Impact Factor ISSN: 1837-9664
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Issue 3; 2026
- Volume 17; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Special Issues
1. Introduction: overview of...
2. Cullins, DNA damage and repair
3. Cullins and cell cycle...
4. Cullins and p53 pathway
5. Cullins and WNT pathways
6. Cullins regulate the...
7. Cullins and apoptosis
8. Cullins and Hypoxia-inducible...
9. Cullins and other pathways...
10. Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(3):233-242. doi:10.7150/jca.11076 This issue Cite
Review
Cullin Family Proteins and Tumorigenesis: Genetic Association and Molecular Mechanisms
Zhi Chen1,*, Jie Sui2,*, Fan Zhang1, Caiguo Zhang3, ![]()
1. Orthopedics Department, Changhai Hospital Affiliated to Second Military Medical University, Shanghai, China, 200433
2. Orthopedics Department, 102 Hospital of People's Liberation Army, Changzhou, Jiangsu, China, 213003
3. Department of Biochemistry and Molecular Genetics, University of Colorado School of Medicine, Aurora, CO, USA, 80045
* These authors contribute equally to this work
Received 2014-11-16; Accepted 2014-12-8; Published 2015-1-16
Abstract
Cullin family proteins function as scaffolds to form numerous E3 ubiquitin ligases with RING proteins, adaptor proteins and substrate recognition receptors. These E3 ligases further recognize numerous substrates to participate in a variety of cellular processes, such as DNA damage and repair, cell death and cell cycle progression. Clinically, cullin-associated E3 ligases have been identified to involve numerous human diseases, especially with regard to multiple cancer types. Over the past few years, our understanding of cullin proteins and their functions in genome stability and tumorigenesis has expanded enormously. Herein, this review briefly provides current perspectives on cullin protein functions, and mainly summarizes and discusses molecular mechanisms of cullin proteins in tumorigenesis.
Keywords: Cullins, E3 ligase, Tumorigenesis, DNA damage, apoptosis, cell cycle
1. Introduction: overview of cullins and their functions
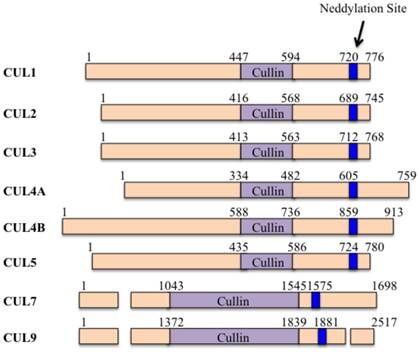
Cullins are evolutionarily conserved family proteins throughout bacteria, plants and mammals [1]. The human genome encodes six cullin proteins characterized by a cullin homology (CH) domain in the carboxyl (C)-terminal, including CUL1, 2, 3, 4A, 4B and 5 (Figure 1) [2]. Moreover, the human genome also encodes another two atypical cullin proteins (CUL7 and CUL9) that consist of additional homology domains [3], such as DOC1 (destruction of cyclin B) homology domain and IBR (in-between-ring) domain [3]. These cullin proteins potentially assemble RING-box protein 1 (RBX1, also known as ROC1) and/or RBX2 (also known as ROC2 or SAG-sensitive to apoptosis gene), forming Cullin-RING ubiquitin ligases (CRLs) [4].
The domain structures of human cullin proteins. Numbers represent the amino acid codons. The blue lines indicate the position of the neddylation site [5].
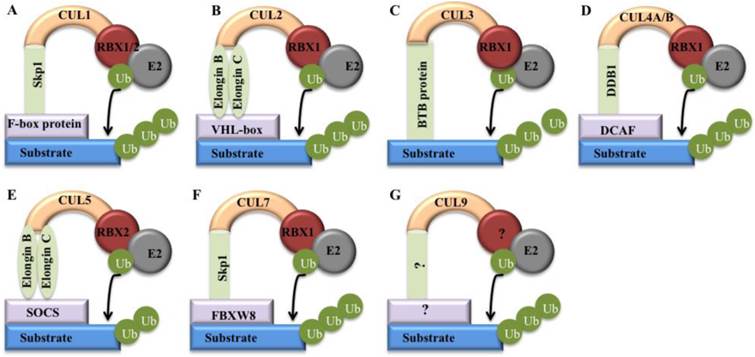
Generally, CRLs consist of four components: cullins, RINGs, adaptor proteins and substrate recognition receptors [5]. The two RING-box components (RBX1 and RBX2) can bind to two zinc atoms via a C3H2C3 motif to form the RING finger domain, which is required for the activity of CRLs [6]. Except for CUL9, all of the other cullin proteins in human can interact with RBX1 or RBX2 through an evolutionarily conserved CH domain at the C-terminus [5]. Moreover, the human genome contains four adaptor proteins, including S-phase kinase-associated protein 1 (Skp1), Elongin B and C, and damaged DNA binding protein 1 (DDB1) [7]. Of them, Skp1 generally interacts with CUL1 and CUL7; Elongin B and C interact with CUL2 and CUL5; and DDB1 interacts with CUL4A and CUL4B (Figure 2) [7]. In addition, BTB (Broad complex, Tramtrack, Bric-a-brac) proteins have been reported to function as adaptors interacting with CUL3 [8]. However, it has not been found adaptor protein that interacts with CUL9 to date (Figure 2). These CRLs have been identified to harbor more than 400 substrate recognition receptors in human genome, including 78 F-box proteins for CRL1, 80 SOCS (suppressor of cytokine signaling) proteins for CRL2/5, ~200 BTB proteins for CRL3, and 90 DCAF (DDB1 and CUL4-associated factors) proteins for CRL4A/B [5, 9]. These substrate recognition receptors further ubiquitinate and degrade thousands of substrates [5], controlling multiple cellular processes, such as cell cycle regulation, gene transcription, apoptosis, DNA damage and repair, chromatin remodeling, oxidative stress response, and signal transduction [5].
Models of cullin-RING E3 ligases. Cullin proteins recruit adaptor proteins (Skp1 for CUL1 and CUL7, Elongin B/C for CUL2 and CUL5, BTB protein for CUL3, and DDB1 for CUL4A and 4B), receptor proteins (F-box proteins for CUL1, VHL-box for CUL2, DCAFs for CUL4A and 4B, SOCS for CUL5, and FBXW8 for CUL7), and RING protein (RBX1/2) to form CRL E3 ligases that promote ubiquitin to transfer from RBX1/RBX2-bound E2 to a substrates [5]. (A) CRL1; (B) CRL2; (C) CRL3; (D) CRL4; (E) CRL5; (F) CRL7; (G) CRL9.
