Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(10):984-989. doi:10.7150/jca.11371 This issue Cite
Research Paper
Mutation Analysis of Nine Chordoma Specimens by Targeted Next-Generation Cancer Panel Sequencing
Carina Fischer1#, Susanne Scheipl3,5#, Agnes Zopf1, Norbert Niklas1, Alexander Deutsch4, Mette Jorgensen3, Birgit Lohberger5, Elke Verena Froehlich5, Andreas Leithner5, Christian Gabriel1, Bernadette Liegl-Atzwanger6, Beate Rinner2 ![]()
1. Red Cross Transfusion Service for Upper Austria, Krankenhausstraße 7, 4020 Linz, Austria.
2. Center for Medical Research, Cell Culture Facility, Medical University of Graz, Stiftingtalstrasse 24, 8010 Graz, Austria.
3. University College London Cancer Institute, 72 Huntley Street, London, WC1 6BT, UK.
4. Division of Hematology, Department of Internal Medicine, Medical University of Graz, Auenbruggerplatz 38D, 8036 Graz, Austria.
5. Department of Orthopaedic Surgery, Medical University of Graz, Auenbruggerplatz 5, 8036 Graz, Austria.
6. Institute of Pathology, Medical University of Graz, Auenbruggerplatz 25, 8036 Graz, Austria
# C.F. and S.S. contributed equally to this paper
Received 2014-12-17; Accepted 2015-7-19; Published 2015-8-20
Abstract
Background: Chordoma is a rare primary malignant bone tumour. Treatment options are mainly restricted to surgical excision, since chordomas are largely resistant to conventional ionising radiation and chemotherapy. Thus, there is a strong need to gain more thorough insights into the molecular biology and genetics of chordoma to allow for the development of new therapeutic options. We performed an ultra-deep sequencing analysis to find novel mutations in cancer associated genes in chordomas to date unseen with Sanger sequencing.
Material and Methods: Nine chordomas (skull base (n=3), mobile spine (n=4), and sacrum/coccyx (n=2) were screened for mutations in 48 cancer genes using the Hot Spot Cancer Panel (Illumina). All putative mutations were compared against multiple databases (e.g. NCBI, COSMIC, PolyPhen, EGB, SIFT) and published Copy Number Variation (CNV) data for chordoma.
Results: Our results showed mutations with a frequency above 5% in tumorsuppressor- and onco-genes, revealing new possible driver genes for chordomas. We detected three different variants accounting for 11 point mutations in three cancer associated genes (KIT, KDR and TP53). None of the detected mutations was found in all samples investigated. However, all genes affected interact or are connected in pathway analysis. There were no correlations to already reported CNVs in the samples analysed.
Conclusions: We identified mutations in the associated genes KIT, KDR, and TP53. These mutations have been described previously and have been predicted to be tolerated. Further results on a larger series are warranted. The driver mechanisms of chordoma still have to be identified.
Keywords: chordoma, next-generation sequencing, copy number, somatic mutations, cancer panel
Introduction
Chordoma is a rare primary malignant bone tumour that occurs with a reported incidence of 0.08 per 100,000 (1, 2). This neoplasm mainly arises in the axial skeleton distributed from the base of skull to the coccyx (1-3). Chordomas are thought to develop from notochordal remnants in the axial skeleton (3-5) and usually occur sporadically, although in rare cases they can be familial (6, 7). Even though chordomas are histologically considered low-grade neoplasms (2, 8) the mean survival period after diagnosis is only 7 years (1, 9, 10). Treatment options are mainly restricted to surgical excision (8, 11, 12), as chordomas have proven to be largely resistant to conventional irradiation and chemotherapy (8, 12). Thus, there is a strong need to gain deeper insights into the molecular biology and genetics of chordoma to enable the development of new therapeutic options. Current knowledge about putative factors of this disease is sparse.
