Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(1):32-36. doi:10.7150/jca.13292 This issue Cite
Short Research Communication
Development of Soft Tissue Sarcomas in Ribosomal Proteins L5 and S24 Heterozygous Mice
Shideh Kazerounian1, 2 ![]() , Pedro D.S.C. Ciarlini3, Daniel Yuan1, Roxanne Ghazvinian1, Meritxell Alberich-Jorda4, Mugdha Joshi1, 2, Hong Zhang2, 5, Alan H. Beggs1, 2, Hanna T. Gazda1, 2, 6
, Pedro D.S.C. Ciarlini3, Daniel Yuan1, Roxanne Ghazvinian1, Meritxell Alberich-Jorda4, Mugdha Joshi1, 2, Hong Zhang2, 5, Alan H. Beggs1, 2, Hanna T. Gazda1, 2, 6 ![]()
1. Boston Children's Hospital, Division of Genetics and Genomics, The Manton Center for Orphan Disease Research, Boston, MA, USA
2. Harvard Medical School, Boston, MA, USA
3. University Hospitals Case Medical Center, Cleveland, OH, USA
4. Institute of Molecular Genetics of the ASCR, Prague, Czech Republic
5. Beth Israel Deaconess Medical Center, Hematology/Oncology Division, Boston, MA, USA
6. Broad Institute, Cambridge, MA, USA
Received 2015-7-20; Accepted 2015-10-18; Published 2016-1-1
Abstract
Diamond-Blackfan anemia (DBA) is an inherited bone marrow failure syndrome associated with ribosomal protein (RP) gene mutations. Recent studies have also demonstrated an increased risk of cancer predisposition among DBA patients. In this study, we report the formation of soft tissue sarcoma in the Rpl5 and Rps24 heterozygous mice. Our observation suggests that even though one wild-type allele of the Rpl5 or Rps24 gene prevents anemia in these mice, it still predisposes them to cancer development.
Keywords: Ribosomal proteins RPL5 and RPS24, Diamond-Blackfan anemia, Soft tissue sarcoma, Rpl5 and Rps24 heterozygous mice
Introduction
Diamond-Blackfan anemia (DBA) is a hereditary red blood cell aplasia that usually presents within the first year of life. Heterozygous point mutations and large deletions in 16 ribosomal protein (RP) genes, RPS19, RPS24, RPS17, RPL5, RPL11, RPL35A, RPS7, RPS10, RPS26, RPL26, RPL15, RPS28, RPS29, RPL31, RPS27, and RPL27 have been considered the underlying cause of disease in about 65% of patients [1-5]. DBA is also associated with physical abnormalities with varying severity such as craniofacial, upper limb, heart, and urinary-system defects in about 30-50% of patients [3, 6]. However, the severity of these abnormalities varies among patients [3]. A potential link between ribosomal protein deficiency and risk of tumor formation has also been demonstrated in human and zebrafish [7-10]. Vlachos et al. reported that a percentage of patients registered in the Diamond-Blackfan Anemia Registry of North America developed a variety of cancer including acute myeloid leukemia, colon carcinoma, osteogenic and soft tissue sarcomas at the median age of 41 [11]. The risk of cancer in these patients was 5.4 fold higher than that of the general population [11]. Current therapeutic approaches for DBA have been focused on increasing the level of red blood cells through bone marrow transplantation, glucocorticoids, and red blood cell transfusion in steroids-resistant patients [6, 12]. L-leucine has also been considered as an alternative therapy [6, 12].
Over the past decade, many research teams have established both in vitro and in vivo models of DBA to better understand the pathological and molecular mechanisms of ribosomal protein deficiency [3, 13, 14]. To date, very few approaches have been taken to address the mechanism of increased cancer predisposition associated with this disease. Recently, we reported the detection of Rps24 and RpL5 mutations in patients with DBA [15, 16], and have generated mouse models to further address the effect of these mutations in developing anemia and cancer. In this study, we characterized the Rpl5+/- and Rps24+/- mice models for DBA. Even though these mice did not exhibit anemia, two Rpl5+/- and one Rps24+/- mice developed soft tissue sarcoma. By taking advantage of our murine models, we hope to gain insight into the molecular mechanism of ribosomal protein deficiency and cancer development.