Interestingly, all of these CRLs share a similar core architecture, in which a cullin protein serves as a scaffold that binds to an adaptor protein or a substrate receptor protein at the N-terminus, and interacts with a RING protein at the C-terminus (Figure 2) [5]. The substrate specificity of CRLs is determined by substrate recognition receptors such as F-box proteins and DCAF proteins, whereas the activity of core ligases is controlled by CUL-RBX1 (or RBX2) complex (Figure 2) [2]. Clinically, deregulation of CRLs is linked to uncontrolled proliferative diseases such as cancer [5]. However, only a few components in these hundreds of CRLs are well studied for their involvement in cancer [5]. Generally, cancer-associated CRL components can be categorized into two classes: oncogenes and tumor suppressors [5]. The oncogenes such as Skp2 (S-phase kinase-associated protein 2) and CUL4A are frequently overexpressed in cancers [10]. Tumor suppressors such as FBXW7 (F-box/WD repeat-containing protein 7) and VHL (Von Hippel-Lindau) are always mutated and inactivated in human cancers [11].
CRLs are the largest family of E3 ubiquitin ligases and they are responsible for the ubiquitination of about 20% of intracellular proteins through ubiquitin-proteasome system (UPS) [5]. Cancer cells utilize the UPS for their increased growth and decreased apoptotic cell death [12]. The UPS consists of six components: ubiquitin (Ub), the Ub-activating enzyme (E1), a variety of Ub-conjugating enzymes (E2s), numerous Ub ligases (E3s), the proteasome and the deubiquitinases (DUBs) [12]. Protein degradation by the UPS mainly includes two distinct steps: (1) ubiquitination, which adds a mono- or poly-Ub-tag to the targeted proteins, and (2) proteasomal degradation, which degrades the Ub-tagged proteins into oligopeptides [12]. Two major ubiquitin-ligase complexes, including Skp1-CUL1-F-box (SCF) complex and Anaphase Promoter Complex or Cyclosome (APC/C), are responsible for ubiquitin-mediated proteolysis in mammals [13]. Previous studies have revealed that UPS contributes to oncogenesis through a variety of mechanisms, such as controlling cell proliferation and survival, regulating the turnover of key proteins (cyclins, p27 and p53) involved in cell cycle progression, and regulating the NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) pathway [12]. Moreover, cancer cells can utilize the UPS to achieve aberrant growth and resistance to apoptosis [12].
The cullin family proteins are also known as substrates of NEDD8 (neural precursor cell expressed, developmentally down-regulated 8), an ubiquitin-like protein [14, 15]. The NEDD8-associated ubiquitination includes several critical steps: (1) NEDD8 is activated by NEDD8-activating enzyme E1 (also known as NAE1); (2) the activated NEDD8 is further transferred to ubiquitin-conjugating enzyme E2 (UBC12 or UBE2F) [15]; and (3) NEDD8-conjugating enzyme E2 collaborates with NEDD8-E3 ligase, conjugating NEDD8 to its target substrates [15]. The NEDD8 pathway is critical in mediating the ubiquitination of numerous CRL substrate proteins, such as DNA replication factor CDT1 (cdc10-dependent transcript 1), the NF-κB transcription factor inhibitor pIκBα, and the cell cycle regulators cyclin E and p27 [16, 17].
2. Cullins, DNA damage and repair
DNA damage is one of the most pervasive characteristics of tumor cells [18]. Numerous biotic and abiotic stresses can result in DNA damage, such as genotoxic chemicals, ionizing radiation (IR) and ultraviolet (UV) radiation [19, 20]. DNA damage mainly includes DNA double-strand breaks (DSBs), DNA lesions, single-strand breaks, DNA base mismatches, and DNA crosslinks [21, 22, 23]. Organisms have evolved sophisticated mechanisms to repair damaged DNA and maintain genome integrity. Generally, DSBs are repaired through nonhomologous end joining (NHEJ), alternative NHEJ, or homologous recombination (HR) [19, 24]. UV-induced DNA lesions and other DNA bulks are repaired by nucleotide excision repair (NER) [19, 25]. Small and non-helix-distorting base lesions are repaired by base-excision repair (BER) [19]. Single-strand breaks and DNA base mismatches are corrected by mismatch repair (MMR), while DNA crosslinks are repaired by Fanconi anemia (FA) pathway [19].
DNA damage can induce cascades of posttranslational modifications, such as phosphorylation, ubiquitination and sumoylation [19]. The biochemical and molecular studies have indicated that ubiquitination regulates and coordinates various pathways of DNA damage recognition, signaling and repair [19, 26]. Two cullin proteins (CUL4A and CUL4B) associate with DDB1 and DCAF-type substrate receptors, forming numerous E3 ligases to promote substrate ubiquitination in response to DNA damage [27, 28]. The DDB1-CUL4-DCAFs complexes can ubiquitinate histone H2A, H3 and H4 at sites of UV-induced DNA damage, thereby facilitating their removal from the nucleosome and promoting subsequent DNA repair [29]. In different organisms, a variety of DCAFs have been reported to interact with CUL4-DDB1 in response to DNA damage and repair, such as DDB2 [30], cockayne syndrome A (CSA) [31], two CSA homologues in Arabidopsis (CSAat1A and CSAat1B) [32], transducin-like enhancer of split 1 to 3 (TLE1-3) [33], WD40 repeat protein 5 (WDR5) [27], Drosophila lethal (2) denticleless protein (L2DTL) (also known as CDT2) and the Polycomb-group protein EED (also known as ESC) [33].
In human, CUL4A overexpression has been widely found in multiple cancer types, including breast cancer [34], ovarian cancer [35], hepatocellular carcinomas [36], adrenocortical carcinomas [37], and childhood medulloblastoma [38]. However, the relevance between CUL4B and cancer remains to be determined. Mutations of the CUL4B gene are causally associated with human X-linked mental retardation [39]. CRL4 ubiquitin ligase complex recognizes a variety of substrates involved in DNA damage and repair, such as DDB2, XPC (xeroderma pigmentosum complementation group C), H2A, H3 and H4, UNG2 (uracil-N-glycosylase 2) and SMUG1 (single-strand-selective monofunctional uracil-DNA glycosylase 1) [10]. DDB2 acts as a substrate receptor in complex with CUL4A and DDB1 to target another NER damage sensor (XPC) for ubiquitination, which can increase its affinity to DNA [10]. Downregulation of DDB2 and its substrates limits NER activity, whereas enhanced DDB2 expression or knockout of CUL4A results in the increase of DNA repair efficiency following genotoxic insults, thereby protecting genome from carcinogenesis [10]. Moreover, CRL4DDB2 E3 ligase also can target histone H2A, H3 and H4, facilitating the initiation of NER [29, 40]. Both UNG2 and SMUG1 function to prevent mutagenesis by eliminating uracil from DNA molecules via cleaving the N-glycosylic bond and initiating the BER pathway [41, 42]. CRL4 ubiquitin ligase can mediate UNG2 and SMUG1 degradation, thereby maintaining genome integrity [43, 44].
