose contributions were not cited due to space limitations.
Competing Interests
The authors declare no conflict of interest. This article does not contain any studies with human or animal subjects performed by the authors.
References
1. Su Z, Yang Z, Xu Y. et al. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. 2015;14(1):48
2. Sun L, Peng Q, Qu L. et al. Anticancer agent icaritin induces apoptosis through caspase-dependent pathways in human hepatocellular carcinoma cells. Mol Med Rep. 2015;11:3094-100
3. Zhang C, Liu Y. Targeting cancer with sesterterpenoids: the new potential antitumor drugs. J Nat Med. 2015;69:255-66
4. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495-516
5. Zhang C, Zhang F. The Multifunctions of WD40 Proteins in Genome Integrity and Cell Cycle Progression. J Genomics. 2015;3:40-50
6. Troy CM, Jean YY. Caspases: therapeutic targets in neurologic disease. Neurotherapeutics. 2015;12:42-8
7. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5:a008656
8. Brady SW, Zhang J, Tsai MH. et al. PI3K-independent mTOR activation promotes lapatinib resistance and IAP expression that can be effectively reversed by mTOR and Hsp90 inhibition. Cancer Biol Ther. 2015;16:402-11
9. Huang J, Lyu H, Wang J. et al. MicroRNA regulation and therapeutic targeting of survivin in cancer. Am J Cancer Res. 2015;5:20-31
10. Mobahat M, Narendran A, Riabowol K. Survivin as a preferential target for cancer therapy. Int J Mol Sci. 2014;15:2494-516
11. Saleem M, Qadir MI, Perveen N. et al. Inhibitors of apoptotic proteins: new targets for anticancer therapy. Chem Biol Drug Des. 2013;82:243-51
12. Jarrin M, Mansergh FC, Boulton ME. et al. Survivin expression is associated with lens epithelial cell proliferation and fiber cell differentiation. Mol Vis. 2012;18:2758-69
13. Ambrosini G, Adida C & Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917-21
14. Fong WG, Liston P, Rajcan-Separovic E. et al. Expression and genetic analysis of XIAP-associated factor 1 (XAF1) in cancer cell lines. Genomics. 2000;70:113-22
15. LaCasse EC, Baird S, Korneluk RG. et al. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene. 1998;17:3247-59
16. Li J, Feng Q, Kim JM. et al. Human ovarian cancer and cisplatin resistance: possible role of inhibitor of apoptosis proteins. Endocrinology. 2001;142:370-80
17. Lin JH, Deng G, Huang Q. et al. KIAP, a novel member of the inhibitor of apoptosis protein family. Biochem Biophys Res Commun. 2000;279:820-31
18. Tamm I, Kornblau SM, Segall H. et al. Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin Cancer Res. 2000;6:1796-803
19. Hunter AM, LaCasse EC, Korneluk RG. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis. 2007;12:1543-68
20. Luk SU, Xue H, Cheng H. et al. The BIRC6 gene as a novel target for therapy of prostate cancer: dual targeting of inhibitors of apoptosis. Oncotarget. 2014;5:6896-908
21. Kusner LL, Ciesielski MJ, Marx A. et al. Survivin as a potential mediator to support autoreactive cell survival in myasthenia gravis: a human and animal model study. PLoS One. 2014;9:e102231
22. Gu Y, Jin S, Wang F. et al. Clinicopathological significance of PI3K, Akt and survivin expression in gastric cancer. Biomed Pharmacother. 2014;68:471-5
23. Sierra JR, Cepero V, Giordano S. Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy. Mol Cancer. 2010;9:75
24. Carter BZ, Mak DH, Schober WD. et al. Regulation of survivin expression through Bcr-Abl/MAPK cascade: targeting survivin overcomes imatinib resistance and increases imatinib sensitivity in imatinib-responsive CML cells. Blood. 2006;107:1555-63
25. Anandharaj A, Cinghu S, Park WY. Rapamycin-mediated mTOR inhibition attenuates survivin and sensitizes glioblastoma cells to radiation therapy. Acta Biochim Biophys Sin (Shanghai). 2011;43:292-300