In order to screen for novel hotspot mutations in chordomas we performed targeted analysis of a set of known hotspot cancer-related genes by using massive parallel sequencing (MPS), also known as next generation sequencing (NGS), for the first time. The TruSeq Amplicon Cancer Panel® (Illumina, San Diego, CA, USA) allows detection of mutations in 212 hotspot amplicons of 48 cancer-associated genes. These mutations include single nucleotide variants (SNV), multiple nucleotide variants (MNV) and small insertions and deletions. Simultaneous multiplexed sequencing in a single reaction enables a cheap and reliable detection of mutations. In the present study we aim to apply the TruSeq Amplicon Cancer Panel to nine chordoma samples in order to screen for novel mutations in cancer-related genes and to identify potentially treatable targets.
Materials and Methods
Study population
The Caucasian study population consisted of nine chordoma specimens that were obtained from five male and four female patients. At the time of diagnosis the patients' age ranged from 25 to 75 years (mean 60.3). The tumours were located in the skull base, the mobile spine and the sacrum/coccyx in three (33.3%), four (44.4%) and two (22.2%) cases, respectively. The tumour volume, available in seven patients, ranged from 1.5 to 668.2 cm3 (mean 122.44 cm3). Seven tumours were of classic and two of chondroid differentiation. All patients had been treated surgically by an intralesional resection in seven cases (77.8%), while wide margins could only be obtained in two (22.2%) of them. The follow-up period ranged from 2 to 113 months (mean 50.4 months). At last follow-up, five patients (55.5%) had died of disease (DOD), three (33.3%) were alive with disease (AWD), in the last examination and another patient (11.1%) had died of another cause (DOC).
The study was approved by the Ethics Committee of the Medical University of Graz, Graz, Austria. The study protocol and the informed consent of patients were approved by the ethics committee from the Medical University Graz (vote # 18-192ex06/07; valid until 17.04.2015).
Immunohistochemistry
HE-sections were generated from formalin-fixed paraffin-embedded tissues. All cases were reviewed by a pathologist (B.LA) and the diagnosis of chordoma was confirmed prior to inclusion in the study. Immunohistochemical (IHC) stains were performed with established antibodies against S-100 (Dako, Glostrup, Denmark), CK (Dako), Vimentin (Linaris, Wertheim, Germany), EMA (Dako) and brachyury (Santa Cruz, Santa Cruz, CA, USA). Adequate positive and negative controls were included. The antibodies, clones, dilutions, pretreatment conditions and sources used are enlisted in Table S1.
TruSeq Amplicon Cancer Panel
We extracted genomic DNA from nine fresh frozen tissue samples using the QIAmp DNA Mini Kit (Qiagen, Hilden, Germany) and determined the DNA concentration via photometric measurement (Eppendorf, Hamburg, Germany). We then enriched, amplified, purified and normalized 212 amplicons of 48 cancer genes (Table S2) using the TruSeq Amplicon Cancer Panel (Illumina Inc., San Diego, CA, USA) according to the manufacturer's protocol. For target enrichment and amplification, 250 ng of each sample DNA were amplified with a Verity thermocylcer (Life Technologies, Foster City, CA, USA). Samples were marked with patient specific barcode sequences of 8 base-pairs (bp). Dual-indexing strategy was applied where each sample is identified by a combination of two different barcodes. We verified the success of amplification with a DNA 1000 kit (Qiagen, Hilden, Germany) using a Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA). Prior to library normalization we purified the amplicons using Ampure Beads XP (Beckman Coulter, Brea, CA, USA). Subsequently, the amplified samples were normalized according to the manufacturer's protocol. We then pooled 5 µl of normalized solution of each sample as well as 10 pM of PhiX control (Illumina) to create the final library. We finally performed paired-end sequencing using the MiSeq system and the MiSeq reagent kit v2 (both Illumina) reading 2x150 bp. All samples were sequenced in one sequencing run, which generated 1.52 Gbases resulting in an average coverage of 1.12 x 106 reads. The library included an external control PhiX to evaluate run quality, this resulted in an error rate of 0.275%.
