Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(8):671-678. doi:10.7150/jca.7118 This issue Cite
Research Paper
Aspirin Blocks EGF-stimulated Cell Viability in a COX-1 Dependent Manner in Ovarian Cancer Cells
May Cho1, Syeda M. Kabir2, Yuanlin Dong2, Eunsook Lee3, Valerie Montgomery Rice4, Dineo Khabele5, Deok-Soo Son2 ![]()
1. Department of Internal Medicine, Barnes-Jewish hospital, Saint Louis, MO 63110, USA.
2. Department of Biochemistry and Cancer Biology, Meharry Medical College, Nashville, TN 37208, USA.
3. Department of Physiology, Meharry Medical College, Nashville, TN 37208, USA.
4. Department of Medicine, Morehouse College of Medicine, Atlanta, GA 30033-4097, USA.
5. Department of Obstetrics and Gynecology, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Received 2013-7-8; Accepted 2013-9-20; Published 2013-9-27
Abstract
Objective: Although aspirin has been associated with a reduction of the risk of cancer when used as a nonsteroidal anti-inflammatory drug, its use to reduce the risk of ovarian cancer is controversial. Ovarian cancer cells usually express high levels of cyclooxygenase-1 (COX)-1. Because aspirin is a rather selective inhibitor of COX-1, the ability of aspirin to reduce the risk of ovarian cancer may be dependent on the level of COX-1 expression in those cells. Furthermore, epidermal growth factor receptor (EGFR) is frequently overexpressed in the malignant phenotype of ovarian cancer leading to increased cell proliferation and survival. Here we investigated if aspirin attenuates EGFR-activated ovarian cancer cell growth in a COX-1 dependent manner.
Methods: Cell viability assays and Western blot analyses were used to determine the effect of aspirin on EGF-stimulated cell proliferation. Gene silencing and gene expression techniques were employed to knockdown or to express COX-1, respectively.
Results: Aspirin inhibited cell viability induced by EGF in a dose dependent manner in COX-1 positive ovarian cancer cells. On the other hand, aspirin had no effect on cell viability in COX-1 negative ovarian cancer cells. In particular, aspirin decreased phosphorylated Akt and Erk activated by EGF. COX-1 silencing in COX-1 positive cells attenuated the inhibitory effect of aspirin on EGF-stimulated cell viability. Furthermore, we developed a COX-1 expressing cell line (SKCOX-1) by stably transfecting COX-1 expression vector into COX-1 negative SKOV-3 cells. SKCOX-1 cells were more responsive to aspirin when compared to cells transfected with empty vector, and decreased EGF-activated Akt and Erk as well as cell viability.
Conclusions: Taken together, aspirin inhibits viability of ovarian cancer cells by blocking phosphorylation of Akt and Erk activated by EGF. Thus it may potentiate the therapeutic efficacy of drugs used to treat COX-1 positive ovarian cancer subsets.
Keywords: ovarian cancer, aspirin, COX-1, EGF, Erk, Akt, cell viability.
Introduction
Ovarian cancer is the deadliest gynecologic malignancy and is the fifth cause of cancer death in women in the United States [1, 2]. Ovarian cancer typically is asymptomatic until tumors have spread far beyond the ovaries. Unfortunately to date, there are no reliable methods to detect early stage disease. Thus, it is not surprising that the 5-year survival rate for ovarian cancer has not changed much over the past several decades despite the introduction of intensive surgical treatments and advances in the use of novel therapeutic agents [2]. Therefore new diagnostic biomarkers and therapeutic options are needed to reduce the morbidity and mortality observed with advanced stage ovarian cancer.
Chronic inflammation has been proposed as a risk factor for ovarian cancer [3, 4]. Aspirin (acetylsalicylate) is one of the most commonly used nonsteroidal anti-inflammatory drugs in the United States [5], and its use has increased significantly over the last 5 years [6]. Although the accumulated evidence shows that aspirin use is associated with a reduced risk of prostate [7, 8], breast [9], colorectal [9, 10] and endometrial cancer [11], the relationship between aspirin and ovarian cancer risk remains controversial. Some investigators have found no association [9, 12-14] while others reported an inverse association between the use of aspirin and ovarian cancer [15-17]. Our group and others have demonstrated that most epithelial ovarian cancer cells express high levels of cyclooxygenase-1 (COX)-1 rather than COX-2 [18, 19]. Therefore, COX-1 could be a potential therapeutic target for the prevention and/or treatment of ovarian cancer [20, 21].
