Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(7):632-635. doi:10.7150/jca.11779 This issue Cite
Short Research Communication
Restriction-based Multiple-fragment Assembly Strategy to Avoid Random Mutation during Long cDNA Cloning
Shang Wang1,2#, Wen Chen1,2#, Kai Zhang1,2#, Peng Jiao1,2, Lihua Mo1,2, Xiaoxu Yang1,2, Xiang Hu1,2, Jian Zhang1,2, Chenxi Wei1,2 ![]() , Shuanglin Xiang1,2
, Shuanglin Xiang1,2 ![]()
1. Key Laboratory of Protein Chemistry and Developmental Biology of Education Ministry of China, College of Life Sciences, Hunan Normal University, Changsha 410081, China;
2. The Cooperative Innovation Center of Engineering and New Products for Developmental Biology of Hunan Province (20134486), Changsha 410081, China
# These authors contributed equally to this work.
Received 2015-2-3; Accepted 2015-4-15; Published 2015-5-25
Abstract
Long fragment cloning is a challenge for its difficulty in accurate amplifying and tendency to get unwanted mutation. Here we discuss Restriction-based Multiple-fragment Assembly Strategy's advantages and limitations. In this strategy, rather than PCR amplifying the entire coding sequence (CDS) at one time, we amplified and sequenced smaller fragments which are shorter than 1.5kb spanning the CDS. After that, the sequence-proved fragments were assembled by digestion-ligation cloning to the target vector. We test its universality in our script programmed in Python. Our data shows that, among the entire human and mouse CDS, at least 70% of long CDS cloning will benefit from this strategy.
Keywords: PCR, ligation, cloning strategy, CDS, endonuclease.
Introduction
Classical PCR-ligation strategy is the most familiar one to clone for its convenience and efficiency. In long CDS cloning, PCR and sequencing are the most challenging steps. DNA is not easily amplified from complementary DNA (cDNA) for many reasons, e.g. cDNA may not be in good quality, primers are easier to miss-prime during long extending time, GC-rich region will stop DNA polymerizing. On the other hand, fidelity is far from our satisfactory. In addition, long fragment sequencing will be troublesome for both time and money cost on intermediated sequencing and primer synthesis. That's why large gene, in many cases, have to be interrupted into several domains to imitate the whole gene function [1]. To solve the problem, artificial gene synthesis is a very direct way to save effort [2-4]. But not many scientists are willing to pay that much on a plasmid. Somebody will turn to Golden Gate ligation system. Because the cutting sequences of the two enzymes are not restricted, the overhanging 4 nucleotides could give at most 256 different patterns of cohesive ends. This dramatically increases the choices for our consideration to assemble small fragments into a long one [5-7]. However, if our target long CDS contains BbsI or BsaI restriction sites, golden-gate ligation system will not work well. Besides, this strategy requires elite skills on cohesive end selecting and designing, it will not be favored by many scientists even though it is widely used in Talen tandem assembly [8].
We found a strategy to clone large fragments of DNA by dividing them to short PCR fragment cloning, followed by restriction digestion and ligation cloning [9]. This strategy shows dominance in amplifying efficiency, it helps overcome PCR problem, and constructs very big clones without turning to complex reagents and technologies, like Red/ET system[10] or TAR system [11, 12]. This strategy shows potential in handling long CDS clones, including introducing mutation, deleting and inserting domains.
Here, we name it Restriction-based Multiple-fragment Assembly Strategy. A slight modification was added to make it more compatible to sequencing system and easier for us to handle random mutation. A series of experiments were done to judge its efficiency and universality. In this modified strategy, instead of PCR-amplifying the entire CDS, we amplify short fragments, which are no longer than 1,500bp, spanning the whole CDS. Since Sanger sequencing's read length is 800bp, 1,500bp is very friendly to bi-directional sequencing strategy. Fragments were ligated to KSII and followed by Sanger sequencing. Then, mutated clones were discarded. Sequence-proved fragments were released by restriction digestion and assembled into our target vectors in a ligation system. Since the digestion and ligation sub-cloning does not introduce random mutation, and restriction sites were found in the sequence to be cloned, ligation will not break open reading frame (ORF).