Sanger sequencing
All non-synonymous sequence variants observed by Illumina sequencing were manually checked for conflicts and re-evaluated by using Sanger sequencing. Polymerase chain reaction (PCR) products were purified with an ExoSAP-IT kit (Affymetrix Inc., Santa Clara, CA, USA) according to the manufacturer's protocol. We created capillary electrophoresis sequencing reactions with the BigDye Terminator v1.1 Ready Reaction Cycle Sequencing kit (Life Technologies, Carlsbad, CA, USA). The sequencing reactions were purified using Sephadex plates (Sigma-Aldrich, Vienna, Austria) and run on an ABI 3130xl sequencer (Life Technologies). Data files were analysed using ABI SeqScape v2.5 software (Life Technologies).
Data analysis, variant detection and annotation for NGS
After sequencing data were subsequently demultiplexed by MiSeq Reporter v2.3.0.8 (Illumina). Reads were mapped against the human genome hg19 with Genomics Workbench 6.5.1 (CLCbio, Aarhus, Denmark) and a quality based variant detection was conducted. To exclude false variants, which may result from a deficient polymerase activity during the amplification or during the sequencing run, we considered a minimum of 5% variation frequency as a cut-off. All annotated variants were checked in the following databases: NCBI (National Center for Biotechnology Information; http://www.ncbi.nlm.nih.gov/), COSMIC (Catalogue of Somatic Mutations in Cancer; http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/), PolyPhen (Polymorphism Phenotyping; http://genetics.bwh.harvard.edu/pph2/), EGB (Ensembl Genomes Browser; http://www.ensembl.org/Homo_sapiens/Info/Index), and SIFT (Sorting Intolerant from Tolerant; http://sift.jcvi.org/). Two of these databases, namely PolyPhen and SIFT, allow for prediction whether an amino acid substitution affects the structure and function of a human protein and can thus be classified as benign or pathologic.
Correlation of hotspot mutations and copy number state
All nine chordoma samples had been tested for chromosomal abnormalities, or copy number variations (CNV) using Affymetrix 6.0 CNV/SNP arrays by Rinner et al. 2013 (data not shown) (13). Mutations found in sequencing were compared to these data in order to correlate gene alterations with chromosomal CNVs to check as to whether similar loci were altered.
Results
Immunohistochemistry
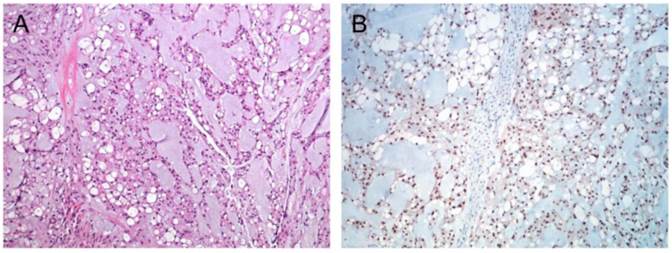
Immunhistochemistry confirmed chordoma diagnosis in all nine samples included in the present study. All samples showed distinct nuclear staining for brachyury (Figure 1) and were also positive for S-100 and Pan-CK (data not shown).
Immunohistochemical staining of chordoma. (A) HE staining of a classic chordoma (B) Chordoma with expression of brachyury.
Screening for hotspot mutations using the TruSeq Amplicon Cancer Panel reveals alterations in eight cancer genes
We detected 48 mutations in the nine chordoma samples. 37 mutations found in GNA11 (guanine nucleotide-binding protein subunit alpha-11; ENSP00000465935: p.His175Pro, ENSP00000465935: p.Val179Ala, ENSP00000465935: p.Pro181Gln), ERBB2 (v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2; ENSP00000463002: p.Phe154Cys) and HNF1A (hepatocyte nuclear factor 1-alpha; ENSP00000443964: p.Gly226Ala) were manually excluded because of misalignment.
Thus, three different point mutations within coding sequences of three oncogenes remained (Table 1). The oncogenes affected were KDR (kinase insert domain receptor; also known as VEGFR-2 n=2/11, 18%), KIT (v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog; n=1/11, 9%) and TP53 (tumor protein p53; n=8/11, 73%).