Aspirin is a relatively selective COX-1 inhibitor [5] and has been shown to suppress cell growth in COX-1 expressing ovarian cancer cells [18-20, 22]. Our group has shown in a previous study that aspirin potentiates the effectiveness of histone deacetylase inhibitors by upregulating cell cycle arrest protein p21 in COX-1 positive ovarian cancer cells, but not in COX-1 negative cells [23]. These findings support the concept that the effectiveness of aspirin may be linked to the degree of COX-1 expression in ovarian cancer cells.
Ovarian cancer has been associated with many genetic and epigenetic changes that occur during the progression from a benign to a malignant stage. One of the alterations in high-grade malignant ovarian cancer is overexpression of the epidermal growth factor receptor (EGFR) [24, 25]. EGFR inhibitors are potentially useful therapeutic agents in patients with advanced or recurrent ovarian cancers [26-28]. However, clinical trials have been disappointing. Here we asked if modulating COX-1 could be a method for improving upon current EGFR targeted therapy. To our knowledge, little is known about the effects of aspirin on EGFR signaling in COX-1 abundant ovarian cancer cells. In the present study, we demonstrated that aspirin attenuates EGFR-activated ovarian cancer cell viability in a COX-1 dependent manner. These results have important implications for defining COX-1 positive ovarian cancer subsets and considering aspirin for enhancing the therapeutic efficacy of EGFR inhibitors.
Materials and Methods
Reagents
Aspirin (acetylsalicylate) was purchased from Sigma (St. Louis, MO). Recombinant human EGF was obtained from R&D Systems (Minneapolis, MN). Antibodies for COX-1, COX-2 and β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and antibodies for Erk, p38, SAPK/JNK, Akt, EGFR and their phosphorylated forms were obtained from Cell Signaling Technology (Danvers, MA). Chemiluminescent detection kits came from GE Healthcare. All liquid culture media were acquired from Invitrogen Life Technologies (Grand Island, NY). Pre-designed siRNAs for nontargeting controls and COX-1 experiments were purchased from Dharmacon (Lafayette, CO).
Cell culture
The human ovarian cancer cell lines (OVCAR-3, SKOV-3, TOV-21G and CaOV-3) and human THP-1 monocyte line were purchased from the American Type Culture Collection and the A2780 human ovarian cancer cell line was provided by Dr. Andrew Godwin (University of Kansas, Kansas City, KS). Cells (approximately 5 X 104 cells/ml) were cultured at 37°C in a water-saturated atmosphere of 95% air and 5% CO2 with 10% FBS containing RPMI medium with penicillin/streptomycin. After an overnight culture to allow cellular attachment to the plates, the medium was removed and fresh medium without FBS was added to remove the effects of serum, per se. Where indicated, cells were treated with aspirin, EGF, or a combination of both, as detailed under Results.
Cell viability assays
Cell viability assays were performed using the cleavage of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to a colored product. After a 48 hour incubation in a 24-well plate, each well was washed twice with phenol red-free media and then MTT solution (1 mg/ml of phenol red-free media: PBS = 4:1) was added. The plates were incubated for 3 hours with protection from light. The MTT solution was removed and 500 µl of isopropanol was added. The plates were placed on a shaker for 10 minutes at room temperature to thoroughly dissolve the MTT color product. Optical density was measured at 595 nm using a microplate reader (Bio-Rad, Hercules, CA).
Western blot
Whole-cell lysates were prepared, fractionated on SDS-polyacrylamide gels and transferred to nitrocellulose membranes according to established procedures. Blocking of nonspecific proteins was performed by incubation of the membranes with 5% nonfat dry milk in Tris-buffered saline Tween-20 (TBST containing 10 mM Tris, 150 mM phosphate buffered saline, 0.05% Tween 20, pH 8.0) for 2 hours at room temperature. The following primary antibodies were used: COX-1 and COX-2 (Santa Cruz Biotechnology, Santa Cruz, CA) and EGFR, Erk, p38, SAPK/JNK, Akt and their phosphorylated forms (Cell Signaling Technology, Beverly, MA). After an overnight incubation at 4ºC, the membranes were washed 3 times with TBST for 10 minutes, followed by incubation for 1 hour with horseradish peroxidase conjugated secondary antibody according to each primary antibody. The membranes were then rinsed 3 times with TBST for 10 minutes, and the bands were visualized by enhanced chemiluminescence detection kits from GE Healthcare (Piscataway, NJ). After membrane stripping for 10 min with methanol containing 3% H2O2, β-actin (Santa Cruz Biotechnology, Santa Cruz, CA) was detected in order to serve as an internal loading control of cell lysates.
