Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(2):184-191. doi:10.7150/jca.12841 This issue Cite
Research Paper
Adenovirus-Mediated Angiotensin II Type 2 Receptor Overexpression Inhibits Tumor Growth of Prostate Cancer In Vivo
Jinlong Li1,*, Jie Luo1,*, Dongsheng Gu5,*, Feilong Jie1,*, Nana Pei6, Andrew Li3, Xinglu Chen1, Yanling Zhang1, Hongyan Du1, Baihong Chen1, Weiwang Gu2, ![]() , Colin Sumners4, Hongwei Li1,
, Colin Sumners4, Hongwei Li1, ![]()
1. School of Biotechnology, Southern Medical University, Guangzhou, Guangdong, China
2. Institute of Comparative Medicine and Center of Laboratory Animals, Southern Medical University, Guangzhou, Guangdong, China;
3. Department of Biomedical Engineering, The Johns Hopkins University School of Medicine, Baltimore, USA;
4. Department of Physiology and Functional Genomics, University of Florida, Gainesville, Florida, USA; and
5. Department of Urology, the 421st Hospital of PLA, Guangzhou, Guangdong, China;
6. Department of Clinical Pathology, The First Affiliated Hospital of Jinan University, Guangzhou, Guangdong, China
* These authors contributed equally to this work.
Received 2015-6-1; Accepted 2015-10-17; Published 2016-1-1
Abstract
The renin-angiotensin system (RAS) plays important roles in tumorigenesis and is involved with several hallmarks of cancer. Evidence shows that angiotensin II (AngII) type 1 receptor (AT1R) blockers may be associated with improved outcome in prostate cancer patients. Furthermore, our previous studies indicate that increased expression of Ang II type 2 receptor (AT2R) alone induced apoptosis in human prostate cancer lines, an effect that did not require Ang II. This study aimed to investigate the effects of AT2R on tumor growth in vivo and we hypothesized that AT2R over-expression would inhibit proliferation and induce apoptosis in vivo. Human prostate cancer DU145 xenograft mouse model was used to assess the effect of AT2R on tumor growth in vivo. Mice bearing a palpable tumor were chosen and divided randomly into three treatment groups: AT2R, GFP, and PBS. Then we directly injected into the xenograft tumors of the mice every three days with recombinant adenoviruses encoding AT2R (Ad5-CMV-AT2R-EGFP), EGFP (Ad5-CMV-EGFP) and PBS, respectively. The tumor sizes of the tumor bearing mice were then measured. Immunohistochemical Ki-67 staining and TUNEL assay were performed to examine the inhibitory effect of AT2R on tumor cell proliferation. The results showed that AT2R overexpression can inhibit tumor growth of prostate cancer in vivo by inhibiting proliferation and inducing apoptosis of tumor cells. GADD45A is involved in the AT2R-induced antitumor activity. This suggests that AT2R is a potentially useful gene for prostate gene therapy.
Keywords: AT2R, Adenovirus, prostate cancer, tumor growth, apoptosis.
Introduction
Prostate cancer (PCa) is among the most prevalent malignancies in older men and a frequent cause of death. Most of patients will eventually progress to a hormone-refractory state, making them unresponsive to further hormonal manipulation and resistant to chemotherapy. Thus there is an urgent need to develop new therapeutic strategies for PCa treatment.
Accumulating evidence suggests that angiotensin II (Ang II) and its receptor, AT2R, are involved in tumorigenesis. It is well-established that AT2R signaling has anti-proliferative effects through apoptosis induction in numerous cell lines such as those for lung adenocarcinoma, colorectal cancer, hepatocellular carcinoma, insulinoma and prostate cancer [1-5]. In vivo studies have revealed that AT2R deficiency significantly enhanced chemical carcinogen-induced tumorigenesis in mouse colon [6] and lung [7] and stimulates the growth of murine pancreatic carcinoma grafts [8]. More direct studies on AT2R effects showed that intratracheal administration of a nanoparticle-based therapy with the AT2R gene attenuates lung cancer growth even more so than TRAIL treatment [9]. With regard to PCa, AT2R was localized at the level of the acinar epithelial layer and its expression was decreased in cancers with a Gleason score 6 or higher [10]. Functional AT2R receptors are present and have the capacity to inhibit EGF-induced prostate cancer cell growth in LNCaP and fast growing androgen-independent PC3 cell lines [11]. Our previous studies indicate that increased expression of AT2R alone induced apoptosis in the prostate cancer lines [4]. These findings led to the hypothesis that the induction of AT2R expression in PCa tumor tissue stimulates apoptosis in vivo and that AT2R is a potential therapeutic gene for controlling prostate cancer.