Predicted malignancy of mutations based on COSMIC, PolyPhen and SIFT databases (Note: n.DB; not in database)
| Prediction of Databases | ||||||||
|---|---|---|---|---|---|---|---|---|
| Transcript ID | Protein ID, Amino Acid Change | Gene | # samples | PolyPhen DB | SIFT | |||
| Biotype | Novel/Identifier | HumDiv | HumVar | |||||
| ENST00000263923:c.1416A>T | ENSP00000263923:p.Gln472His | KDR | Protein coding | COSM149673, rs1870377 | 2/9 | 0,003 | 0,011 | tolerated |
| ENST00000288135:c.1621A>C | ENSP00000288135:p.Met541Leu | KIT | Protein coding | rs3822214 | 1/9 | 0,009 | 0,005 | tolerated |
| ENST00000269305:c.215C>G | ENSP00000269305:p.Pro72Arg | TP53 | Protein coding | rs1042522 | 8/9 | 0,000 | 0,000 | tolerated |
The mutations were distributed across eight patients. One sample did not reveal any mutations at all (Table 1).
Screening for hotspot mutations in chordomas using the TruSeq Amplicon Cancer Panel reveals alterations in known cancer genes
All mutations detected involved protein coding genes (Table 1). Already described mutations included point mutation c.1621A>C in KIT in one patient (9%), as well as point mutations in KDR (c.1416A>T) in two patients (18%), and in TP53 (c.215C>G), which was seen in eight different patients (89%) (Table 2). Neither of these mutations is reported to promote tumour progression. Data analysis did not reveal any multiple nucleotide variants (two or more consecutive variants), deletions or insertions in any of our samples tested.
Mutations found in nine chordoma samples sequenced for 48 cancer-related genes.
| Cancer-related hot spot genes | |||||
|---|---|---|---|---|---|
| Sample ID | Gender | Tumor location | KDR | KIT | TP53 |
| 1 | male | soft tissue | p.Pro72Arg (100%) Exon 4 | ||
| 2 | female | bone | p.Pro72Arg (53%) Exon 4 | ||
| 3 | male | soft tissue | p.Gln472His (50.59%) Exon 11 | p.Pro72Arg (100%) Exon 4 | |
| 4 | female | clivus | |||
| 5 | female | clivus | p.Met541Leu (50.86%) Exon 10 | p.Pro72Arg (100%) Exon 4 | |
| 6 | male | sacrum | p.Gln472His (96.12%) Exon 11 | p.Pro72Arg (52.95%) Exon 4 | |
| 7 | female | bone | p.Pro72Arg (44.05%) Exon 4 | ||
| 8 | male | bone | p.Pro72Arg (99.46%) Exon 4 | ||
| 9 | female | bone | p.Pro72Arg (99.41%) Exon 4 | ||
| Total | 2/9 | 1/9 | 8/9 | ||
| % | 18 | 9 | 89 | ||
Hotspot mutated genes are not correlated with CNVs
NGS identified only one nucleotide alteration located in gain or loss regions as identified by Rinner et al 2013 via Affymetrix SNP arrays. Thus, we found no direct correlations of hotspot mutations detected in NGS to CNVs (Table 3).
Comparison of copy number state and nucleotide variant analysis
| Samples | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene (Mutation analysis) | Chr | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| KIT | 4 | loss | normal | normal | normal | normal | normal | loss | normal | loss |
| KDR | 4 | loss | normal | normal | normal | normal | normal | loss | normal | loss |
| TP53 | 14 | loss | normal | normal | normal | normal | normal | normal | normal | normal |
Note: Three states (normal, gain or loss) represent copy number variations in the target gene. A mutation detected by NGS analysis is marked in bold.
Discussion
Chordoma is a rare disease with a high incidence of recurrence and progression with shortened patient survival and impaired quality of life (14). The pathogenesis of chordoma is not fully elucidated and the identification of molecular genetics and mechanisms involved in cancer biology could lead to modulating therapeutic approaches.
Loss of heterozygosity (LOH) of the retinoblastoma tumour suppressor gene RB1 and TP53 mutations were reported to be associated with aggressive growth and significantly shorter recurrence-free survival in skull base chordomas (15, 17). However, somatic mutations have not been found in mutation „hotspots“ of genes known to be involved in cancer development: Le LP et al 2011 failed to show somatic mutations in thirteen „hotspots“ including APC, CTNNB1, BRAF, EGFR, FLT3, JAK2, KIT, KRAS, NOTCH1, NRAS, PIK3CA, PTEN and TP53 in 21 tumour samples.