Results and Discussion
Generation of Rps24 and Rpl5 Heterozygous mice
Generation of the C57BL/6 Rpl5+/- and Rps24+/- mice was carried out by InGenious Targeting Laboratory (iTL; Ronkonkoma, NY, USA), and all animal studies were approved by Boston Children's Hospital's Institutional Animal Care and Use Committee. A pGK-gb2 loxP/FRT-flanked Neomycin cassette was inserted in embryonic stem cells to replace 383 bp of the Rps24 gene, including exons 2-3, or 8.11 kb of the Rpl5 gene, including exons 1-8. These cells were injected into C57BL/6 blastocysts, and the chimeric animals were mated to generate heterozygous mice. All the Rps24-/- and Rpl5-/- mice died by E11-12 despite there being a normal Mendelian distribution of heterozygous, homozygous, and wild-type blastocysts. Heterozygous mice were born at the expected frequency of about two-thirds (given the embryonic lethality of homozygous KO mice) and appeared clinically normal. In particular, heterozygotes of both genotypes did not develop hematological phenotypes that have been detected in patients with DBA, such as anemia. No changes were detected in the complete blood cell count (CBC) (Table S1) as well as the in vitro colony forming unit-erythroid (CFU-E), burst forming unit-erythroid (BFU-E) and colony forming unit-granulocyte /macrophage (CFU-GM) assays (Figure S1). Real-time PCR and immunoblot analysis also showed similar expression levels of Rpl5 and Rps24 mRNA, and RPL5 and RPS24 proteins in both heterozygous and wild-type mice (Table S2 and Figure S2). These observations are similar to previously reported findings for Rps19+/- mice [17] suggesting that one wild-type copy of these ribosomal proteins is sufficient to prevent the development of anemia.
Detection of Tumors in Rps24 and Rpl5 Heterozygous Mice
We also investigated the risk of cancer development in aging Rpl5+/- and Rps24+/- male and female mice by monitoring them for up to 36 months after birth (Table S3). Out of 21 Rpl5+/- mice monitored between 15 to 36 months of age, two male mice each developed a 0.5 cm tumor at 22 months of age (Figure S3). Similarly, out of 23 Rps24+/- mice (between15 and 30 months of age), one female mouse developed a 2 cm tumor spanning from upper left ear to the head/neck region at 17 months of age (Figure S3). However, none of the 31 control wild-type mice, ranging from 18 to 30 months of age, developed any type of tumor. We preformed statistical analysis, and the development of sarcoma was not statistically significant. The low number of cancer occurrence in mice matches the low incident of cancer in patients with DBA. One possible explanation for late occurrence of cancer development in mice (around 2 years) compared to patients with DBA (median age of 41) is that mice compensate for the loss of one Rpl5 or Rps24 allele possibly by either producing more Rpl5 and Rps24 mRNA from a single allele or increasing the stability of Rpl5 and Rps24 mRNA. However, over time, having only one allele may activate a new set of signaling pathways that may reduce the effect of the compensatory pathway and promote the development of late onset cancer. In contrast, ribosomal protein gene mutations in zebrafish have resulted in developmental defects of varying degrees similar to the symptoms detected in patients with DBA [18-20]. Similar to mice, some of these mutations have predisposed fish to cancer development by 2 years of age, which is considered late in the life span of zebrafish [9]. Together, these observations further support the possibility that prolonged ribosomal protein deficiency accumulation can increase the risk of cancer, as has been observed in patients [11].