In this paper, high AT2R expression was obtained by direct intratumoral injection of AT2R recombinant adenoviral vector. The antitumoral effect of the highly-expressed AT2R was examined in a PCa xenograft tumor model and the possible underlying mechanisms were characterized. Our results showed that the adenovirus-mediated high AT2R expression efficiently inhibited the xenograft tumor growth in mice. Apoptosis and inhibition of proliferation were observed with no upregulation of VEGF in the tumors. These results suggest that AT2R overexpression represents a new approach towards PCa treatment.
Materials and Methods
Cell line
Human prostate cancer DU145 cell lines were obtained from the American Type Culture Collection (ATCC, Rockville, MD) and were cultured in EMEM medium supplemented with 10% FBS under 5.0% CO2. Sera and media were purchased from Life Technologies-Invitrogen and ATCC.
Recombinant adenoviral construction
Recombinant adenoviral vectors were constructed as previously detailed [12]. An adenoviral vector containing genomic AT2R (G-AT2R) DNA with introns 1 and 2, the encoding region, and the enhanced green fluorescent protein (EGFP) gene controlled by cytomegalovirus promoters (Ad-G-AT2R-EGFP) was constructed. An adenoviral vector containing just the EGFP gene controlled by a cytomegalovirus promoter (Ad-CMV-EGFP) was used as a vector control. The recombinant viruses were produced, purified and titrated as described previously [12].
Cell Transduction and AT2R Immunostaining
For viral transduction, DU145 cells (4 × 105) were seeded into six-well Nunc tissue culture plates. On the following day, cells were transduced with Ad-G-AT2R-EGFP or the control vector Ad-CMV-EGFP. At 48hr after viral transduction, cells were washed briefly with Dulbecco's PBS and then fixed for 10 min at 4°C with cold methanol. Immunostaining was then performed on the fixed cells as described previously (4) using a goat anti-AT2R receptor polyclonal antibody (1:200; Santa Cruz Biotechnology) followed by Alexa Fluor 594 goat anti-rabbit IgG (1:1,000; Life Technologies-Invitrogen) as the secondary antibody. AT2R immunoreactivity (red) and green fluorescence (from GFP) were detected using an Olympus BX41 fluorescence microscope.
Xenograft tumor model
BALB/c nude mice at 4 to 5 weeks of age were purchased from the Experimental Animal Center of the Southern Medical University and were maintained under standard pathogen-free conditions. Experiments were performed in accordance with the guidelines and approval of the local Institutional Animal Experimentation Ethics Committee. Mice were anaesthetized by chloral hydrate (3.5%) and a total of 5 × 106 (100 μl) human prostate carcinoma DU145 cells were injected subcutaneously at the left axilla of mice. When palpable tumors formed, the mice were randomly divided into three groups. A dose of 109 infectious units (ifu) of Ad-CMV-EGFP or Ad-G-AT2R-EGFP virus suspended in 100 μl of PBS or 100 μl PBS alone was administrated via intratumoral injection to each of the assigned mice. Injections were given once every 3 day for 15 days (5 injections in total). Before each injection, the length and width of the tumors were measured by using a Vernier caliper.
At the end of experiments, mice were killed by cervical dislocation, and the tumors were collected, weighed, and photographed. To confirm the transgene expression within the tumor, a series of 7-μM-thick fresh-frozen sections of the samples were made using a microtome at low-temperature and observed under fluorescence microscopy. The tumor samples were preserved in liquid nitrogen for further study.
Immunohistochemical staining
Formalin-fixed paraffin-embedded tumor samples were cut into 5μm-thick sections and stained with hematoxylin and eosin (H&E) to determine cell morphology. The expression of Ki-67 in tumors tissues were examined using an Immunostain SP Kit (Zhongshan Gene Bridge Biotechnology Company, Beijing, China). The anti-Ki-67 primary antibody was obtained from Millipore (Merck KGaA, Darmstadt, Germany). The immunohistochemically stained samples were assessed independently by two researchers blinded from the samples. The final staining score was calculated according to the intensity of the dye color and the percentage of positive cells as detailed previously described [13].