Even though previous attempts have failed to identify common mutations in cancer-related genes in chordoma, more data is required to allow final conclusions. In order to verify previous results and to contribute additional data to the mutation screening in chordoma, we analysed nine chordoma patients to identify mutations (single variants, deletions, insertions) in hotspot cancer genes by Illumina Cancer Panel sequencing. By using deep sequencing strategy we were able to increase the sensitivity of mutation detection to a frequency as low as 5%, compared to a 20% threshold for Sanger-based sequencing.
Whereas Rinner et al 2013 (13) studied chromosomal abnormalities in ten chordoma samples, we have now generated a genomic molecular profile of nine of these chordomas using the remaining sample material. Since those analyses were carried out on the same study group this allows for a comparison of the data. In total, we were able to detect three different variants which accounted for 11 point mutations in three cancer-related genes (KDR, KIT and TP53). These point mutations affected eight patients, whereas one patient did not show any mutations at all.
Two patients revealed mutations in KDR. KDR is also known as vascular endothelial growth factor receptor 2 (VEGFR2), which is one of the two receptors of VEGF. We detected one KDR-mutation in two of our patients. The mutation had already been reported previously in chordoma and is supposed to be tolerated (13). According to the literature, VEGF has been shown to be expressed in 77.8% of 28 sacral chordomas (17, 18). Akhavan-Sigari et al 2013 (19) reported low expression of VEGFR-2 in 145 clivus chordomas, yet high expression of VEGFR-2 was correlated with a poor prognosis in their samples. In addition, a recent report has described a durable stabilization of chordomas of the skull base and the sacrum, respectively, after treatment with Erlotinib, an EGFR antagonist given in combination with the antiangiogenetic agent Bevacizumab (20). This supports a potential role of EGFR- and VEGFR-inhibition in the treatment of chordoma.
Another patient showed a mutation in the KIT gene. In previous studies imatinib and sunitinib, both tyrosine receptor kinase inhibitors known also to target KIT, have shown promising effects against chordoma in clinical trials (21, 22). Although this mutation wasn't recurrent in our samples it had been reported previously in chordoma as shown by the databases applied. In combination with the encouraging data from early phase clinical trials this supports KIT to be kept in mind as a potential target for chordoma therapy. It may furthermore be speculated that more KIT mutations might be found in larger sequencing studies. Interestingly, eight (89%) out of nine tested chordoma patients showed a mutation in TP53. TP53 has repeatedly been linked to chordoma biology e.g. by Hallor et al 2008 (23) who reported TP53 copy number losses in more than five out of 30 tumour samples which had been obtained from 26 patients as identified by CGH (microarray-based comparative genomic hybridization). Expression of p53 was recently also described as being more common in paediatric compared to adult chordomas (24). Furthermore, p53 expression has been reported to be associated with aggressive growth and significantly shorter recurrence-free survival in skull base chordomas (15, 16). However, the specific TP53 mutation found in our samples (p.Pro72Arg) is a well-known germline mutation and predicted to be benign by databases. Brunotto et al 2014 (25) reported that there was no significant association between different allelic variants of Arg72Pro (rs1042522) in head and neck cancer, thus supporting the benign nature of this mutation.
Our study could contribute another, not published study with two datasets (50 chordoma samples and 18 chordoma samples) of a chordoma sequencing project, therefore we thougt to identify mutations to better understand chordoma research (26). However there are very limited data regarding the mutational spectrum of chordoma but every mutation which is present in a chordoma tumor can be critical to identify and/or develop effective treatment strategies.
In conclusion, our deep sequencing mutation analysis by quantitative assessment of mutant alleles revealed three hotspot mutations in the cancer genes KDR, KIT and TP53. Although these mutations have been reported to be “tolerated” and thus not altering protein functions, all three genes involved have been linked to chordoma pathogenesis and thus might form potential treatment targets. Apart from a tolerated mutation of TP53, which was observed in a high percentage of our samples, the mutations in KDR and KIT only affected one and two patients, however, this mutations have well-known biological functions in cancer. This is well in line with other sequencing projects so far failing to eludicate recurrent mutations occurring in a majority of chordomas (27). Yet particularly this heterogeneity in potential genetic alterations in chordomas might indicate a screening for hot spot mutations in cancer associated genes prior to a personalised treatment approach in patients with limited therapeutic options.