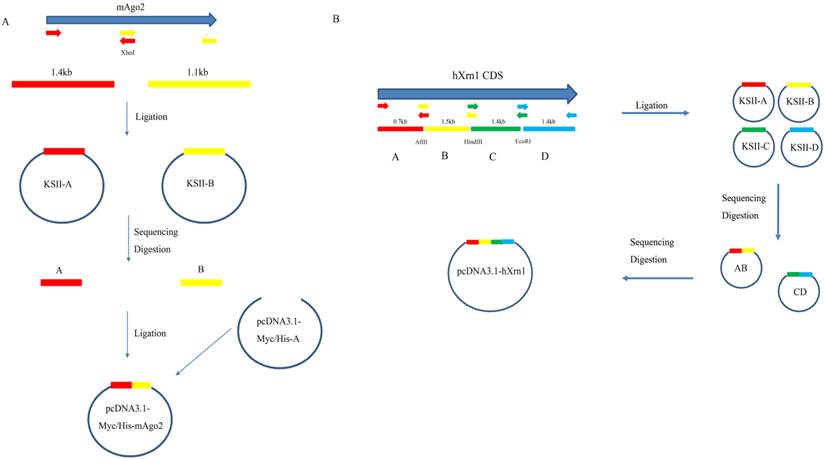
In this study, mouse Ago2 was constructed as examples to elaborate this strategy. mAgo2 (NM_153178.4) was submitted to NEBcutter V2.0 (http://nc2.neb.com/NEBcutter2/) [13] for restriction analysis. Results showed mAgo2 contains an XhoI restriction site which breaks mAgo2 CDS into 1.4kb and 1.1kb two fragments. Each interrupted fragments were PCR-amplified from N2A cDNA using primers mAgo2F1 ( 5'-ACG GAT CCG CCA CCA TGT ACT CGG GAG CCG GCC CCG TTC-3'), mAgo2R1 (5' -ACT TGC ATA CAC AGG AGT T-3'), mAgo2F2 ( 5'-AGC GCC AGT GTA CAG AAG TC-3') mAgo2R2 (5'-ACG AAT TCA GCA AAG TAC ATG GTG CGC AG-3') by KOD-FX (Cat: KFX-101). PCR was processed 28 cycles on Eppendorf thermos-cycler (Eppendorf AG 22331 Hamburg) with the denaturing temperature at 94℃ for 30seconds, annealing temperature at 58℃ for 30seconds, followed by extending temperature at 68℃ for 1min/kb. PCR product was purified (Guangzhou Dongsheng Biotech) and ligated (Fermentas # K1423) to pBlueScript-KSII that had been digested with EcoRV to give blunt end. Ligation product was transformed into Top 10 competent E. coli. and plated onto LB agar plate supplemented with X-gal, IPTG and Ampicillin. Blue White Screen was used to help check the insert [14]. The inserted clones were sent for sequencing using M13F/M13R sequencing primers. The sequence-proved clones were digested to release favored fragments. Purified fragments were ligated to pcDNA3.1-Myc/His A that had been digested by BamHI and EcoRI in one ligation system. Ligation systems were transformed. Clones were picked for enzyme digestion screen and followed by Sanger sequencing. So pcDNA3.1-Myc/His-mAgo2 was completed (Figure. 1A).
Detailed Strategy to clone mAgo2(A) and hXrn1(B). Fragments were first amplified and ligated to pBS-KSII vector for sequencing. The correct clones were then subcloned into the target vector. For too many fragments, like hXrn1, two or more rounds of assembly are recommended.
Full-length and fragments PCR of mAgo2 (A) and hXrn1 (B). Full length of PCR amplifying was either too low effecient (mAgo2) or totally a failure (hXrn1). While fragments amplification enhanced both accuracy and success rate.