Histological and Immunohistochemical Analysis of Tumors Isolated from Rps24 and Rpl5 Heterozygous Mice
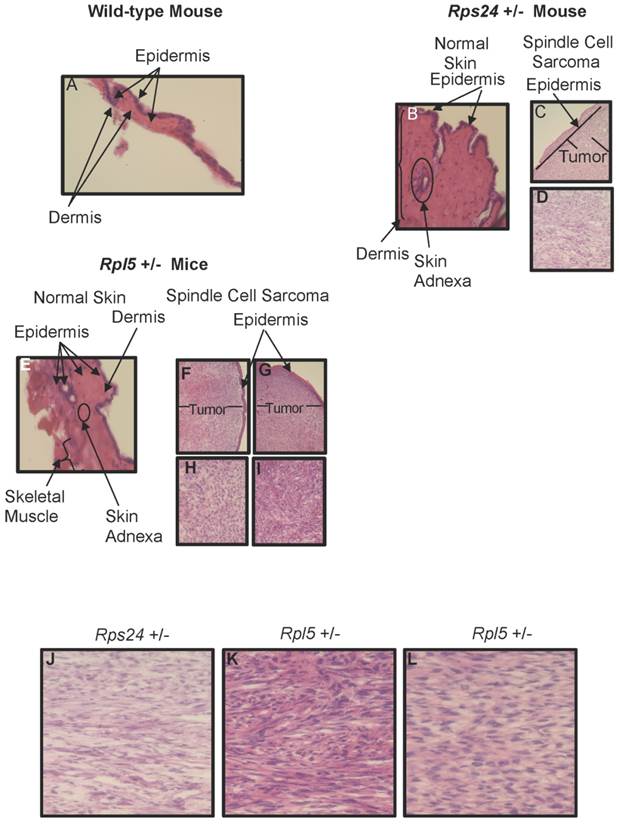
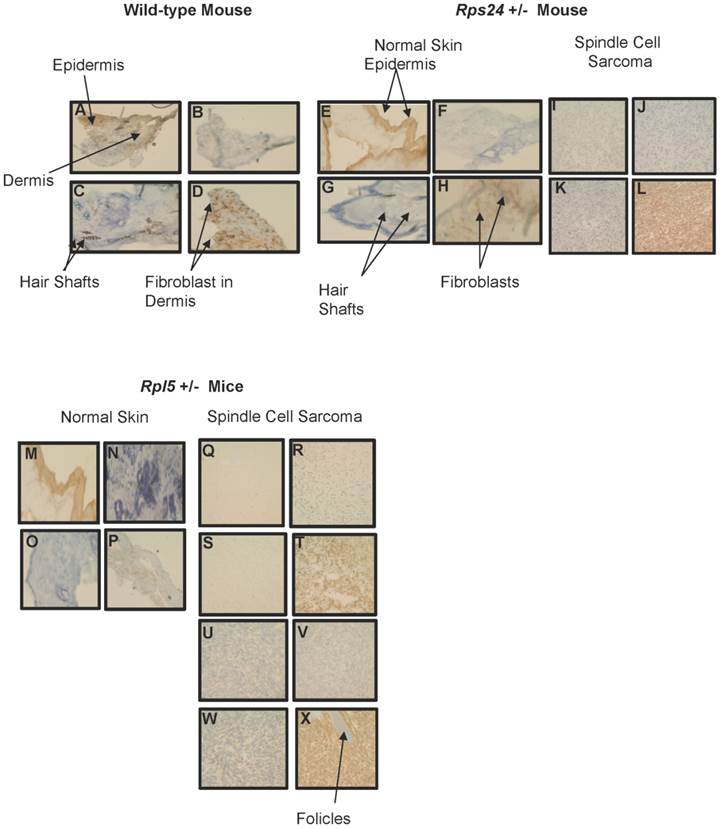
To determine the nature of tumors, we performed histological and immunohistochemical studies on tumor and normal skin tissues from Rpl5+/- and Rps24+/- mice as well as normal skin tissue of wild-type mice (Figure 1). Histological sections from all tumors showed that the tumors involved the dermis, were densely cellular, and composed of predominantly atypical spindle cells arranged in intersecting fascicles with brisk mitotic activity (Figures 1C-1D and 1F- 1I). The overlying epidermis showed no evidence of dysplasia and was not associated with the tumors (Figure 1). Immunohistochemical studies of tumor cells showed strong cytoplasmic reactivity for vimentin, a mesenchymal marker commonly expressed in sarcomas, and negative staining for pan-keratin, LCA, and S100, which excludes diagnosis of carcinoma, lymphoma, and melanoma, respectively (Figure 2). These overall findings were consistent with the characteristics of a high-grade spindle cell soft tissue sarcoma. In all the studies, wild-type, Rpl5+/-, and Rps24+/- normal skin tissues were histologically unremarkable (Figure 1). Due to the low incident of sarcoma formation in our mouse model, we performed PubMed and two public databases searches and to our knowledge, there are no records of spontaneous sarcoma formation specifically on the C57BL/6 wild-type mice [21, 22]. Also, we did not find any records indicating a correlation between the age of the mouse and the formation of sarcoma in C57BL/6 strain [21, 22].