Gene silencing with small interfering RNA transfection
Gene expression level of COX-1 was selectively knocked down by using pre-designed siRNAs (ON-TARGET plus Duplex PTGS1) obtained from Dharmacon (Lafayette, CO). A non-targeting siRNA (NT1) was used as a negative control. OVCAR-3 cells were transiently transfected with 10 nmol/L siRNA (final concentration of maximum knock-down) for 72 hours using the transfection reagent. After adding new media, cells were treated as described in Results and the cell proliferation assay was performed 48 hours after addition of EGF. Western blots were performed to validate the silencing of protein expression.
Generation of a COX-1 stable ovarian cancer cell line
COX-1 null SKOV-3 cells were stably transfected with a pcDNA based COX-1 expression vector which was a kind gift from Dr. S.K. Dey (Cincinnati Children's Hospital Medical Center, Cincinnati, OH) followed by G418 (Sigma) selection. Cell transfection was performed following the protocol of Lipofectamine 2000 (Invitrogen). The empty vector transfected cells were used as controls (SKpcDNA cells). After 12 days, resistant colonies were selected to confirm COX-1 expression (SKCOX-1 cells).
Statistics
Each experiment was repeated at least in triplicate. Data were analyzed by the paired Student's t-test and one-way analysis of variance (ANOVA) as appropriate. If statistical significance (p ≤ 0.05) was determined by ANOVA, the data were further analyzed by Tukey's pairwise comparisons to detect specific differences between treatments.
Results
Aspirin inhibits COX-1 positive ovarian cancer cell viability but not in COX-1 negative cells
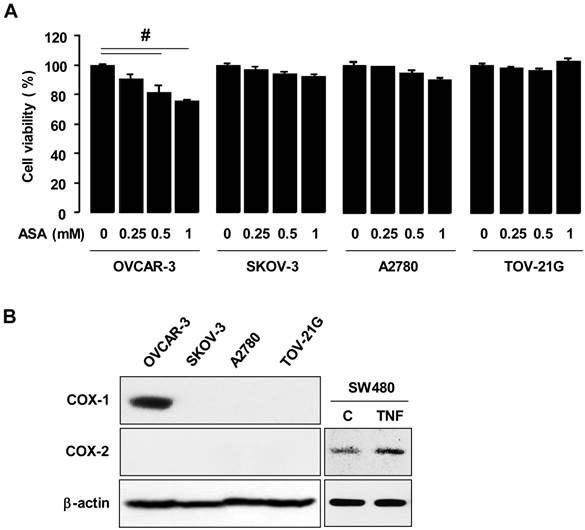
We employed the ovarian cancer cell lines OVCAR-3, SKOV-3, A2780 and TOV-21G to examine effects of aspirin on cell proliferation. We selected 1 mmol/L of aspirin as a maximum concentration for treatment, based on the mean maximum concentration 10.4-12.2 mg/dL at 180 minutes after dosing humans with 1,950 mg of aspirin per mean weight of 73.3 kg [29]. Furthermore, we found that a higher concentration than 1 mmol/L of aspirin was not suitable for cell incubations because of a resultant pH change in the culture media. Results indicated that aspirin inhibited cell viability in OVCAR-3 cells in a dose-dependent manner but not in SKOV-3, A2780 and TOV-21G cells (figure 1A). Since aspirin is a relatively selective COX-1 inhibitor compared to COX-2 [5], the expression status of COX-1 and COX-2 proteins were compared in all ovarian cancer cells tested. Only OVCAR-3 cells expressed COX-1 whereas other cancer cells did not (figure 1B), as previously published [23]. COX-2 was not expressed in any of the ovarian cancer cells tested (figure 1B).
Aspirin differentially regulates cell viability in COX-1 positive versus negative ovarian cancer cells. (A) Effects of aspirin on cellular proliferation in ovarian cancer cell lines OVCAR-3, SKOV-3, A2780 and TOV-21G. Cells were treated for 48 hours with aspirin (0.25, 0.5 and 1 mmol/L) in a dose-dependent manner. The cell viability assay was performed by using MTT, and values were normalized to untreated controls. Experiments were performed in triplicate and all data are shown as mean ± SE. # Indicates a significant decrease (p≤0.05) using ANOVA and Tukey's pairwise analyses. (B) COX-1 and COX-2 protein expressions were evaluated in ovarian cancer cell lines by western blot. β-actin was used as a loading control. As a positive control of COX-2, SW480 cells were used at 30 minute TNF (10 ng/ml) post-treatment.