26. Sehara Y, Sawicka K, Hwang JY. et al. Survivin Is a transcriptional target of STAT3 critical to estradiol neuroprotection in global ischemia. J Neurosci. 2013;33:12364-74
27. Tang C, Lu YH, Xie JH. et al. Downregulation of survivin and activation of caspase-3 through the PI3K/Akt pathway in ursolic acid-induced HepG2 cell apoptosis. Anticancer Drugs. 2009;20:249-58
28. Tamm I, Wang Y, Sausville E. et al. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 1998;58:5315-20
29. Altieri DC. Cytokinesis, apoptosis and survivin: three for tango? Cell Death Differ. 2001;8:4-5
30. Wang Z, Fukuda S, Pelus LM. Survivin regulates the p53 tumor suppressor gene family. Oncogene. 2004;23:8146-53
31. Zhang L, Yan R, Zhang Q. et al. Survivin, a key component of the Wnt/beta-catenin signaling pathway, contributes to traumatic brain injury-induced adult neurogenesis in the mouse dentate gyrus. Int J Mol Med. 2013;32:867-75
32. Bai H, Ge S, Lu J. et al. Hypoxia inducible factor-1alpha-mediated activation of survivin in cervical cancer cells. J Obstet Gynaecol Res. 2013;39:555-63
33. Chen Y, Li D, Liu H. et al. Notch-1 signaling facilitates survivin expression in human non-small cell lung cancer cells. Cancer Biol Ther. 2011;11:14-21
34. Chen W, Zhong X, Wei Y. et al. TGF-beta Regulates Survivin to Affect Cell Cycle and the Expression of EGFR and MMP9 in Glioblastoma. Mol Neurobiol. 2015 DOI 10.1007/s12035-015-9121-6
35. Chen Z, Sui J, Zhang F. et al. Cullin family proteins and tumorigenesis: genetic association and molecular mechanisms. J Cancer. 2015;6:233-42
36. Zhang C. Essential functions of iron-requiring proteins in DNA replication, repair and cell cycle control. Protein Cell. 2014;5:750-60
37. Asanuma H, Torigoe T, Kamiguchi K. et al. Survivin expression is regulated by coexpression of human epidermal growth factor receptor 2 and epidermal growth factor receptor via phosphatidylinositol 3-kinase/AKT signaling pathway in breast cancer cells. Cancer Res. 2005;65:11018-25
38. Zhu H, Zhang G, Wang Y. et al. Inhibition of ErbB2 by Herceptin reduces survivin expression via the ErbB2-beta-catenin/TCF4-survivin pathway in ErbB2-overexpressed breast cancer cells. Cancer Sci. 2010;101:1156-62
39. Vaira V, Lee CW, Goel HL. et al. Regulation of survivin expression by IGF-1/mTOR signaling. Oncogene. 2007;26:2678-2684
40. Li Y, Xia ZL, Chen LB. HIF-1-alpha and survivin involved in the anti-apoptotic effect of 2ME2 after global ischemia in rats. Neurol Res. 2011;33:583-92
41. Muller GA, Engeland K. The central role of CDE/CHR promoter elements in the regulation of cell cycle-dependent gene transcription. FEBS J. 2010;277:877-93
42. Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003;3:46-54
43. Misso G, Di Martino MT, De Rosa G. et al. Mir-34: a new weapon against cancer? Mol Ther Nucleic Acids. 2014;3:e194
44. O'Connor DS, Wall NR, Porter AC. et al. A p34(cdc2) survival checkpoint in cancer. Cancer Cell. 2002;2:43-54
45. Zhao J, Tenev T, Martins LM. et al. The ubiquitin-proteasome pathway regulates survivin degradation in a cell cycle-dependent manner. J Cell Sci. 2000;113(Pt 23):4363-71
46. Hakonen E, Ustinov J, Palgi J. et al. EGFR signaling promotes beta-cell proliferation and survivin expression during pregnancy. PLoS One. 2014;9:e93651
47. Karar J, Maity A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front Mol Neurosci. 2011;4:51
48. Fernandez JG, Rodriguez DA, Valenzuela M. et al. Survivin expression promotes VEGF-induced tumor angiogenesis via PI3K/Akt enhanced beta-catenin/Tcf-Lef dependent transcription. Mol Cancer. 2014;13:209