Histology of Wild-type, Rps24+/-, and Rpl5+/- Normal Skin and Spindle Cell Sarcoma Tissues. Hematoxylin and Eosin staining showed normal epidermis of wild-type (A), Rps24+/- (B), and Rpl5+/- (E). However, there was a uniform localization of spindle tumor cells beneath the epidermis from Rps24+/- mouse (C and D) and Rpl5+/- mice (F, H, G, and I) with Rps24+/- tumor cells (Figure S3 and J) and tumor cells from Rpl5+/- with smaller tumor (Figure S3 and K) having very similar morphological appearances. In contrast, tumor cells from the Rpl5+/- mouse with a larger tumor (Figure S3 and L) were more rounded and had clear vacuoles, lesser degree of fascicular architecture and nuclear pleomorphism. All images are at 40X magnification.
Immunohistochemical Comparison of Wild-type, Rps24+/-, and Rpl5+/- Normal Skin with Tumor Tissues. Pan-keratin staining was detected throughout the epidermis with no detectable staining in the dermis of wild-type (A), Rps24+/- (E), and Rpl5+/- (M) skin sections, and was also negative in all the tumor tissues (I, Q, and U). Negative staining for both LCA (B, F, J, N, R, V) and S100 (C, G, K, O, S, W) was observed in the dermis of all tissue sections. Vimentin staining throughout the dermis in wild-type (D) and Rps24+/- (H) normal skin tissues corresponded to fibroblasts. In contrast, all the tumor tissues stained very strongly for vimentin (L, T, and X). All images are taken at 40X magnification.
The proposed mechanisms for sarcoma development are either a mutation in the p53 gene, which is considered to be a low incidence in sarcoma, or an overexpression of one of the p53 inhibitors such as MDM2 [23-26]. Recent in vitro and in vivo models of ribosomal protein deficiencies have also demonstrated the role of ribosomal proteins in regulating p53 stabilization and activity [10, 27, 28]. According to these studies, MDM2 interaction with ribosomal proteins dissociates it from p53, resulting in an increase in p53 expression and activation. To investigate if the presence of a mutation in p53 gene or a change in p53 expression level was the potential mechanism for sarcoma formation in our mouse model, we performed DNA sequencing of the Tp53 gene in DNA isolated from tumors and normal skin (control). However, no mutations were detected. This could be due to the low number of tumors studied in this experiment or a low incidence of p53 mutations in sarcomas. Moreover, there was a similar fold change in the expression level of p53 protein in Rpl5+/- tumor compared with Rpl5+/- normal skin and wild-type skin tissues (Figure S4). In contrast, the level of p53 protein was lower in Rps24+/- tumors and normal skin compared with Rpl5+/- tumor and normal skin and wild-type skin tissues (Figure S4). As it has been reported previously, ribosomal proteins use different signaling pathways to regulate p53 expression and activity, which may account for the differences detected in the p53 expression between the Rps24+/- and Rpl5+/- tissues [19, 27, 28]. Another possibility is that these ribosomal proteins exert different functions depending on cell types and tissues. Interestingly, in their recent article, Wang et al. also reported that knock-down of RPS24 in human colon cancer cells in vitro significantly decreased cell proliferation and migration and induced cell cycle arrest, which suggested the possible role of RPS24 in cell growth possibly through regulating the cell cycle [29]. Therefore, further experiments are required to investigate the effect of RPS24 and RPL5 proteins on p53 expression in our mouse models. In conclusion, even though Rpl5+/- and Rps24+/- mice did not have anemia, they became more susceptible to cancer development when compared with wild-type mice.