TUNEL staining
Apoptosis in the tumor tissues was assessed by an in situ TUNEL staining kit (Roche Applied Science, Indianapolis IN, USA) according to the manufacturer's protocol. TUNEL signals were generated by horseradish peroxidase (HRP) labelling and visualized with DAB color formation using a standard protocol. Slides were counterstained with hematoxylin. At least 5 randomly selected fields were viewed in each section under a 10×40 microscope to calculate the positive cells. Data are presented as a percent of the total number of cells on the dish.
Real-time PCR
Frozen tissue samples were cryopulverized in liquid nitrogen with mortar and pestle. Total RNA was extracted with TRIzol reagent (Life Technologies-Invitrogen) according to the manufacturer's protocol. The isolated RNA was then converted into cDNA with Oligo dT and PrimeScript Reverse Transcriptase (TAKARA, Dalian, China) regents. Quantitative real-time PCR was performed in triplicate by using iQ SYBR Green Supermix (BIO-RAD, Hercules, CA). The samples were analyzed using an ABI 7500 real-time PCR system (Life Technologies -Applied Biosystems) and the mRNA level was normalized by using GAPDH as the house-keeping gene. The oligonucleotide sequences of forward and reverse primers used for real-time PCR are shown in Table 1.
Primer sequences used for real-time PCR
| Gene | Sense | Sequence | Product size(bp) |
|---|---|---|---|
| AT2R | Forward | 5'-CCGCATTTAACTGCTCACACA-3' | 169 |
| Reverse | 5'-ATCATGTAGTAGAGAACAGGAATTGCTT-3' | ||
| GADD45A | Forward | 5'-GCTCTCTCCCTGGGCGACCT-3' | 86 |
| Reverse | 5'-TCGGGGTCGCTTTCGGTCTT-3 | ||
| TRAIL-R2 | Forward | 5'-CCCAGCTGTGGAGGAGACGGT-3' | 82 |
| Reverse | 5'-ACTACGGCTGCAACTGTGACTCCT-3' | ||
| VEGF | Forward | 5'-GGAGCACTTGGGTTGGATA-3' | 95 |
| Reverse | 5'-GCGATGAATGGGCCATAGT-3' | ||
| GAPDH | Forward | 5'-ACGGATTTGGTCGTATTGGG-3' | 175 |
| Reverse | 5'-CGCTCCTGGAAGATGGTGAT-3' |
Data Analysis
Data are presented as mean ± standard deviation (SD) from at least 3 independent experiments. Statistical analysis was performed with SPSS 13.0 software. Repeated measures ANOVA was performed to analyze the difference in tumor volume or weight between the AT2R-EGFP, EGFP and PBS groups. P<0.05 was considered statistically significant.
Results
1. Ad-G-AT2R-EGFP injection inhibits tumor growth in vivo
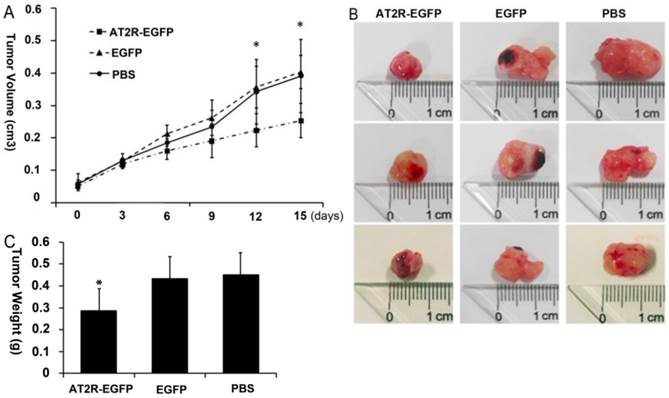
Two weeks after tumor cell injections, palpable tumors formed in the injection site. Then multi-point intratumoral injections of viruses were given once every 3 days. Throughout this experiment, the mice were given 5 injections in total. During the 15 days after the first injection, the tumors grew progressively in the PBS and EGFP control virus group, reaching a tumor volume of 0.391±0.103 cm3 in the PBS group and 0.4034±0.05 cm3 in the EGFP group at the end of experiment. In contrast, after the 4th virus injection (9 days after injection) of Ad-G-AT2R-EGFP, the tumor growth was much slower than in PBS and EGFP virus groups, with a much smaller tumor volume (0.2531±0.053 cm3) at the end of experiment (p<0.05) (Fig.1a). The appearance and weight of the tumors at the time of sacrifice after treatment with Ad-G-AT2R-EGFP, Ad-CMV-EGFP or PBS are shown in Figures 1b and 1c, respectively. These results indicate that AT2R overexpression via Ad-G-AT2R-EGFP treatment was associated with inhibition of tumor growth in vivo.