Supplementary Material
Table S1 and Table S2.
Acknowledgements
The samples used for the research project were provided by the Biobank Graz.
Competing Interests
The authors have declared that no competing interests exist.
References
1. Smoll NR, Gautschi OP, Radovanovic I, Schaller K, Weber DC. Incidence and relative survival of chordomas: the standardized mortality ratio and the impact of chordomas on a population. Cancer. 2013;119(11):2029-2037
2. Walcott BP, Nahed BV, Mohyeldin A, Coumans JV, Kahle KT, Ferreira MJ. Chordoma: current concepts, management, and future directions. Lancet Oncol. 2012;13(2):e69-76
3. Nibu Y, Jose-Edwards DS, Di Gregorio A. From notochord formation to hereditary chordoma: the many roles of Brachyury. Biomed Res Int. 2013;2013:826435
4. Yamaguchi T, Watanabe-Ishiiwa H, Suzuki S, Igarashi Y, Ueda Y. Incipient chordoma: a report of two cases of early-stage chordoma arising from benign notochordal cell tumors. Mod Pathol. 2005;18(7):1005-1010
5. Chugh R, Tawbi H, Lucas DR, Biermann JS, Schuetze SM, Baker LH. Chordoma: the nonsarcoma primary bone tumor. Oncologist. 2007;12(11):1344-1350
6. Pillay N, Plagnol V, Tarpey PS, Lobo SB, Presneau N, Szuhai K, Halai D, Berisha F, Cannon SR, Mead S, Kasperaviciute D, Palmen J, Talmud PJ, Kindblom LG, Amary MF, Tirabosco R, Flanagan AM. A common single-nucleotide variant in T is strongly associated with chordoma. Nat Genet. 2012;44(11):1185-1187
7. Yang XR, Ng D, Alcorta DA, Liebsch NJ, Sheridan E, Li S, Goldstein AM, Parry DM, Kelley MJ. T (brachyury) gene duplication confers major susceptibility to familial chordoma. Nat Genet. 2009;41(11):1176-1178
8. Sciubba DM, Chi JH, Rhines LD, Gokaslan ZL. Chordoma of the spinal column. Neurosurg Clin N Am. 2008;19(1):5-15
9. McMaster ML, Goldstein AM, Bromley CM, Ishibe N, Parry DM. Chordoma: incidence and survival patterns in the United States, 1973-1995. Cancer Causes Control. 2001;12(1):1-11
10. Whelan J, McTiernan A, Cooper N, Wong YK, Francis M, Vernon S, Strauss SJ. Incidence and survival of malignant bone sarcomas in England 1979-2007. Int J Cancer. 2012;131(4):E508-17
11. Casali PG, Stacchiotti S, Sangalli C, Olmi P, Gronchi A. Chordoma. Curr Opin Oncol. 2007;19(4):367-370
12. Muro K, Das S, Raizer JJ. Chordomas of the craniospinal axis: multimodality surgical, radiation and medical management strategies. Expert Rev Neurother. 2007;7(10):1295-1312
13. Rinner B, Weinhaeusel A, Lohberger B, Froehlich EV, Pulverer W, Fischer C, Meditz K, Scheipl S, Trajanoski S, Guelly C, Leithner A, Liegl B. Chordoma characterization of significant changes of the DNA methylation pattern. PLoS One. 2013;8(3):e56609
14. Diaz RJ, Guduk M, Romagnuolo R, Smith CA, Northcott P, Shih D, Berisha F, Flanagan A, Munoz DG, Cusimano MD, Pamir MN, Rutka JT. High-resolution whole-genome analysis of skull base chordomas implicates FHIT loss in chordoma pathogenesis. Neoplasia. 2012;14(9):788-798
15. Pallini R, Maira G, Pierconti F, Falchetti ML, Alvino E, Cimino-Reale G, Fernandez E, D'Ambrosio E, Larocca LM. Chordoma of the skull base: predictors of tumor recurrence. J Neurosurg. 2003;98(4):812-822