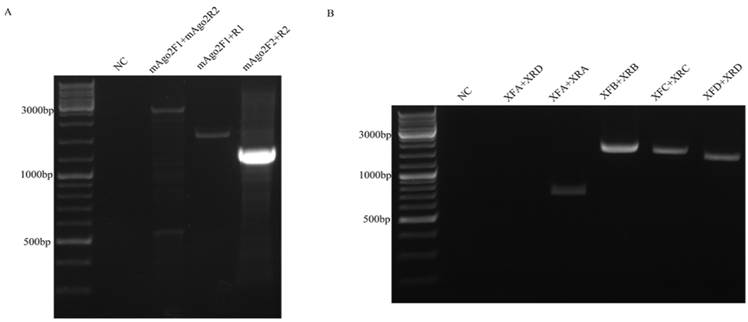
This strategy to clone long DNA fragment takes advantage of the dominance of short fragment PCR. mAgo2 fragments were easily amplified using mAgo2F1/R1 and mAgo2F2/R2. Also it could be successfully amplified directly using mAgo2F1/R2. But, full-length PCR product showed smear at 500bp and 1,000bp (Figure. 2 A). In order to apply this strategy to a more complex CDS, we tried it on hXrn1, whose CDS is 5.4 kb. After our analysis, hXrn1 CDS would be divided into 4 short fragments (A, B, C, D) with their lengths at 0.7kb, 1.5kb, 1.4kb, 1.4kb adjoined by endonucleases AflII, HindIII, EcoRI. We firstly assembled fragments A and B to get KSII-AB. At the same time we got KSII-CD. Again, we repeated the processes above and got pcDNA3.1-Myc/His-hXrn1 (Figure. 1B). hXrn1 full length PCR showed no band at all; while fragments PCR shows clear and bright bands (Figure. 2B). This means that short fragments are much more easily amplified and products are more accurate.
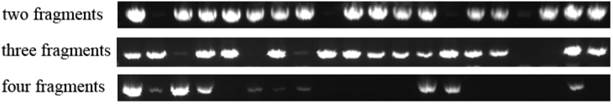
Next, hXrn1 4 fragments were used to measure one round assembling efficiency of 2 fragments (A/B), 3 fragments (A/B/C) and 4 fragments (A/B/C/D). Fragments were ligated to pre-digested pcDNA3.1 vector. Ligation products were transformed into top10 competent cells. After overnight growth, 20 clones of each plated were picked for colony PCR. In 2 fragments system, 16 positive clones were checked, in 3 fragments system, 15 positive clones were checked, in 4 fragments system, only 6 positive clones were checked (Figure 3). Results showed that the more fragments are tried in an assembly system; the lower success rate is checked. To overcome the efficiency problem, we recommend two or more round of assembly, as it has been processed on pcDNA3.1-hXrn1.
Colony PCR to measure ligation efficiency. The more fragments in a ligation system, the smaller chance will be in producing a complete construct. In four fragments ligation system, only 6 positive clones were checked.
In order to know how universal this strategy is, we searched the entire human and mouse CDS in CCDS database [15] to see how many of them will benefit from our strategy. Sequences were checked in our Python Script (supplementary material S1) if they were longer than 1,500bp, if 15 candidate restriction sites (ApaI, BamHI, BglII, EcoRI, HindIII, KpnI, NcoI, NdeI, NheI, NotI, SacI, SalI, SphI, XbaI, XhoI) could be found only once on a CDS, and then if the distance between adherent restriction sites are no longer than 1,500nt. Our data showed 12118 in 29064 human CDSs and 9479 in 23874 mouse CDSs were longer than 1,500 (supplementary material S2). These CDS possess nearly 40% of all CDS. Further, 8304 in 12118 in human and 6678 in 9479 mouse meet our requirements (supplementary material S3). Sequences meet our requirement are good candidates for Restriction-based Multiple-fragment Assembly Strategy. This data strongly suggests our strategy is potential to solve most long CDS cloning problem. Furthermore, we can increase the number by referring more restriction sites or turning to other ligation free system [16].