49. Wang H, Gambosova K, Cooper ZA. et al. EGF regulates survivin stability through the Raf-1/ERK pathway in insulin-secreting pancreatic beta-cells. BMC Mol Biol. 2010;11:66
50. Carter BZ, Milella M, Altieri DC. et al. Cytokine-regulated expression of survivin in myeloid leukemia. Blood. 2001;97:2784-90
51. Zhang C, Zhang F. Iron homeostasis and tumorigenesis: molecular mechanisms and therapeutic opportunities. Protein Cell. 2015;6:88-100
52. Aoki Y, Feldman GM, Tosato G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood. 2003;101:1535-42
53. Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87
54. Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005;5:876-85
55. Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798-811
56. Ott M, Robertson JD, Gogvadze V. et al. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci U S A. 2002;99:1259-63
57. Brentnall M, Rodriguez-Menocal L, De Guevara RL. et al. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013;14:32
58. Small S, Keerthivasan G, Huang Z. et al. Overexpression of survivin initiates hematologic malignancies in vivo. Leukemia. 2010;24:1920-6
59. Chandele A, Prasad V, Jagtap JC. et al. Upregulation of survivin in G2/M cells and inhibition of caspase 9 activity enhances resistance in staurosporine-induced apoptosis. Neoplasia. 2004;6:29-40
60. Wright ME, Han DK, Hockenbery DM. Caspase-3 and inhibitor of apoptosis protein(s) interactions in Saccharomyces cerevisiae and mammalian cells. FEBS Lett. 2000;481:13-8
61. Song Z, Yao X, Wu M. Direct interaction between survivin and Smac/DIABLO is essential for the anti-apoptotic activity of survivin during taxol-induced apoptosis. J Biol Chem. 2003;278:23130-40
62. Singh M, Chaudhry P, Fabi F. et al. Cisplatin-induced caspase activation mediates PTEN cleavage in ovarian cancer cells: a potential mechanism of chemoresistance. BMC Cancer. 2013;13:233
63. Liu T, Brouha B, Grossman D. Rapid induction of mitochondrial events and caspase-independent apoptosis in Survivin-targeted melanoma cells. Oncogene. 2004;23:39-48
64. Mita AC, Mita MM, Nawrocki ST. et al. Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res. 2008;14:5000-5
65. Beardmore VA, Ahonen LJ, Gorbsky GJ. et al. Survivin dynamics increases at centromeres during G2/M phase transition and is regulated by microtubule-attachment and Aurora B kinase activity. J Cell Sci. 2004;117:4033-42
66. Dai D, Liang Y, Xie Z. et al. Survivin deficiency induces apoptosis and cell cycle arrest in HepG2 hepatocellular carcinoma cells. Oncol Rep. 2012;27:621-7
67. Suzuki A, Hayashida M, Ito T. et al. Survivin initiates cell cycle entry by the competitive interaction with Cdk4/p16(INK4a) and Cdk2/cyclin E complex activation. Oncogene. 2000;19:3225-34
68. Chumakov PM. Function of the p53 gene: choice between life and death. Biochemistry (Mosc). 2000;65:28-40
69. Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22:9030-40
70. Zhang C, Liu G, Huang M. Ribonucleotide reductase metallocofactor: assembly, maintenance and inhibition. Front Biol. 2014;9:104-13
71. Lai YJ, Lin CI, Wang CL. et al. Expression of survivin and p53 modulates honokiol-induced apoptosis in colorectal cancer cells. J Cell Biochem. 2014;115:1888-99
72. Hoffman WH, Biade S, Zilfou JT. et al. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247-57
73. Mirza A, McGuirk M, Hockenberry TN. et al. Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene. 2002;21:2613-22
74. Ulasov IV, Tyler MA, Zhu ZB. et al. Oncolytic adenoviral vectors which employ the survivin promoter induce glioma oncolysis via a process of beclin-dependent autophagy. Int J Oncol. 2009;34:729-42
75. Mirza A, McGuirk M, Hockenberry TN. et al. Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene. 2002;21:2613-22
76. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11-26
77. Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. 2008;4:68-75
78. Stewart DJ. Wnt signaling pathway in non-small cell lung cancer. J Natl Cancer Inst. 2014;106:djt356
79. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393-410
80. Penn JS, Madan A, Caldwell RB. et al. Vascular endothelial growth factor in eye disease. Prog Retin Eye Res. 2008;27:331-71
81. Ziello JE, Jovin IS, Huang Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J Biol Med. 2007;80:51-60
82. Wang YF, Ma SR, Wang WM. et al. Inhibition of survivin reduces HIF-1alpha, TGF-beta1 and TFE3 in salivary adenoid cystic carcinoma. PLoS One. 2014;9:e114051
83. Chen YQ, Zhao CL, Li W. Effect of hypoxia-inducible factor-1alpha on transcription of survivin in non-small cell lung cancer. J Exp Clin Cancer Res. 2009;28:9
84. Qiao Q, Nozaki Y, Sakoe K. et al. NF-kappaB mediates aberrant activation of HIF-1 in malignant lymphoma. Exp Hematol. 2010;38:1199-208
85. Benekli M, Baer MR, Baumann H. et al. Signal transducer and activator of transcription proteins in leukemias. Blood. 2003;101:2940-54
86. Carpenter RL, Lo HW. STAT3 Target Genes Relevant to Human Cancers. Cancers (Basel). 2014;6:897-925
87. Wang H, Holloway MP, Ma L. et al. Acetylation directs survivin nuclear localization to repress STAT3 oncogenic activity. J Biol Chem. 2010;285:36129-37
88. Allenspach EJ, Maillard I, Aster JC. et al. Notch signaling in cancer. Cancer Biol Ther. 2002;1:466-76
89. Lobry C, Oh P, Aifantis I. Oncogenic and tumor suppressor functions of Notch in cancer: it's NOTCH what you think. J Exp Med. 2011;208:1931-35
90. Chen Y, Li D, Liu H. et al. Notch-1 signaling facilitates survivin expression in human non-small cell lung cancer cells. Cancer Biol Ther. 2011;11:14-21
91. Lee CW, Raskett CM, Prudovsky I. et al. Molecular dependence of estrogen receptor-negative breast cancer on a notch-survivin signaling axis. Cancer Res. 2008;68:5273-81
92. Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117-29
93. Wahdan-Alaswad RS, Song K, Krebs TL. et al. Insulin-like growth factor I suppresses bone morphogenetic protein signaling in prostate cancer cells by activating mTOR signaling. Cancer Res. 2010;70:9106-17
94. Song K, Shankar E, Yang J. et al. Critical role of a survivin/TGF-beta/mTORC1 axis in IGF-I-mediated growth of prostate epithelial cells. PLoS One. 2013;8:e61896
95. Yang J, Song K, Krebs TL. et al. Rb/E2F4 and Smad2/3 link survivin to TGF-beta-induced apoptosis and tumor progression. Oncogene. 2008;27:5326-38
96. Bates DJ, Lewis LD. Manipulating the apoptotic pathway: potential therapeutics for cancer patients. Br J Clin Pharmacol. 2013;76:381-95
97. Pennati M, Folini M, Zaffaroni N. Targeting survivin in cancer therapy: fulfilled promises and open questions. Carcinogenesis. 2007;28:1133-9
98. Pennati M, Colella G, Folini M. et al. Ribozyme-mediated attenuation of survivin expression sensitizes human melanoma cells to cisplatin-induced apoptosis. J Clin Invest. 2002;109:285-6
99. Yamaki H, Nakajima M, Shimotohno KW. et al. Molecular basis for the actions of Hsp90 inhibitors and cancer therapy. J Antibiot (Tokyo). 2011;64:635-44
100. Schmidt SM, Schag K, Muller MR. et al. Survivin is a shared tumor-associated antigen expressed in a broad variety of malignancies and recognized by specific cytotoxic T cells. Blood. 2003;102:571-6
101. Arber C, Feng X, Abhyankar H. et al. Survivin-specific T cell receptor targets tumor but not T cells. J Clin Invest. 2015;125:157-68
Author contact
![]() Corresponding authors: zhangwentao1975com and caiguo.zhangedu
Corresponding authors: zhangwentao1975com and caiguo.zhangedu