Anti-tumor effect of Ad-G-AT2R-EGFP treatment on DU145 tumor growth in nude mice. A human prostate cancer xenografts model was established by subcutaneously injection of DU145 cells at the left axilla of nude mice. The length and width of the tumors were measured to calculate the volumes every 3 days, just before each virus injection. (A) The mean volume of the subcutaneous xenograft tumors was calculated for 6 mice in each group. A significant difference was observed between the results of Ad-G-AT2R-EGFP and Ad-CMV-EGFP or PBS treatment (*p<0.05 by repeated measurement of ANOVA). (B and C) Appearance and weight of the tumors at the time of sacrifice after treatment with Ad-G-AT2R-EGFP and Ad-CMV-EGFP or PBS (*p<0.05).
2. Ad-G-AT2R-EGFP induces AT2R overexpression in vivo and in vitro
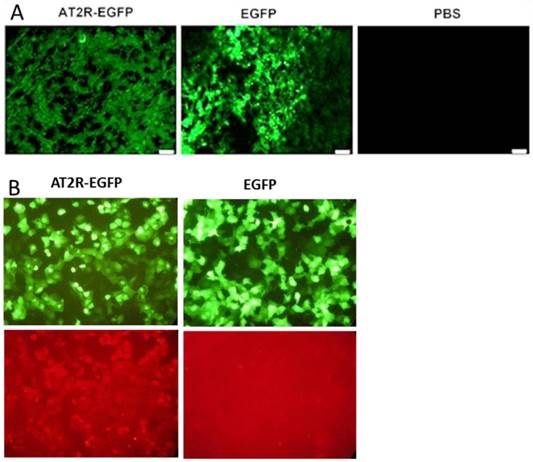
Strong EGFP fluorescence was detected in fresh frozen sections from AT2R-EGFP and EGFP injected tumors, but not in PBS injected tumors demonstrating the ability of these adenoviral vectors to transduce human prostate cells in vivo (Fig.2A). In vivo Ad-G-AT2R-EGFP-mediated AT2R overexpression in tumor cells can be inferred from the EGFP expression since AT2R and EGFP were located in two expression cassettes on the same vector. This is further supported by the presence of AT2R mRNA in the AT2R-EGFP injected group, but not in the EGFP and PBS groups, as shown by real-time PCR (Fig.5a).
Adenoviral vector-mediated EGFP or AT2R expression in prostate cancer cells in vivo (A) and in vitro (B). (A) EGFP expression in tumors. At the end of experiments (3 days after the fifth virus injection), mice were sacrificed to obtain the tumors. EGFP expression in fresh-frozen tumor sections was observed under a fluorescent microscope. (B) Representative fluorescence micrographs from transduced cells showing EGFP fluorescence (Upper panels) and AT2R immunostaining (Bottom panels). Ad-G-AT2R-EGFP-mediated AT2R expression in DU145 cells. DU145 cells were transduced with either Ad-CMV-EGFP or Ad-G-AT2R-EGFP as described in Materials and Methods. Incubations were followed by detection of EGFP fluorescence and AT2R immunoreactivity using an anti-AT2R antibody.
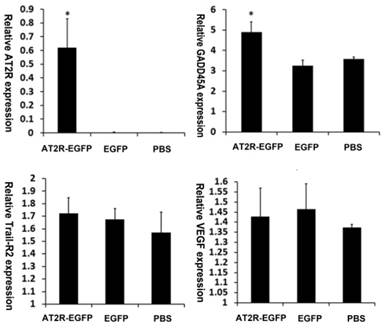
AT2R over-expression upregulates GADD45A mRNA level. AT2R, GADD45A, Trail-R2 and VEGF mRNA levels in the tumor tissues were examined by real-time PCR according to the materials and methods. Data are presented as mean ± SD (*P<0.05).
Ad-G-AT2R-EGFP also proved to be effective in eliciting AT2R expression in human prostate cancer cells in vitro. The fluorescence micrographs presented in Fig. 2B show that incubation of DU145 cells with Ad-G-AT2R-EGFP produced a high level of AT2R immunoreactivity when compared with DU145 cells infected with Ad-CMV-EGFP.