3. Cullins and cell cycle progression
Cells that progress through the cell cycle unchecked may eventually develop as malignant tumors [45]. Cell cycle progression is controlled by a series of cyclin-dependent kinases (CDKs), proteins interacting with cyclins and CDK inhibitors (CKIs) [46]. The activity of CDKs is regulated by several mechanisms, including association with regulatory subunits, protein modification such as phosphorylation and dephosphorylation, and interaction with CKIs [46, 47]. The levels of cyclins, CKIs and many other cell cycle regulators oscillate during the cell cycle as a result of periodic proteolysis through UPS [48]. Two major ubiquitin ligases, APC/C and SCF, are responsible for the periodic proteolysis of many cell cycle regulators [49]. The APC/C is required for the transition from M phase into G1, and this process is mediated by the ubiquitination of anaphase inhibitors known as securins and mitotic cyclins, respectively [50]. The SCF complex targets cyclins and CKIs for ubiquitination, functioning in most phases of the cell cycle [48]. The SCF complex can bind to multiple F-box proteins functioning as receptor components and three of these F-box proteins including Skp2, FBXW7, and β-transducin repeat-containing protein (β-TrCP, also known as FBXW1) are well characterized and implicated in cell cycle regulation [51]. Clinical evidence suggests that dysregulation of these F-box proteins contributes to human cancers [15].
Skp2 behaves as an oncogene in cell systems and overexpression of Skp2 is frequently observed in human cancer progression and metastasis [52]. Multiple cancer types, including lymphomas, prostate cancer, melanoma, nasopharyngeal carcinoma, pancreatic cancer and breast carcinomas, have been reported to associate with Skp2 overexpression [11]. Biochemically, Skp2 functions as the substrate-recruiting component of SCF complex, and SCFSkp2 can recognize and promote the degradation of several negative cell cycle regulators, including p27, p21, p130 and p57 [53]. The expression of Skp2 is significantly increased in many transformed cells during S and G2 phases of the cell cycle, but not in G1 [54]. Additionally, Skp2 mediates the ubiquitination of the p27, which is degraded via the UPS in G1 phase [48]. The CDK inhibitor p27Kip1 is also involved in cancer, playing roles in inhibiting cyclin E-CDK2 complex [55]. Levels of p27Kip1 are regulated by SCFSkp2 complex during cell cycle through proteasome system [55].
FBXW7 protein is a well-established cell cycle regulator and functions as tumor suppressor required for substrate recognition in an SCF complex [15]. SCFFBXW7 can ubiquitinate several substrates involved in oncogenesis, including cyclin E, c-Myc (myelocytomatosis oncogene), c-Jun (jun proto-oncogene) and Notch, thereby promoting cells to exit from the cell cycle [10]. Interestingly, FBXW7 binds to each of these substrates through a conserved phosphorylated domain known as the CDC4 phosphodegron [56]. The in vitro analysis of FBXW7 gene silencing enhances the expression of c-Myc and cyclin E genes, and promotes cell proliferation [57]. Thus, FBXW7 is crucial for preventing carcinogenesis as its important roles in cell cycle regulation [56].
β-TrCP is emerging as an important protein in carcinogenesis because of the deregulated proteolysis of its substrates [52]. Previous studies have identified numerous substrates of β-TrCP, and these substrates can be divided into two main groups: cell cycle regulators such as CDC25A (cell division cycle 25 homolog A), CDC25B, WEE1, IκBα (nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha), IκBβ, IκBε, p100 and p105, and pro-apoptotic regulators such as pro-caspase, p63, STAT1 (signal transducers and activator of transcription 1) [52]. Importantly, β-TrCP plays critical roles in regulating cell cycle checkpoints, and the increased activities of the SCFβ-TrCP complex may promote tumor cell growth and/or genetic instability by accelerating cell cycle events [58]. In response to DNA damage, β-TrCP contributes to CDK1 dysfunction by mediating the degradation of CDC25A in collaboration with Chk1 (checkpoint kinase 1), thereby preventing cell cycle progression before the completion of DNA repair [59].
Moreover, other cullin proteins such as CUL3, CUL4A and CUL5 also have been demonstrated to contribute to cell cycle progression in cancer. CUL3 forms a complex known as BTB-CUL3-RBX1 (BCR) ubiquitin ligase, targeting cyclin E for ubiquitination and controlling S phase in mammalian cells [60]. Overexpression of CUL3 in mice increases ubiquitination of cyclin E, but not other cyclins [60]. Conversely, the deletion of CUL3 gene results in increased accumulation of cyclin E protein, and has cell-type-specific effects on S-phase regulation [60]. CRL4 interacts with the substrate receptor CDT2, promoting protein ubiquitination in S phase and after DNA damage [61]. The CRL4CDT2 complex can target a variety of substrates involved in cell cycle regulation, such as CDT1 [62], p21 [63], and Chk1 [64]. The CUL4A-DDB1 E3 complex can target tumor suppressor RASSF1A (RAS association domain family 1, isoform A) for degradation during mitosis [65]. CUL4A depletion results in increased levels of RASSF1A protein, whereas overexpression of CUL4A and DDB1 significantly enhances RASSF1A protein ubiquitination and results in reduced RASSF1A protein levels [65]. Overexpression of CUL4A antagonizes the ability of RASSF1A to induce M-phase cell cycle arrest [65]. CUL5 is a target of microRNA-7 (miR-7) [66]. Downregulation of miR-7 increases the expression of CUL5 to facilitate G1/S transition in human hepatocellular carcinoma cells [66].
4. Cullins and p53 pathway
p53 functions as a tumor suppressor through several mechanisms: (1) activating DNA repair proteins when DNA has sustained damage; (2) arresting cell cycle at the G1/S regulation point on DNA damage recognition; and (3) initiating cancer cell apoptosis [67]. In normal cells, p53 is generally kept at low level by proteasome-mediated degradation, allowing cell cycle progression and cell proliferation [68]. Under stress conditions, p53 is stabilized and activated through posttranslational modifications of itself and its regulatory proteins, eventually inducing the expression of downstream genes that are responsible for cell cycle arrest, DNA repair and apoptosis [69]. E3 ligases are also involved in p53 pathway, and over 20 different E3 ligases have been identified to play roles in the control of stability, oligomerization, nuclear export, translocation of p53 to chromatin or nuclear foci [70]. Interestingly, cullin proteins have been reported to directly interact with p53, or regulate p53 expression and its protein stability [71, 72, 73].