Aspirin inhibits EGFR-activated cell viability in COX-1 positive ovarian cancer cells
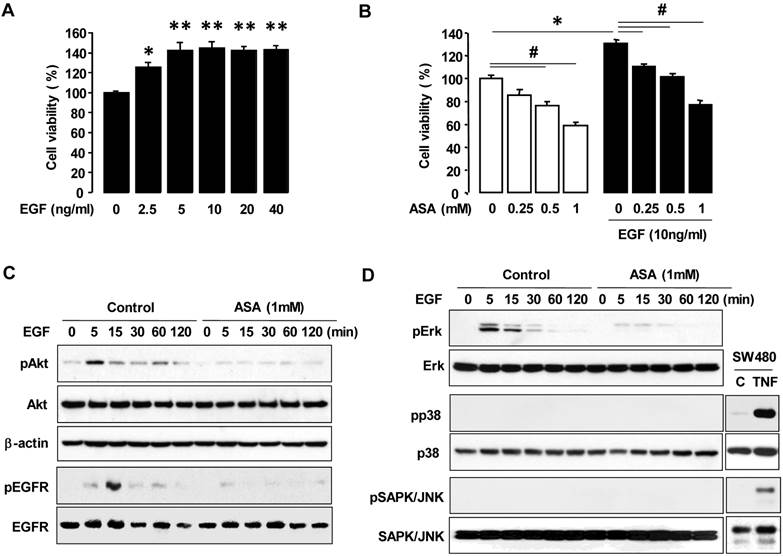
High-grade malignant ovarian cancers frequently overexpress EGFR, which contributes to cellular proliferation, survival and motility [24, 25, 30]. First, we checked a dose-dependent effect of EGF on cell viability in OVCAR-3 cells. EGF increased maximally cell viability at 5 ng/ml in OVCAR-3 cells (figure 2A). So we used 10 ng of EGF/ml of medium as a final concentration and examined if aspirin affected EGFR-activated ovarian cancer cell viability. EGF increased cell viability in OVCAR-3 cells and aspirin blocked the EGFR-activated cell viability in a dose-dependent manner (figure 2B). We also investigated the effect of aspirin on the PI3K/Akt pathway which is also known as a primary downstream signaling pathway linked to EGFR activation. EGF induced Akt phosphorylation and aspirin inhibited EGFR-activated Akt in OVCAR-3 cells (figure 2C). Furthermore, EGF activated Erk within 5-15 minutes and pre-addition of aspirin inhibited the EGFR-activated Erk in OVCAR-3 cells. However, EGF did not activate p38 MAPK or SAPK/JNK (figure 2D). EGFR activation, upstream of Akt and Erk signaling, also was inhibited by aspirin (figure 2C).
ASA decreases EGFR-activated cell viability by blocking phosphorylation of Erk and Akt. (A) A dose-dependant effect of EGF on cell viability in COX-1 positive OVCAR-3 cells. Cells were treated for 48 hours with EGF (0-40 ng/ml). *, ** Indicates a significant increase (p≤0.05) using ANOVA and Tukey's pairwise analyses. (B) Effects of aspirin on EGFR-activated cell viability in COX-1 positive OVCAR-3 cells. Cells were treated for 48 hours with aspirin (0.25, 0.5 and 1 mmol/L) in the absence or presence of EGF (10 ng/ml). The cell proliferation assay was performed using MTT, and values were normalized to untreated controls. Experiments were performed in triplicate and all data are shown as means ± SE. *, # Indicate a significant increase or decrease (p≤0.05), respectively, by Student's-t test. The inhibitory effect of aspirin on EGFR, Akt (C) Erk (D) activation in OVCAR-3 cells by Western blot. Cells were pretreated with aspirin (1 mmol/L) for 24 hours followed by EGF (10 ng/ml) treatment for 0-120 minutes as shown. pEGFR (tyr1068), pAkt, pErk, pp38 and pSAPK/JNK indicate phosphorylated EGFR, Akt, Erk, p38, and SAPK/JNK, respectively. β-Actin was used as a loading control. As positive controls for phosphorylated p38 and SAPK/JNK, SW480 cells were used at 30 minute TNF (10 ng/ml) post-treatment.