3. AT2R overexpression inhibits tumor cell proliferation in vivo
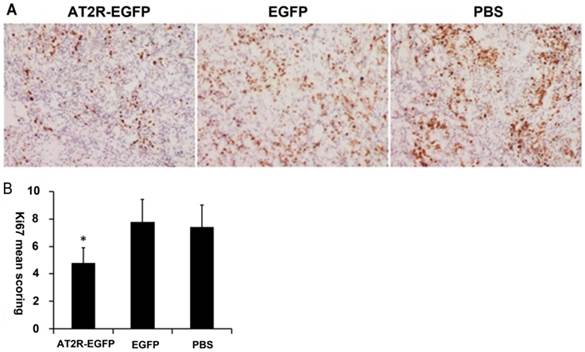
As a nuclear marker of cell proliferation, Ki-67 is overexpressed in many types of tumor cells. To examine the proliferation profiles of the xenograft tumor cells, Ki-67 levels were analyzed by immunohistochemical staining. The representative micrographs in the EGFP and PBS groups demonstrate that many cells are Ki-67 positive, appearing as brown particles (Figure 3A), and quantification revealed that there was no difference in the number of Ki-67 positive cells between the two groups (P>0.05). However, Figures 3A and 3B demonstrate that in the AT2R group, the percentage of Ki-67 positive cells present was dramatically lower compared with the EGFP and PBS groups (P<0.05) .
AT2R over-expression inhibits Ki-67 expression in vivo. At the end of experiments, mice were sacrificed to obtain the tumors. (A) Proliferation of the tumor cells were assessed by Ki-67 immunohistochemistry staining. (B) The immunohistochemistry staining scoring from different groups. Data are presented as mean ± SD. (*P<0.05)
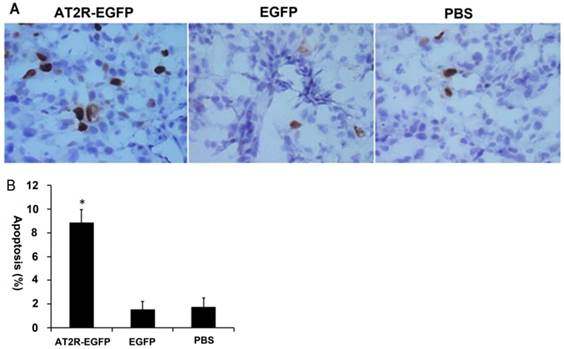
4. AT2R over-expression induces apoptosis in prostate cancer xenografts
In vitro cell culture experiments have demonstrated that AT2R overexpression induces apoptosis in many types of tumor cells, including DU145 cells [4,14]. In order to confirm whether it has the same effect in vivo, we performed TUNEL assay to examine the apoptosis in the tumor tissues. As shown in Fig.4, AT2R over-expression induced apoptosis in more than 8% of the DU145 cells, while in the EGFP and PBS groups, the percentage was lower than 2%. These results indicate that high expression of AT2R in vivo inhibits proliferation and induces apoptosis in tumor cells.
AT2R over-expression induces apoptosis in vivo. At the end of experiments, mice were sacrificed to obtain the tumors. (A) In situ TUNEL staining was performed to detect the apoptosis of tumor cells. (B) The percentage of apoptotic cells from different groups. Data are presented as mean ± SD. (*P<0.05).
5. AT2R over-expression up-regulates GADD45A but not VEGF expression within the tumor
Previously, we have demonstrated that TRAIL-R2 and GADD45A play important roles in AT2R-induced apoptosis in vitro. AT2R over-expression leads to up-regulation of TRAIL-R2 and GADD45A in cultured DU145 cells [15]. It was reported that AT2R was associated with increased VEGF secretion at low Ang-II concentrations [16] and our in vivo study also found that moderately increasing AT2R expression could increase the growth of HCC tumors [2]. To characterize these effects in this in vivo experiment, we analyzed the mRNA levels from the xenograft tumors. First, we confirmed AT2R mRNA overexpression in the tumors after virus injection. GADD45A mRNA levels were increased in AT2R-EGFP injected tumors while TRAIL-R2 and VEGF mRNA showed no significant difference among these three groups (Fig.5)
Discussion
The renin-angiotensin system (RAS) is usually associated with cardiovascular homeostasis, with the much focus on hypertension. It is now generally acknowledged that Ang II /AT1R signaling induces cell growth while activation of AT2R causes the opposing effect of growth inhibition and apoptosis via either Ang II-dependent or Ang II-independent regulation [17]. In our previous studies, AT2R over-expression by an adenoviral approach induced apoptosis in cancer cells [2,4,15]. In this study, we applied intratumoral injection to obtain high levels of AT2R expression in vivo (Fig.2 and Fig.5). Our results demonstrated that high AT2R expression can inhibit tumor growth in vivo (Fig.1).