Supplementary Material
Supplementary methods, supplementary tables and figures.
Acknowledgements
These studies were funded in part by generous support from the Mauch Family, a Manton Center for Orphan Disease Research Junior Investigator Award, and by NIH R01 HL107558 and NIH K02 HL111156 to HTG. MAJ was supported by MSMT Navrat grant LK21307.
Authors' Contribution
SK and PD performed the experiments, analyzed the data, and wrote the manuscript. DY, RG, MJ, MAJ, and HZ performed the experiments and edited the manuscript. AHB and HTG edited the manuscript and advised with experiments.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gripp KW, Curry C, Olney AH, Sandoval C, Fisher J, Chong JX. et al. Diamond-Blackfan anemia with mandibulofacial dystostosis is heterogeneous, including the novel DBA genes TSR2 and RPS28. Am J Med Genet A. 2014;164A:2240-9
2. Horos R, Ijspeert H, Pospisilova D, Sendtner R, Andrieu-Soler C, Taskesen E. et al. Ribosomal deficiencies in Diamond-Blackfan anemia impair translation of transcripts essential for differentiation of murine and human erythroblasts. Blood. 2012;119:262-72
3. Lipton JM, Ellis SR. Diamond-Blackfan anemia: diagnosis, treatment, and molecular pathogenesis. Hematol Oncol Clin North Am. 2009;23:261-82
4. Mirabello L, Macari ER, Jessop L, Ellis SR, Myers T, Giri N. et al. Whole-exome sequencing and functional studies identify RPS29 as a novel gene mutated in multicase Diamond-Blackfan anemia families. Blood. 2014;124:24-32
5. Wang R, Yoshida K, Toki T, Sawada T, Uechi T, Okuno Y. et al. Loss of function mutations in RPL27 and RPS27 identified by whole-exome sequencing in Diamond-Blackfan anaemia. Br J Haematol. 2015;168:854-64
6. Narla A, Vlachos A, Nathan DG. Diamond Blackfan anemia treatment: past, present, and future. Semin Hematol. 2011;48:117-23
7. Gazda HT, Kho AT, Sanoudou D, Zaucha JM, Kohane IS, Sieff CA. et al. Defective ribosomal protein gene expression alters transcription, translation, apoptosis, and oncogenic pathways in Diamond-Blackfan anemia. Stem Cells. 2006;24:2034-44
8. Lipton JM, Federman N, Khabbaze Y, Schwartz CL, Hilliard LM, Clark JI. et al. Osteogenic sarcoma associated with Diamond-Blackfan anemia: a report from the Diamond-Blackfan Anemia Registry. J Pediatr Hematol Oncol. 2001;23:39-44
9. Amsterdam A, Sadler KC, Lai K, Farrington S, Bronson RT, Lees JA. et al. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2004;2:E139
10. MacInnes AW, Amsterdam A, Whittaker CA, Hopkins N, Lees JA. Loss of p53 synthesis in zebrafish tumors with ribosomal protein gene mutations. Proc Natl Acad Sci U S A. 2008;105:10408-13
11. Vlachos A, Rosenberg PS, Atsidaftos E, Alter BP, Lipton JM. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood. 2012;119:3815-9
12. Horos R, von Lindern M. Molecular mechanisms of pathology and treatment in Diamond Blackfan Anaemia. Br J Haematol. 2012;159:514-27
13. McGowan KA, Mason PJ. Animal models of Diamond Blackfan anemia. Semin Hematol. 2011;48:106-16
14. Taylor AM, Zon LI. Modeling Diamond Blackfan anemia in the zebrafish. Semin Hematol. 2011;48:81-8
15. Gazda HT, Grabowska A, Merida-Long LB, Latawiec E, Schneider HE, Lipton JM. et al. Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2006;79:1110-8