An F-box protein JFK (just one F-box and Kelch domain-containing protein) promotes p53 degradation through the formation of an SCF complex in which JFK serves as a substrate receptor [74]. The CUL1 knockout mouse is lethal at E6.5 before the onset of gastrulation, associating with increased p53 levels and apoptosis in the embryonic ectoderm [73]. The BZLF1 (BamHI Z fragment leftward open reading frame 1) protein directly functions as an adaptor component of the ECS (Elongin B/C-CUL2/5-SOCS-box protein) E3 ligase complex, targeting the phosphorylated p53 for degradation [75]. Deletion of CUL3 gene in liver cells causes a persistent and massive expansion phenotype, which is dependent on differentiation and undergoes remarkable DNA damage, thereby leading to cell death [73]. Interestingly, this process appears to be controlled by p53, as simultaneous loss of CUL3 and p53 in hepatic progenitors transforms these cells to highly malignant tumor-initiating cells that form largely undifferentiated tumors in nude mice [72, 73]. CDT2 and PCNA (proliferating cell nuclear antigen) interact with p53, and regulate p53 poly-ubiquitination and protein stability through both MDM2 (mouse double minute 2 homolog) and CUL4A-DDB1 complexes in unstressed human cells [76]. Moreover, PCNA and DDB1-CUL4A complex also regulate the proteolysis of MDM2 in response to UV-irradiation [76]. Cells deleting CUL4A result in proliferation defects and genome instability, and the decrease of p53 levels [77]. Moreover, expression of CUL4A increases the decay-rate of p53 and delays the accumulation of p53 in response to DNA damage [78]. Recently, CUL4B has been reported to directly interact with p53 and induce its poly-ubiquitination [79]. VACM-1 (Vasopressin-activated Ca2+-mobilizing 1, also known as CUL5) inhibits cell growth by a mechanism that involves MAPKs (mitogen activated protein kinases) and p53 signaling pathways [80]. Additionally, the degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving an ECS E3 ligase [81]. The cytoplasmic localized ubiquitin ligase CRL7 can bind to p53 and promote cell growth by antagonizing p53 function [71]. Biochemically, CUL7 only causes mono- or di-ubiquitination of p53 in vitro, whereas MDM2 polyubiquitinates p53 in the same condition [71]. In a proteomic screen for p53 interactors, CUL7 was also identified to efficiently associate with p53 [82]. The functional analysis indicated that induction of CUL7 by DNA damage attenuated p53 function [82].
5. Cullins and WNT pathways
WNTs and their downstream effectors regulate various cellular processes that are important for carcinogenesis and tumor progression, including tumor initiation, tumor growth, cell senescence, cell death, differentiation and metastasis [83]. Cullin proteins, such as CUL1, CUL3 and CUL4, have been identified to involve WNT pathways [84, 85].
SCFβ-TrCP E3 ligase can specifically regulate the stability of β-catenin, thereby having a significant impact on cancer genesis [85]. Recently, FAS-associated factor 1 (FAF1) has been reported to function as a scaffold protein and form a complex with SCFβ-TrCP E3 ligase, antagonizing WNT signaling by stimulating β-catenin degradation [86]. Moreover, an F-box/WD40 protein, FWD1, form a complex with SCF, mediating ubiquitin-dependent proteolysis of β-catenin [87].
KLHL12 (Kelch-like protein 12) is a substrate-specific adapter of a BCR E3 ligase complex [84]. The BCRKLHL12 ubiquitin ligase negatively regulates the WNT-β-catenin pathway by targeting and degrading Dishevelled, which is a conserved protein that interprets signals received by Frizzled receptors [84].
The DDB1-CUL4 complex has been demonstrated to be a WNT-induced ubiquitin ligase [88]. The CDK inhibitor p27KIP1 is tightly linked with human cancer [88]. Interestingly, the WNT-induced p27KIP1 turnover is independent from classical SCFSkp2-mediated degradation in both mouse and human cells [88]. Instead, the turnover requires CUL4A and CUL4B [88]. Additionally, DDB1-CUL4B complex can negatively regulate β-catenin [89]. The knockdown of CUL4B in mouse results in an increase in β-catenin levels [89]. Immunoprecipitation experiments suggest that CUL4B and β-catenin form a signal transduction complex in Drosophila, mouse and human [89].
6. Cullins regulate the expression or stability of oncogenes
Cullin proteins are also involved in regulating the expression or stability of several oncogenes, such as c-Myc, c-Jun and IκBα.
c-Myc is a transcription factor that plays important roles in cell cycle progression, apoptosis and cellular transformation [90]. Malfunctions in c-Myc have also been found in carcinoma of the cervix, colon, breast, lung and stomach cancer [91]. c-Myc enhances the expression of CUL1 gene, promoting ubiquitin-dependent proteolysis and cell cycle progression through SCFSkp2 E3 ligase [92]. TRUSS (tumor necrosis factor receptor-associated ubiquitous scaffolding and signaling protein) is a receptor protein of DDB1-CUL4 complex [93]. The suppression of DDB1-CUL4-TRUSS E3 ligase stabilizes c-Myc protein in cancer cells [93]. CUL7 can function as an oncogene that cooperates with c-Myc in transformation by blocking c-Myc-induced apoptosis in a p53-dependent manner [94]. The MM-1 (Myc modulator 1) protein binds to c-Myc and represses transcriptional activity of c-Myc [95]. Further studies indicate that MM-1 facilitates c-Myc degradation by recruiting proteasome and the ECSSkp2 E3 ligase [95]. Interestingly, Skp2 interacts with c-Myc and participates in its ubiquitylation and proteasomal degradation [96]. Surprisingly, Skp2 acts as a positive cofactor for c-Myc-regulated transcription [96].
c-Jun is the firstly demonstrated as an oncogenic transcription factor, and it is required at the early stage of tumor development [97]. Deletion of c-Jun can significantly suppress tumor formation [98]. The human De-etiolated-1 (hDET1) promotes ubiquitination and degradation of c-Jun by assembling DDB1-CUL4A-RBX1 E3 ligase transcription [99]. Ablation of DDB1, CUL4A or RBX1 by RNA interference stabilizes c-Jun and increases c-Jun-activated transcription [99]. Biochemically, RBX2 regulates skin carcinogenesis by targeting c-Jun/AP1 (activator protein 1) and IκBα/NF-κB in a stage-dependent manner [100]. RBX2 promotes ubiquitination and degradation of c-Jun to inhibit AP1 activity and AP1-induced neoplastic transformation in a mouse epidermal cell model [100]. It appears that RBX2 inhibits tumor formation at the early stage by targeting c-Jun/AP1 and promotes tumor growth at the later stage by targeting IκBα/NF-κB [100].
IκBα functions to inhibit the NF-κB transcription factor [101]. Biochemically, IκBα can be ubiquitinated by a SCF-like complex containing Skp1, CUL1, β-TrCP1 and β-TrCP2 [102]. All these components are cooperatively recruited to bind to a phosphorylated IκBα (pIκBα) produced by tumor necrosis factor-α (TNF-α) stimulation [102]. IκBα-E3 ligase binds to pIκBα and catalyzes the ubiquitination of pIκBα in the presence of ATP, Ub, and E1-activating and E2-conjugating enzymes [102]. Further studies indicate that NEDD8 modification of CUL1 activates SCFβ-TrCP dependent ubiquitination of IκBα [103].
7. Cullins and apoptosis
Studies have revealed a high frequency of apoptosis in spontaneously regressing tumors and in tumors treated with cytotoxic anticancer agents [104]. The cullin proteins such as CUL1, CUL3 and CUL4 are also involved in apoptosis.