It is now well established that binding of Ang II to AT1R contributes to malignant progression in many type of tumors [16,18-20]. AT1R signal blocking strategies are shown to be effective in inhibiting tumor growth [18,19]. In human hormone-refractory PCa, AT1R blockade could exert preventive effects through the inhibition of tumor angiogenesis [20]. However, the role of the AT2R in tumorigenesis remained controversial. Several studies report that AT2R, similar to AT1R, is over-expressed in gastric cancer [18,21] and renal clear-cell carcinoma [19] tumor cells. These studies suggest that, similar to AT1R, AT2R contribute to tumor malignant transformation and blocking strategies should be considered to inhibit cancer growth. On the contrary, there are results showing that AT2R expression exhibited an inverse correlation with tumor stage and liver metastasis in colorectal cancer [16]. In PCa, AT2R expression was decreased in cancers with a Gleason score 6 or higher [10]. The effect of AT2R on tumor progression remains to be elucidated, especially in in vivo studies.
A recent study notes that AT2R overexpression significantly attenuates the growth of fast-growing Lewis lung carcinoma (LLC) tumors, suggesting AT2R as a potential gene target for lung cancer therapy [9]. In addition, adenovirus-mediated AT2R overexpression induced apoptosis in hepatocellular carcinoma cells in vitro, however, the same vector actually increased hepatocellular tumor growth in an intrahepatic tumor model [2]. It has also been suggested that AT2R is associated with increased VEGF secretion at low Ang II concentrations, whereas high Ang II condition might evoke the antitumoral role of AT2R [16]. Thus, there is the possibility that moderately increased AT2R expression will up-regulate VEGF secretion so as to promote tumor growth while high AT2R expression may exert an anticancer effect in vivo.
In this study, AT2R over-expression decreased Ki-67 expression in DU145-derived xenograft tumor cells (Fig.3). Moreover, TUNEL assay revealed a proapoptotic effect of AT2R on tumor cells (Fig.4). Ki-67 is a nuclear non-histone protein that is increased in proliferating cells. Ki-67 expression is frequently seen in a variety of malignant tissues and is associated with worse survival. High Ki-67 expression was observed in 33% of patients with PCa [22]. It has also been shown that there is a strong relationship between Ki-67 and Gleason grade, with a continuous increase in the proliferative activity from low-grade to high-grade tumors [23]. Our results indicate that AT2R could inhibit Ki-67 expression and cellular proliferation and induce apoptosis of prostate carcinoma in vivo, which is consistent with our previous in vitro experiments.
GADD45A is a sensor protein produced in response to physiological and environmental stress, which can function to either promote or suppress tumor development. In PCa, GADD45A proteins and their signaling targets represent a novel class of molecules for therapeutic intervention [24]. Elevation of androgen receptor expression induced GADD45 expression and resulted in a reduction of PCa tumor growth [25]. Fucoxanthin, a marine carotenoid found in brown algae, was shown to induce GADD45A expression in LNCap PCa cells which couples with SAPK/JNK activation to result in an anticancer effect [26]. In our previous in vitro study, AT2R over-expression leads to up-regulation of GADD45A. Furthermore, down-regulation of GADD45A by siRNA reduced the AT2R-induced apoptosis. In this study, GADD45A was up-regulated by AT2R over-expression in vivo tumor models, consistent to our previous in vitro results (Fig.5). These results indicate that GADD45A plays important roles in the AT2R-induced apoptosis in PCa. The function of GADD45A is mediated via a complex interplay with other proteins, notably p38 and JNK stress response kinases [27]. In our previous in vitro studies, AT2R induces apoptosis via an extrinsic cell death signaling pathway that is dependent on activation of p38 mitogen-activated protein kinase and JNK [2,4]. Therefore, the p38 and JNK stress signal pathways should be modulated by AT2R over-expression in this in vivo experiment. Any approaches targeting these stress signals may have a synergistic inhibitory effect with AT2R on tumor growth.