16. Gazda HT, Sheen MR, Vlachos A, Choesmel V, O'Donohue MF, Schneider H. et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am J Hum Genet. 2008;83:769-80
17. Matsson H, Davey EJ, Draptchinskaia N, Hamaguchi I, Ooka A, Leveen P. et al. Targeted disruption of the ribosomal protein S19 gene is lethal prior to implantation. Mol Cell Biol. 2004;24:4032-7
18. Uechi T, Nakajima Y, Chakraborty A, Torihara H, Higa S, Kenmochi N. Deficiency of ribosomal protein S19 during early embryogenesis leads to reduction of erythrocytes in a zebrafish model of Diamond-Blackfan anemia. Hum Mol Genet. 2008;17:3204-11
19. Uechi T, Nakajima Y, Nakao A, Torihara H, Chakraborty A, Inoue K. et al. Ribosomal protein gene knockdown causes developmental defects in zebrafish. PLoS One. 2006;1:e37
20. Zhang Y, Ear J, Yang Z, Morimoto K, Zhang B, Lin S. Defects of protein production in erythroid cells revealed in a zebrafish Diamond-Blackfan anemia model for mutation in RPS19. Cell Death Dis. 2014;5:e1352
21. The Mouse Phenome Database. http://phenome.jax.org
22. The Mouse Tumor Biology. http://www.nih.gov/science/models/mouse/resources/mtbdb.html
23. Hupp TR, Hayward RL, Vojtesek B. Strategies for p53 reactivation in human sarcoma. Cancer Cell. 2012;22:283-5
24. Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC. et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591-602
25. Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F. et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004;36:55-62
26. Post SM. Mouse models of sarcomas: critical tools in our understanding of the pathobiology. Clinical sarcoma research. 2012;2:20
27. McGowan KA, Pang WW, Bhardwaj R, Perez MG, Pluvinage JV, Glader BE. et al. Reduced ribosomal protein gene dosage and p53 activation in low-risk myelodysplastic syndrome. Blood. 2011;118:3622-33
28. Lindstrom MS, Nister M. Silencing of ribosomal protein S9 elicits a multitude of cellular responses inhibiting the growth of cancer cells subsequent to p53 activation. PLoS One. 2010;5:e9578
29. Wang Y, Sui J, Li X, Cao F, He J, Yang B. et al. RPS24 knockdown inhibits colorectal cancer cell migration and proliferation in vitro. Gene. 2015
Author contact
![]() Corresponding author: Shideh Kazerounian, Ph.D., Boston Children's Hospital, Harvard Medical School, Genetics/Genomics CLSB 15030.20, 3 Blackfan Circle, Boston, MA 02115. shideh.kazerounianharvard.edu; Phone: 617-355-7748; Fax: 617-730-0253 or Hanna T. Gazda, M.D., Ph.D., Boston Children's Hospital, Harvard Medical School, Genetics/Genomics CLSB 15023, 3 Blackfan Circle, Boston, MA 02115. hanna.gazdaharvard.edu; Phone: 617-919-4587; Fax: 617-730-0253
Corresponding author: Shideh Kazerounian, Ph.D., Boston Children's Hospital, Harvard Medical School, Genetics/Genomics CLSB 15030.20, 3 Blackfan Circle, Boston, MA 02115. shideh.kazerounianharvard.edu; Phone: 617-355-7748; Fax: 617-730-0253 or Hanna T. Gazda, M.D., Ph.D., Boston Children's Hospital, Harvard Medical School, Genetics/Genomics CLSB 15023, 3 Blackfan Circle, Boston, MA 02115. hanna.gazdaharvard.edu; Phone: 617-919-4587; Fax: 617-730-0253