CDK-associated CUL1 can promote cell proliferation and inhibit cisplatin-induced apoptosis in the AGS gastric cancer cell line via modulating the expression of p53, BCL2 (B-cell lymphoma 2) and BAX (BCL2 associated X protein) [105]. CUL3-based poly-ubiquitination and p62-dependent aggregation of caspase-8 can mediate extrinsic apoptosis signaling [106]. Caspase-8 is a critical initiator of the extrinsic apoptosis pathway, and the stimulation of death receptors 4 and 5 (DR4 and 5) induces CUL3/RBX1-dependent poly-ubiquitination of caspase-8 [106]. Further, the Ub-binding protein p62 promotes caspase-8 aggregation, augmenting the activation and full processing of the enzyme and thereby leading to robust stimulation of effector caspases and apoptosis [106]. CUL4-DDB1CDT2 and CUL4-DDB1DDB2 E3 ligase complexes have been implicated in the proteolysis of cell cycle inhibitor p21, which can inhibit apoptosis following DNA damage [107]. Moreover, cells lacking CDC7 expression undergo apoptosis after mild DNA damage, where CUL4-DDB1CDT2 E3 ligase induces Tob (Transducer of ErbB-2) ubiquitination and its subsequent degradation [108]. However, CDC7 phosphorylates and interacts with Tob to inhibit the CUL4-DDB1CDT2-dependent Tob degradation in normal cells [108]. Additionally, CUL4B promotes proliferation and inhibits apoptosis of human osteosarcoma cells [109]. In the osteosarcoma SAOS-2 cells harboring decreased CUL4B gene expression by RNA interference (RNAi), cell proliferation rate and clonability are significantly inhibited, whereas the apoptosis rate is greatly increased [109].
8. Cullins and Hypoxia-inducible factor (HIF-1) pathway
HIF-1 is a basic helix-loop-helix-PAS domain transcription factor, and upregulation of HIF-1 is generally associated with tumor cell growth and progression [110]. Some cullin proteins, including CUL2, CUL3 and CLU5, have been demonstrated to involve HIF-1 pathway.
Previous studies have indicated that neddylation plays a major role in modulating the HIF pathway through CUL2 [111]. The von Hippel-Lindau tumor suppressor protein (pVHL) assembles with CUL2 and Elongin B/C, forming a protein complex, namely, CBCVHL [111]. Remarkably, cells lacking pVHL induce the expression of hypoxia-inducible genes, including VEGF (vascular endothelial growth factor), GLUT1 (glucose transporter 1), and PDGF-β (platelet derived growth factor-β) [111], indicating the important roles of CBCVHL in HIF-1 pathway. Recently, inhibition of CUL2 neddylation has been demonstrated to stabilize HIF-1 in mucosal inflammatory responses [112].
A CUL3 substrate adaptor, namely, KLHL20 (kelch-like family member 20), can be induced by HIF-1 [113]. The CUL3-KLHL20 ubiquitin ligase-dependent pathway targets PML (promyelocytic leukemia) to potentiate HIF-1 signaling and prostate cancer progression [113]. Additionally, the PML destruction pathway participates in a feedback mechanism to maximize HIF-1α induction, thereby potentiating multiple tumor hypoxia responses, including metabolic reprogramming, epithelial-mesenchymal transition, migration, tumor growth, angiogenesis, and chemoresistance [113].
RBX2 also can function as a HIF-1 target gene, and it can form a VCB E3 ligase with VHL, CUL5 and Elongin B/C in vivo, promoting VHL-mediated HIF-1α ubiquitination and degradation [114]. Interestingly, the endogenous HIF-1α at both basal and hypoxia-induced levels is significantly increased upon RBX2 gene silencing [114].
9. Cullins and other pathways involved in carcinogenesis
Cullin proteins are also reported to involve some other pathways, such as oxidative stress and chromatin remodeling.
Oxidative stress affects the stability or expression of numerous key signaling proteins, such as nuclear factor erythroid 2-related factor 2 (Nrf2), kelch-like protein 1 (Keap1), Ras, Raf, MAPK, ERK1/2, MEK, p38α, c-Jun N-terminal kinase (JNK), c-Myc and p53 [115]. Of them, Nrf2 and Keap1 can function together with CUL3 and their molecular mechanisms have been well studied [116]. Nrf2 is rapidly degraded through a Keap1-CUL3-RBX1 E3 ligase-dependent mechanism [116]. Keap1 functions as a substrate adaptor, and is responsible for bringing Nrf2 into the E3 complex, in which it utilizes its DGR domain to bind the N-terminal region of CUL3 [116]. RBX1 recruits the catalytic function of E2 by binding to the C-terminal region of CUL3 [116]. E2 catalyzes poly-ubiquitination of Nrf2 protein on the lysine residues of Neh2 domain [116]. Additionally, CUL3 overexpression depletes Nrf2 in breast cancer, which is associated with increased sensitivity to carcinogen, oxidative stress and chemotherapy [117].
All of the human genome is packaged into chromatin. Chromatin remodeling proteins are gatekeepers and function as major determinants of accessory factors to nucleosome DNA, thereby allowing a wide repertoire of biological functions [118]. Thus, aberrant expression or epigenetic modulation of remodeling proteins confers a unique ability to cancer cells, reprograming its genome for the maintenance of oncogenic phenotypes [118]. The CUL4-DDB1-RBX1 E3 ligase can target histones for ubiquitination, and importantly, ubiquitination of histones may facilitate cell in response to DNA damage [119]. Therefore, histone ubiquitination by CRL4 E3 ligase constitutes a novel mechanism through which CUL4 regulates chromatin function and maintains genomic integrity [119]. Knockdown of CUL4A or CUL4B in HeLa cells significantly reduces the levels of H3 and H4 ubiquitination, but has little effects on H2A and H2B ubiquitination [119].
10. Conclusion
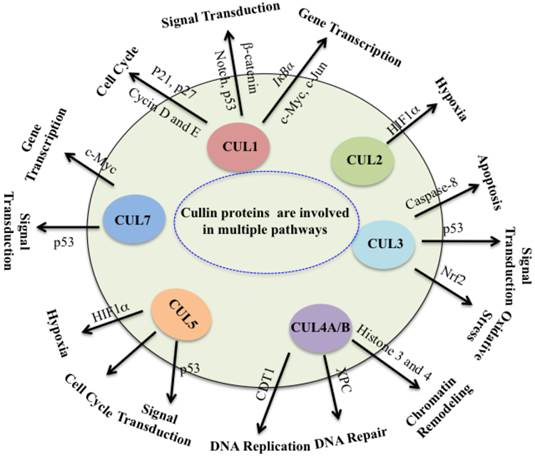
Collectively, cullin proteins play critical roles in cancer through a variety of mechanisms, including DNA damage and repair, cell cycle control, apoptosis, oxidative stresses, chromatin remodeling, regulation of oncogenes, signal transduction involved in p53 and WNT (Figure 3). In which, cullins proteins mainly function as scaffolds to form numerous E3 ligases, and these E3 ligases further recognize different substrates to participate in different cellular processes. Complete inactivation of CRLs causes tumor cell growth suppression, indicating that the major role of CRLs in cancer cells is to target and degrade tumor suppressor proteins in order to sustain an unlimited proliferation [5]. Thus, this unique feature of cancer cells confers them much more vulnerable to be targeted against CRLs in therapy [5]. It is clear that further studies to explore CRL functions and screen substrates of CRLs in different cancer types. To develop potential anti-cancer agents, it is important to focus on the development of CRL inhibitors by targeting cullin neddylation and CRL-mediated ubiquitination in the future.