There are reports that Ang II activation via AT2R receptor increases VEGF secretion. AT2R activation using the agonist CGP42112A increased tumor angiogenesis and VEGF expression in vivo and in vitro [1]. The AT2R antagonist PD123319 reduced tumor microvascular density. Inhibition or deficiency of AT2R was associated with impaired production of VEGF by tumor cells [28]. However, the relation and consequences of AT2R and VEGF secretion is complicated. Despite the pro-angiogenic effect, CGP42112A efficiently inhibited cancer cells proliferation and reduced tumor burden in mouse mode [1]. Recently, a more detailed knockdown study of the AT1R or AT2R together with Ang-II treatment showed that when AT2R were knocked down, the remaining Ang-II/AT1R activation was associated with VEGF secretion in tumor cells in a dose-dependent manner. In contrast, AT1R knockdown showed that the AT2R was associated with increased VEGF secretion at low Ang II concentrations, whereas high concentrations of Ang-II inhibited VEGF secretion [16]. In our studies, moderately increasing AT2R expression increased tumor growth in vivo, possibly through up-regulating of VEGF [2]. However, when AT2R were highly expressed in the tumors, no VEGF up-regulation was observed (Fig.5). Thus, the relation of AT2R and VEGF needs further investigation.
In summary, we demonstrated that high AT2R expression efficiently reduces human PCa tumor growth in a xenograft tumor model. Although the molecular mechanisms and the therapeutic value of AT2R-induced apoptosis in vivo remains to be assessed, more efficient AT2R expression methods or combination strategies with Ang II , AT1R, VEGF inhibition may be promising in future explorations.
Acknowledgements
We acknowledge Renhe Yan, Shanshan Chen, Yingying Mao, Zhibing Liang, Xuansheng Lin and Shiqi Luo for their technical assistance and help with real-time PCR. This work was supported by National Natural Science Foundation of China Grants 81272482 (J.L. ), 81072113 (H.L.) and 81401920 (H.D.), National 863 High Technique Development Project of China Grant 2012AA02A403 (H.L.), Guangdong Province Science and Technology Project 2013B010404026 (H.L.), as well as by awards 2011DFA33290 and 2011CBA01006 to W.G., and 20134433120020 to H.D. from the Chinese government.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ager EI, Chong WW, Wen SW, Christophi C. Targeting the angiotensin II type 2 receptor (AT2R) in colorectal liver metastases. Cancer Cell Int. 2010;10:19
2. Du H, Liang Z, Zhang Y. et al. Effects of angiotensin II type 2 receptor overexpression on the growth of hepatocellular carcinoma cells in vitro and in vivo. PLoS One. 2013;8:e83754
3. Pickel L, Matsuzuka T, Doi C. et al. Over-expression of angiotensin II type 2 receptor gene induces cell death in lung adenocarcinoma cells. Cancer Biol Ther. 2010;9:277-285
4. Li H, Qi Y, Li C. et al. Angiotensin type 2 receptor-mediated apoptosis of human prostate cancer cells. Mol Cancer Ther. 2009;8:3255-3265
5. Liu M, Jing D, Wang Y. et al. Overexpression of angiotensin II type 2 receptor promotes apoptosis and impairs insulin secretion in rat insulinoma cells. Mol Cell Biochem. 2015;400:233-244
6. Takagi T, Nakano Y, Takekoshi S. et al. Hemizygous mice for the angiotensin II type 2 receptor gene have attenuated susceptibility to azoxymethane-induced colon tumorigenesis. Carcinogenesis. 2002;23:1235-1241
7. Kanehira T, Tani T, Takagi T. et al. Angiotensin II type 2 receptor gene deficiency attenuates susceptibility to tobacco-specific nitrosamine-induced lung tumorigenesis: involvement of transforming growth factor-beta-dependent cell growth attenuation. Cancer Res. 2005;65:7660-7665
8. Doi C, Egashira N, Kawabata A. et al. Angiotensin II type 2 receptor signaling significantly attenuates growth of murine pancreatic carcinoma grafts in syngeneic mice. BMC Cancer. 2010;10:67
9. Kawabata A, Baoum A, Ohta N. et al. Intratracheal administration of a nanoparticle-based therapy with the angiotensin II type 2 receptor gene attenuates lung cancer growth. Cancer Res. 2012;72:2057-2067