In this study, first, we discuss Multiple-fragment Assembly Strategy's dominance in PCR amplifying efficiency by amplifying short fragment rather than long one. Second, we clearly answered how this modified strategy solves random mutation problem. Third, we assess the assembly efficiency and give suggestions to solve the problem. Fourth, we programmed a script to test its universality in the entire CDS, and find a great number (~70%) of long CDS problem will benefit from it.
Supplementary Material
Supplementary Material S1-S3.
Acknowledgements
This work was supported by the 973 project of Ministry of Science and Technique of China (No. 2010CB529900), the Scientific Research Fund of Hunan Provincial Education Department (11A072), the Science & Technology Department of Hunan Province (2014FJ2006), and the Cooperative Innovation Center of Engineering and New Products for Developmental Biology of Hunan Province (20134486).
Competing Interests
The authors have declared that no competing interests exist.
References
1. Chang JH, Xiang S, Xiang K, Manley JL, Tong L. Structural and biochemical studies of the 5'-->3' exoribonuclease Xrn1. Nature structural & molecular biology. 2011;18:270-6
2. Boeckle S, Wagner E. Optimizing targeted gene delivery: chemical modification of viral vectors and synthesis of artificial virus vector systems. The AAPS journal. 2006;8:E731-42
3. Tsuchiya Y, Morioka K, Shirai J, Yoshida K, Inumaru S. Comparison of artificial synthesis methods of gene. Nucleic Acids Symp Ser (Oxf). 2006:275-6
4. Wu G, Zheng Y, Qureshi I, Zin HT, Beck T, Bulka B. et al. SGDB: a database of synthetic genes re-designed for optimizing protein over-expression. Nucleic acids research. 2007;35:D76-9
5. Engler C, Kandzia R, Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PloS one. 2008;3:e3647
6. Engler C, Gruetzner R, Kandzia R, Marillonnet S. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PloS one. 2009;4:e5553
7. Weber E, Gruetzner R, Werner S, Engler C, Marillonnet S. Assembly of designer TAL effectors by Golden Gate cloning. PloS one. 2011;6:e19722
8. Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, Zhang F. A transcription activator-like effector toolbox for genome engineering. Nature protocols. 2012;7:171-92
9. Stergiou C, Zisimopoulou P, Tzartos SJ. Expression of water-soluble, ligand-binding concatameric extracellular domains of the human neuronal nicotinic receptor alpha4 and beta2 subunits in the yeast Pichia pastoris: glycosylation is not required for ligand binding. The Journal of biological chemistry. 2011;286:8884-92
10. Wang J, Sarov M, Rientjes J, Fu J, Hollak H, Kranz H. et al. An improved recombineering approach by adding RecA to lambda Red recombination. Molecular biotechnology. 2006;32:43-53
11. Kouprina N, Larionov V. Selective isolation of mammalian genes by TAR cloning. Current protocols in human genetics. 2006 Chapter 5: Unit5 17
12. Kouprina N, Larionov V. TAR cloning: insights into gene function, long-range haplotypes and genome structure and evolution. Nature reviews Genetics. 2006;7:805-12
13. Vincze T, Posfai J, Roberts RJ. NEBcutter: A program to cleave DNA with restriction enzymes. Nucleic acids research. 2003;31:3688-91
14. Xu SY, Fomenkov A. Construction of pSC101 derivatives with Camr and Tetr for selection or LacZ' for blue/white screening. BioTechniques. 1994;17:57
15. Farrell CM, O'Leary NA, Harte RA, Loveland JE, Wilming LG, Wallin C. et al. Current status and new features of the Consensus Coding Sequence database. Nucleic acids research. 2014;42:D865-72
16. Li C, Wen A, Shen B, Lu J, Huang Y, Chang Y. FastCloning: a highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC biotechnology. 2011;11:92
Author contact
![]() Corresponding authors: Tel.: +86 731 88872095/88872916. Fax: 86-731-8887-2905; E-mail address: weicxedu.cn; xshlinedu.cn
Corresponding authors: Tel.: +86 731 88872095/88872916. Fax: 86-731-8887-2905; E-mail address: weicxedu.cn; xshlinedu.cn