Cullin proteins affect multiple pathways involved in cancer. Cullin proteins play critical roles in cancer through a variety of mechanisms, including DNA replication and repair, cell cycle control, apoptosis, oxidative stresses, chromatin remodeling, hypoxia, regulation of oncogenes, and signal transduction involved in p53 and WNT. Proteins participate in these processes are also indicated.
Acknowledgements
We apologize to all authors whose contributions were not cited due to space limitations. We would like to express our gratitude to Dr. Tongwan Li for critically reading the manuscript and for facilitating discussions.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sarikas A, Hartmann T, Pan ZQ. The cullin protein family. Genome Biol. 2011;12:220-232
2. Zimmerman ES, Schulman BA, Zheng N. Structural assembly of cullin-RING ubiquitin ligase complexes. Curr Opin Struct Biol. 2010;20:714-21
3. Sharma P, Nag A. CUL4A ubiquitin ligase: a promising drug target for cancer and other human diseases. Open Biol. 2014;4:130217
4. Wei D, Sun Y. Small RING Finger Proteins RBX1 and RBX2 of SCF E3 Ubiquitin Ligases: The Role in Cancer and as Cancer Targets. Genes Cancer. 2010;1:700-7
5. Zhao Y, Sun Y. Cullin-RING Ligases as attractive anti-cancer targets. Curr Pharm Des. 2013;19:3215-25
6. Sun Y, Li H. Functional characterization of SAG/RBX2/ROC2/RNF7, an antioxidant protein and an E3 ubiquitin ligase. Protein Cell. 2013;4:103-16
7. Jackson S, Xiong Y. CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem Sci. 2009;34:562-70
8. Zhang DD, Lo SC, Cross JV. et al. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941-53
9. Kerzendorfer C, Hart L, Colnaghi R. et al. CUL4B-deficiency in humans: understanding the clinical consequences of impaired Cullin 4-RING E3 ubiquitin ligase function. Mech Ageing Dev. 2011;132:366-73
10. Lee J, Zhou P. Cullins and cancer. Genes Cancer. 2010;1:690-9
11. Wang Z, Gao D, Fukushima H. et al. Skp2: a novel potential therapeutic target for prostate cancer. Biochim Biophys Acta. 2012;1825:11-7
12. Shen M, Schmitt S, Buac D, Dou QP. Targeting the ubiquitin-proteasome system for cancer therapy. Expert Opin Ther Targets. 2013;17:1091-108
13. Rizzardi LF, Cook JG. Flipping the switch from g1 to s phase with e3 ubiquitin ligases. Genes Cancer. 2012;3:634-48
14. Gong L, Yeh ET. Identification of the activating and conjugating enzymes of the NEDD8 conjugation pathway. J Biol Chem. 1999;274:12036-42
15. Wang X, Li L, Liang Y. et al. Targeting the neddylation pathway to suppress the growth of prostate cancer cells: therapeutic implication for the men's cancer. Biomed Res Int. 2014;2014:974309
16. Soucy TA, Smith PG, Rolfe M. Targeting NEDD8-activated cullin-RING ligases for the treatment of cancer. Clin Cancer Res. 2009;15:3912-6
17. Soucy TA, Dick LR, Smith PG. et al. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes Cancer. 2010;1:708-16
18. Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287-94
19. Jackson SP, Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell. 2013;49:795-807
20. Rastogi RP, Richa, Kumar A. et al. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids. 2010;2010:592980
21. Clauson C, Scharer OD, Niedernhofer L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb Perspect Med. 2013;3:a012732
22. Ghosal G, Chen J. DNA damage tolerance: a double-edged sword guarding the genome. Transl Cancer Res. 2013;2:107-29
23. Zhang C. Essential functions of iron-requiring proteins in DNA replication, repair and cell cycle control. Protein Cell. 2014;5:750-60
24. Davis AJ, Chen DJ. DNA double strand break repair via non-homologous end-joining. Transl Cancer Res. 2013;2:130-43
25. D'Errico M, Teson M, Calcagnile A. et al. Apoptosis and efficient repair of DNA damage protect human keratinocytes against UVB. Cell Death Differ. 2003;10:754-6
26. Sato K, Sundaramoorthy E, Rajendra E. et al. A DNA-damage selective role for BRCA1 E3 ligase in claspin ubiquitylation, CHK1 activation, and DNA repair. Curr Biol. 2012;22:1659-66
27. Higa LA, Zhang H. Stealing the spotlight: CUL4-DDB1 ubiquitin ligase docks WD40-repeat proteins to destroy. Cell Div. 2007;2:5
28. Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell. 2006;23:709-21
29. Wang H, Zhai L, Xu J. et al. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol Cell. 2006;22:383-94
30. Scrima A, Konickova R, Czyzewski BK, Kawasaki Y, Jeffrey PD, Groisman R. et al. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell. 2008;135:1213-23
31. Fousteri M, Vermeulen W, van Zeeland AA, Mullenders LH. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell. 2006;23:471-82
32. Zhang C, Guo H, Zhang J. et al. Arabidopsis cockayne syndrome A-like proteins 1A and 1B form a complex with CULLIN4 and damage DNA binding protein 1A and regulate the response to UV irradiation. Plant Cell. 2010;22:2353-69
33. Higa LA, Yang X, Zheng J. et al. Involvement of CUL4 ubiquitin E3 ligases in regulating CDK inhibitors Dacapo/p27Kip1 and cyclin E degradation. Cell Cycle. 2006;5:71-7
34. Chen LC, Manjeshwar S, Lu Y. et al. The human homologue for the Caenorhabditis elegans cul-4 gene is amplified and overexpressed in primary breast cancers. Cancer Res. 1998;58:3677-83
35. Birner P, Schoppmann A, Schindl M. et al. Human homologue for Caenorhabditis elegans CUL-4 protein overexpression is associated with malignant potential of epithelial ovarian tumours and poor outcome in carcinoma. J Clin Pathol. 2012;65:507-11
36. Yasui K, Arii S, Zhao C. et al. TFDP1, CUL4A, and CDC16 identified as targets for amplification at 13q34 in hepatocellular carcinomas. Hepatology. 2002;35:1476-84
37. Dohna M, Reincke M, Mincheva A. et al. Adrenocortical carcinoma is characterized by a high frequency of chromosomal gains and high-level amplifications. Genes Chromosomes Cancer. 2000;28:145-52
38. Michiels EM, Weiss MM, Hoovers JM. et al. Genetic alterations in childhood medulloblastoma analyzed by comparative genomic hybridization. J Pediatr Hematol Oncol. 2002;24:205-10