16. Eisenberg MB, Woloschak M, Sen C, Wolfe D. Loss of heterozygosity in the retinoblastoma tumor suppressor gene in skull base chordomas and chondrosarcomas. Surg Neurol. 1997;47(2):156-60 discussion 160-1
17. Chen KW, Yang HL, Lu J, Wang GL, Ji YM, Wu GZ, Zhu LF, Liu JY, Chen XQ, Gu YP. Expression of vascular endothelial growth factor and matrix metalloproteinase-9 in sacral chordoma. J Neurooncol. 2011;101(3):357-363
18. Li X, Ji Z, Ma Y, Qiu X, Fan Q, Ma B. Expression of hypoxia-inducible factor-1alpha, vascular endothelial growth factor and matrix metalloproteinase-2 in sacral chordomas. Oncol Lett. 2012;3(6):1268-1274
19. Akhavan-Sigari R, Gaab MR, Rohde V, Brandis A, Tezval H, Abili M, von Eckardstein K, Ostertag H. Expression of vascular endothelial growth factor receptor 2 (VEGFR-2), inducible nitric oxide synthase (iNOS), and Ki-M1P in skull base chordoma: a series of 145 tumors. Neurosurg Rev. 2014;37(1):79-88
20. Asklund T, Sandstrom M, Shahidi S, Riklund K, Henriksson R. Durable stabilization of three chordoma cases by bevacizumab and erlotinib. Acta Oncol. 2014
21. Casali PG, Bertulli R, Fumagalli E, Coco P, Grosso F, Stacchiotti S. Some lessons learned from imatinib mesylate clinical development in gastrointestinal stromal tumors. J Chemother. 2004;16(Suppl 4):55-58
22. Stacchiotti S, Marrari A, Tamborini E, Palassini E, Virdis E, Messina A, Crippa F, Morosi C, Gronchi A, Pilotti S, Casali PG. Response to imatinib plus sirolimus in advanced chordoma. Ann Oncol. 2009;20(11):1886-1894
23. Hallor KH, Staaf J, Jonsson G, Heidenblad M, Vult von Steyern F, Bauer HC, Ijszenga M, Hogendoorn PC, Mandahl N, Szuhai K, Mertens F. Frequent deletion of the CDKN2A locus in chordoma: analysis of chromosomal imbalances using array comparative genomic hybridisation. Br J Cancer. 2008;98(2):434-442
24. Yadav R, Sharma MC, Malgulwar PB, Pathak P, Sigamani E, Suri V, Sarkar C, Kumar A, Singh M, Sharma BS, Garg A, Bakhshi S, Faruq M. Prognostic value of MIB-1, p53, epidermal growth factor receptor, and INI1 in childhood chordomas. Neuro Oncol. 2014;16(3):372-381
25. Brunotto M, Zarate AM, Bono A, Barra JL, Berra S. Risk genes in head and neck cancer: a systematic review and meta-analysis of last 5 years. Oral Oncol. 2014;50(3):178-188
26. EMBL-EBI. http://www.ebi.ac.uk/ega/studies/EGAS00001000188
27. Choy E, MacConaill LE, Cote GM, Le LP, Shen JK, Nielsen GP, Iafrate AJ, Garraway LA, Hornicek FJ, Duan Z. Genotyping cancer-associated genes in chordoma identifies mutations in oncogenes and areas of chromosomal loss involving CDKN2A, PTEN, and SMARCB1. PLoS One. 2014(7):e101283
Author contact
![]() Corresponding author: Beate Rinner, PhD, Center for Medical Research, Medical University of Graz, Stiftingtalstrasse 24, 8010 Graz, Austria; Phone: +43/316/385-73524; Fax: +43/316/385-73009; email: beate.rinnerat
Corresponding author: Beate Rinner, PhD, Center for Medical Research, Medical University of Graz, Stiftingtalstrasse 24, 8010 Graz, Austria; Phone: +43/316/385-73524; Fax: +43/316/385-73009; email: beate.rinnerat