10. Guimond MO, Battista MC, Nikjouitavabi F. et al. Expression and role of the angiotensin II AT2 receptor in human prostate tissue: in search of a new therapeutic option for prostate cancer. Prostate. 2013;73:1057-1068
11. Chow L, Rezmann L, Imamura K. et al. Functional angiotensin II type 2 receptors inhibit growth factor signaling in LNCaP and PC3 prostate cancer cell lines. Prostate. 2008;68:651-660
12. Li HW, Gao YX, Raizada MK, Sumners C. Intronic enhancement of angiotensin II type 2 receptor transgene expression in vitro and in vivo. Biochem Biophys Res Commun. 2005;336:29-35
13. Ma Y, Ma L, Guo Q, Zhang S. Expression of bone morphogenetic protein-2 and its receptors in epithelial ovarian cancer and their influence on the prognosis of ovarian cancer patients. J Exp Clin Cancer Res. 2010;29:85
14. Shenoy UV, Richards EM, Huang XC, Sumners C. Angiotensin II type 2 receptor-mediated apoptosis of cultured neurons from newborn rat brain. Endocrinology. 1999;140:500-509
15. Pei N, Jie F, Luo J. et al. Gene expression profiling associated with angiotensin II type 2 receptor-induced apoptosis in human prostate cancer cells. PLoS One. 2014;9:e92253
16. Zhou L, Luo Y, Sato S. et al. Role of Two Types of Angiotensin II Receptors in Colorectal Carcinoma Progression. Pathobiology. 2014;81:169-175
17. De Paepe B, Verstraeten VM, De Potter CR, Bullock GR. Increased angiotensin II type-2 receptor density in hyperplasia, DCIS and invasive carcinoma of the breast is paralleled with increased iNOS expression. Histochem Cell Biol. 2002;117:13-19
18. Huang MM, Guo AB, Sun JF. et al. Angiotensin II promotes the progression of human gastric cancer. Mol Med Rep. 2014;9:1056-1060
19. Dolley-Hitze T, Jouan F, Martin B. et al. Angiotensin-2 receptors (AT1-R and AT2-R), new prognostic factors for renal clear-cell carcinoma? Br J Cancer. 2010;103:1698-1705
20. Kosaka T, Miyajima A, Takayama E. et al. Angiotensin II type 1 receptor antagonist as an angiogenic inhibitor in prostate cancer. Prostate. 2007;67:41-49
21. Carl-McGrath S, Ebert MP, Lendeckel U, Rocken C. Expression of the local angiotensin II system in gastric cancer may facilitate lymphatic invasion and nodal spread. Cancer Biol Ther. 2007;6:1218-1226
22. Pollack A, Cowen D, Troncoso P. et al. Molecular markers of outcome after radiotherapy in patients with prostate carcinoma: Ki-67, bcl-2, bax, and bcl-x. Cancer. 2003;97:1630-1638
23. Bubendorf L, Tapia C, Gasser TC. et al. Ki67 labeling index in core needle biopsies independently predicts tumor-specific survival in prostate cancer. Hum Pathol. 1998;29:949-954
24. Liebermann DA, Hoffman B. Prostate cancer: JunD, Gadd45a and Gadd45g as therapeutic targets. Cell Cycle. 2011;10:3428
25. Tararova ND, Narizhneva N, Krivokrisenko V. et al. Prostate cancer cells tolerate a narrow range of androgen receptor expression and activity. Prostate. 2007;67:1801-1815
26. Satomi Y. Fucoxanthin induces GADD45A expression and G1 arrest with SAPK/JNK activation in LNCap human prostate cancer cells. Anticancer Res. 2012;32:807-813
27. Liebermann DA, Tront JS, Sha X. et al. Gadd45 stress sensors in malignancy and leukemia. Crit Rev Oncog. 2011;16:129-140
28. Clere N, Corre I, Faure S. et al. Deficiency or blockade of angiotensin II type 2 receptor delays tumorigenesis by inhibiting malignant cell proliferation and angiogenesis. Int J Cancer. 2010;127:2279-2291
Author contact
![]() Corresponding authors: Weiwang Gu, MS, Institute of Comparative Medicine and Center of Laboratory Animals, Southern Medical University, 1023 South Shatai Road, Guangzhou, Guangdong 510515, China. Phone: 86-20-61648043, E-mail: guww100com. Hongwei Li, Ph.D., School of Biotechnology, Southern Medical University, 1023 South Shatai Road, Guangzhou, Guangdong 510515, China. Phone: 86-20-61648555, Fax: 86-20-61648555, E-mail: hongwei1com
Corresponding authors: Weiwang Gu, MS, Institute of Comparative Medicine and Center of Laboratory Animals, Southern Medical University, 1023 South Shatai Road, Guangzhou, Guangdong 510515, China. Phone: 86-20-61648043, E-mail: guww100com. Hongwei Li, Ph.D., School of Biotechnology, Southern Medical University, 1023 South Shatai Road, Guangzhou, Guangdong 510515, China. Phone: 86-20-61648555, Fax: 86-20-61648555, E-mail: hongwei1com