39. Tardat M, Brustel J, Kirsh O. et al. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol. 2010;12:1086-93
40. Kapetanaki MG, Guerrero-Santoro J, Bisi DC. et al. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc Natl Acad Sci U S A. 2006;103:2588-93
41. Dingler FA, Kemmerich K, Neuberger MS, Rada C. Uracil excision by endogenous SMUG1 glycosylase promotes efficient Ig class switching and impacts on A:T substitutions during somatic mutation. Eur J Immunol. 2014;44:1925-35
42. Schormann N, Ricciardi R, Chattopadhyay D. Uracil-DNA glycosylases-Structural and functional perspectives on an essential family of DNA repair enzymes. Protein Sci. 2014 doi:10.1002/pro.2554
43. Doseth B, Ekre C, Slupphaug G, Krokan HE, Kavli B. Strikingly different properties of uracil-DNA glycosylases UNG2 and SMUG1 may explain divergent roles in processing of genomic uracil. DNA Repair (Amst). 2012;11:587-93
44. Sharifi HJ, Furuya AK, Jellinger RM. et al. Cullin4A and cullin4B are interchangeable for HIV Vpr and Vpx action through the CRL4 ubiquitin ligase complex. J Virol. 2014;88:6944-58
45. Crawford ED, Scholz MC, Kar AJ. et al. Cell cycle progression score and treatment decisions in prostate cancer: results from an ongoing registry. Curr Med Res Opin. 2014;30:1025-31
46. Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140:3079-3093
47. Cayrol C, Ducommun B. Interaction with cyclin-dependent kinases and PCNA modulates proteasome-dependent degradation of p21. Oncogene. 1998;17:2437-44
48. Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369-81
49. Nakayama KI, Nakayama K. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin Cell Dev Biol. 2005;16:323-33
50. Li M, Zhang P. The function of APC/CCdh1 in cell cycle and beyond. Cell Div. 2009;4:2
51. Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol. 2013;14:369-81
52. Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438-49
53. Jia L, Sun Y. SCF E3 ubiquitin ligases as anticancer targets. Curr Cancer Drug Targets. 2011;11:347-56
54. Egozi D, Shapira M, Paor G. et al. Regulation of the cell cycle inhibitor p27 and its ubiquitin ligase Skp2 in differentiation of human embryonic stem cells. FASEB J. 2007;21:2807-17
55. Lloyd RV, Erickson LA, Jin L. et al. p27kip1: a multifunctional cyclin-dependent kinase inhibitor with prognostic significance in human cancers. Am J Pathol. 1999;154:313-23
56. Takeishi S, Nakayama KI. Role of Fbxw7 in the maintenance of normal stem cells and cancer-initiating cells. Br J Cancer. 2014;111:1054-9
57. Iwatsuki M, Mimori K, Ishii H. et al. Loss of FBXW7, a cell cycle regulating gene, in colorectal cancer: clinical significance. Int J Cancer. 2010;126:1828-37
58. Fuchs SY, Spiegelman VS, Kumar KG. The many faces of beta-TrCP E3 ubiquitin ligases: reflections in the magic mirror of cancer. Oncogene. 2004;23:2028-36
59. Busino L, Donzelli M, Chiesa M. et al. Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature. 2003;426:87-91
60. Singer JD, Gurian-West M, Clurman B, Roberts JM. Cullin-3 targets cyclin E for ubiquitination and controls S phase in mammalian cells. Genes Dev. 1999;13:2375-87
61. Havens CG, Walter JC. Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 2011;25:1568-82
62. Abbas T, Dutta A. CRL4Cdt2: master coordinator of cell cycle progression and genome stability. Cell Cycle. 2011;10:241-9
63. Kim Y, Starostina NG, Kipreos ET. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 2008;22:2507-19
64. Huh J, Piwnica-Worms H. CRL4(CDT2) targets CHK1 for PCNA-independent destruction. Mol Cell Biol. 2013;33:213-26
65. Jiang L, Rong R, Sheikh MS, Huang Y. Cullin-4A.DNA damage-binding protein 1 E3 ligase complex targets tumor suppressor RASSF1A for degradation during mitosis. J Biol Chem. 2011;286:6971-8
66. Ma C, Qi Y, Shao L. et al. Downregulation of miR-7 upregulates Cullin 5 (CUL5) to facilitate G1/S transition in human hepatocellular carcinoma cells. IUBMB Life. 2013;65:1026-34
67. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14:359-70
68. Ponnamperuma RM, King KE, Elsir T. et al. The transcriptional regulatory function of p53 is essential for suppression of mouse skin carcinogenesis and can be dissociated from effects on TGF-beta-mediated growth regulation. J Pathol. 2009;219:263-74
69. Brooks CL, Gu W. New insights into p53 activation. Cell Res. 2010;20:614-21
70. Brooks CL, Gu W. p53 regulation by ubiquitin. FEBS Lett. 2011;585:2803-9
71. Andrews P, He YJ, Xiong Y. Cytoplasmic localized ubiquitin ligase cullin 7 binds to p53 and promotes cell growth by antagonizing p53 function. Oncogene. 2006;25:4534-48
72. Kossatz U, Breuhahn K, Wolf B. et al. The cyclin E regulator cullin 3 prevents mouse hepatic progenitor cells from becoming tumor-initiating cells. J Clin Invest. 2010;120:3820-33
73. Zhou W, Wei W, Sun Y. Genetically engineered mouse models for functional studies of SKP1-CUL1-F-box-protein (SCF) E3 ubiquitin ligases. Cell Res. 2013;23:599-619
74. Sun L, Shi L, Li W. et al. JFK, a Kelch domain-containing F-box protein, links the SCF complex to p53 regulation. Proc Natl Acad Sci U S A. 2009;106:10195-200
75. Sato Y, Kamura T, Shirata N. et al. Degradation of phosphorylated p53 by viral protein-ECS E3 ligase complex. PLoS Pathog. 2009;5:e1000530
76. Banks D, Wu M, Higa LA. et al. L2DTL/CDT2 and PCNA interact with p53 and regulate p53 polyubiquitination and protein stability through MDM2 and CUL4A/DDB1 complexes. Cell Cycle. 2006;5:1719-29
77. Kopanja D, Stoyanova T, Okur MN. et al. Proliferation defects and genome instability in cells lacking Cul4A. Oncogene. 2009;28:2456-65
78. Nag A, Bagchi S, Raychaudhuri P. Cul4A physically associates with MDM2 and participates in the proteolysis of p53. Cancer Res. 2004;64:8152-55
79. Thirunavukarasou A, Singh P, Govindarajalu G. et al. E3 ubiquitin ligase Cullin4B mediated polyubiquitination of p53 for its degradation. Mol Cell Biochem. 2014;390:93-100
80. Van Dort C, Zhao P, Parmelee K. et al. VACM-1, a cul-5 gene, inhibits cellular growth by a mechanism that involves MAPK and p53 signaling pathways. Am J Physiol Cell Physiol. 2003;285:C1386-96
81. Querido E, Blanchette P, Yan Q. et al. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 2001;15:3104-17
82. Jung P, Verdoodt B, Bailey A. et al. Induction of cullin 7 by DNA damage attenuates p53 function. Proc Natl Acad Sci U S A. 2007;104:11388-93
83. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11-26
84. Angers S, Thorpe CJ, Biechele TL. et al. The KLHL12-Cullin-3 ubiquitin ligase negatively regulates the Wnt-beta-catenin pathway by targeting Dishevelled for degradation. Nat Cell Biol. 2006;8:348-57
85. Latres E, Chiaur DS, Pagano M. The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene. 1999;18:849-54
86. Zhang L, Zhou F, Li Y. et al. Fas-associated factor 1 is a scaffold protein that promotes beta-transducin repeat-containing protein (beta-TrCP)-mediated beta-catenin ubiquitination and degradation. J Biol Chem. 2012;287:30701-10
87. Kitagawa M, Hatakeyama S, Shirane M. et al. An F-box protein, FWD1, mediates ubiquitin-dependent proteolysis of beta-catenin. EMBO J. 1999;18:2401-10
88. Miranda-Carboni GA, Krum SA, Yee K. et al. A functional link between Wnt signaling and SKP2-independent p27 turnover in mammary tumors. Genes Dev. 2008;22:3121-34
89. Tripathi R, Kota SK, Srinivas UK. Cullin4B/E3-ubiquitin ligase negatively regulates beta-catenin. J Biosci. 2007;32:1133-8
90. Hoffman B, Liebermann DA. Apoptotic signaling by c-MYC. Oncogene. 2008;27:6462-72
91. Tsai MM, Wang CS, Tsai CY. et al. Potential prognostic, diagnostic and therapeutic markers for human gastric cancer. World J Gastroenterol. 2014;20:13791-803
92. O'Hagan RC, Ohh M, David G. et al. Myc-enhanced expression of Cul1 promotes ubiquitin-dependent proteolysis and cell cycle progression. Genes Dev. 2000;14:2185-91
93. Choi SH, Wright JB, Gerber SA, Cole MD. Myc protein is stabilized by suppression of a novel E3 ligase complex in cancer cells. Genes Dev. 2010;24:1236-41
94. Kim SS, Shago M, Kaustov L. et al. CUL7 is a novel antiapoptotic oncogene. Cancer Res. 2007;67:9616-22
95. Kimura Y, Nagao A, Fujioka Y, Satou A, Taira T, Iguchi-Ariga SM. et al. MM-1 facilitates degradation of c-Myc by recruiting proteasome and a novel ubiquitin E3 ligase. Int J Oncol. 2007;31:829-36
96. von der Lehr N, Johansson S, Wu S. et al. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell. 2003;11:1189-200
97. Vogt PK. Fortuitous convergences: the beginnings of JUN. Nat Rev Cancer. 2002;2:465-9
98. Eferl R, Ricci R, Kenner L. et al. Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell. 2003;112:181-92
99. Wertz IE, O'Rourke KM, Zhang Z. et al. Human De-etiolated-1 regulates c-Jun by assembling a CUL4A ubiquitin ligase. Science. 2004;303:1371-4
100. Gu Q, Bowden GT, Normolle D, Sun Y. SAG/ROC2 E3 ligase regulates skin carcinogenesis by stage-dependent targeting of c-Jun/AP1 and IkappaB-alpha/NF-kappaB. J Cell Biol. 2007;178:1009-23
101. Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the IkappaBalpha/NF-kappaB complex reveals mechanisms of NF-kappaB inactivation. Cell. 1998;95:759-70
102. Suzuki H, Chiba T, Kobayashi M. et al. IkappaBalpha ubiquitination is catalyzed by an SCF-like complex containing Skp1, cullin-1, and two F-box/WD40-repeat proteins, betaTrCP1 and betaTrCP2. Biochem Biophys Res Commun. 1999;256:127-32
103. Read MA, Brownell JE, Gladysheva TB. et al. Nedd8 modification of cul-1 activates SCF(beta(TrCP))-dependent ubiquitination of IkappaBalpha. Mol Cell Biol. 2000;20:2326-33
104. Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis. 2000;21:485-95
105. Zheng Q, Zhao LY, Kong Y. et al. CDK-associated Cullin 1 can promote cell proliferation and inhibit cisplatin-induced apoptosis in the AGS gastric cancer cell line. World J Surg Oncol. 2013;11:5
106. Jin Z, Li Y, Pitti R. et al. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137:721-35
107. Bagchi S, Raychaudhuri P. Damaged-DNA Binding Protein-2 Drives Apoptosis Following DNA Damage. Cell Div. 2010;5:3
108. Suzuki T, Tsuzuku J, Hayashi A. et al. Inhibition of DNA damage-induced apoptosis through Cdc7-mediated stabilization of Tob. J Biol Chem. 2012;287:40256-65
109. Chen Z, Shen BL, Fu QG. et al. CUL4B promotes proliferation and inhibits apoptosis of human osteosarcoma cells. Oncol Rep. 2014;32:2047-53
110. Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13:342-55
111. Liakopoulos D, Busgen T, Brychzy A. et al. Conjugation of the ubiquitin-like protein NEDD8 to cullin-2 is linked to von Hippel-Lindau tumor suppressor function. Proc Natl Acad Sci U S A. 1999;96:5510-5
112. Curtis VF, Ehrentraut SF, Campbell EL. et al. Stabilization of HIF through inhibition of Cullin-2 neddylation is protective in mucosal inflammatory responses. FASEB J. 2014 doi:fj.14-259663
113. Yuan WC, Lee YR, Huang SF. et al. A Cullin3-KLHL20 Ubiquitin ligase-dependent pathway targets PML to potentiate HIF-1 signaling and prostate cancer progression. Cancer Cell. 2011;20:214-28
114. Tan M, Gu Q, He H. et al. SAG/ROC2/RBX2 is a HIF-1 target gene that promotes HIF-1 alpha ubiquitination and degradation. Oncogene. 2008;27:1404-11
115. Sosa V, Moline T, Somoza R. et al. Oxidative stress and cancer: an overview. Ageing Res Rev. 2013;12:376-90
116. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401-26
117. Loignon M, Miao W, Hu L. et al. Cul3 overexpression depletes Nrf2 in breast cancer and is associated with sensitivity to carcinogens, to oxidative stress, and to chemotherapy. Mol Cancer Ther. 2009;8:2432-40
118. Nair SS, Kumar R. Chromatin remodeling in cancer: a gateway to regulate gene transcription. Mol Oncol. 2012;6:611-9
119. Dai Q, Wang H. Cullin 4 makes its mark on chromatin. Cell Div. 2006;1:14
Author contact
![]() Corresponding author: caiguo.zhangedu
Corresponding author: caiguo.zhangedu