Silencing COX-1 abrogates inhibitory effect of aspirin on cell viability in OVCAR-3 cells
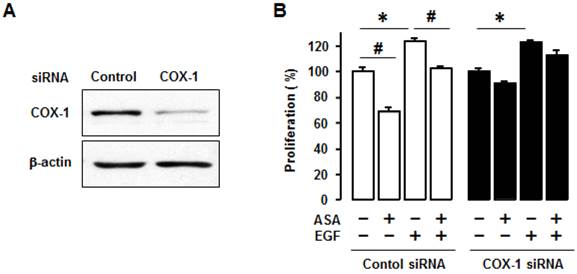
We used COX-1 siRNA to knockdown COX-1 to help determine whether COX-1 activity, per se, contributed to the inhibitory effect of aspirin on cell viability. We first confirmed COX-1 knockdown by COX-1 siRNA (figure 3A). We found that COX-1 knockdown blocked the inhibitory effect of aspirin on both basal and EGF-induced cell viability in OVCAR-3 cells when compared to control siRNA (figure 3B).
Silencing COX-1 with a small interfering RNA blocks inhibitory effect of aspirin on cell viability in OVCAR-3 cells. (A) Confirmation of COX-1 knockdown in OVCAR-3 cells by using COX-1 siRNA. Whole cell lysates were prepared and a western blot was carried out using COX-1 specific antibody. β-Actin was used as a loading control. (B) Effects of COX-1 siRNA on the inhibitory effect of aspirin on basal and EGF-stimulated cell viability in OVCAR-3 cells. Cells were transiently transfected with Control or COX-1 siRNAs (final concentration 10 nmol/L) for 48 hours followed by treatment for 48 hours with EGF (10 ng/mL). A cell viability assay was performed using MTT and values were normalized to untreated controls. Experiments were performed in triplicate and all data are shown as means ± SE. *, # Indicate a significant increase or decrease (p≤0.05), respectively, by Student's-t test.
COX-1 expressing cells contribute to cell viability when compared to COX-1 null cells
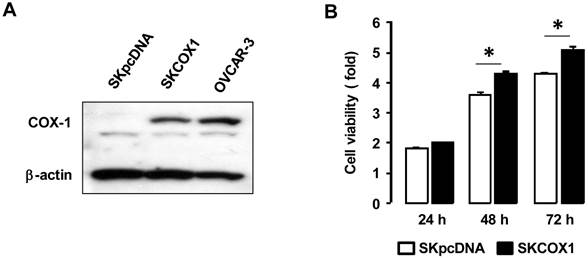
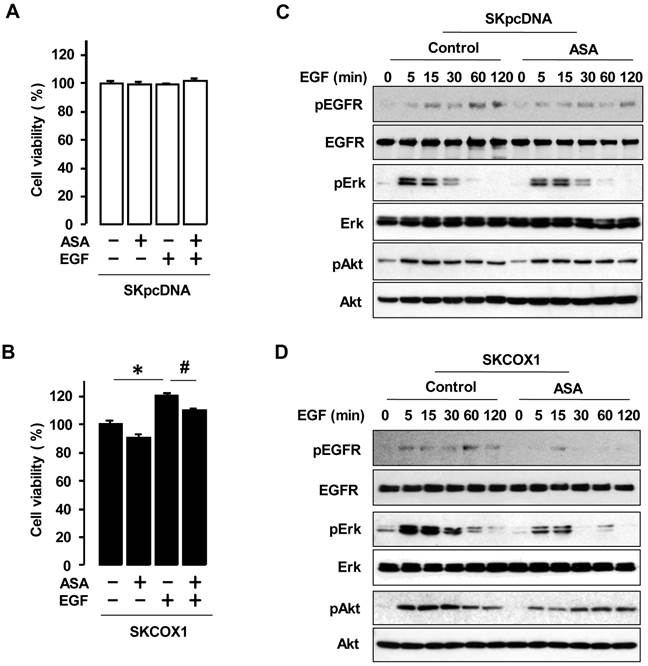
We made COX-1 expressing cells by stably transfecting COX-1 expression vector into the COX-1 null parental SKOV-3 cell line to produce SKCOX-1 (COX-1 vector) cells. SkpcDNA (empty vector) transfected cells served as a control. We confirmed COX-1 expression in SKCOX-1 cells (figure 4A). We then compared cell viability in SkpcDNA versus SKCOX-1 cells. Cell viability was significantly greater in COX-1 expressing SKCOX-1 cells compared to COX-1 null SKpcDNA cells (figure 4B) when examined after 48 and 72 hours of incubation (figure 4B).
COX-1 contributes to cell viability in ovarian cancer cells. (A) Confirmation of COX-1 expression in COX-1 stable (SKCOX1) transfected cells. Whole cell lysates were prepared from each cell line and a western blot was carried out using a COX-1 specific antibody. β-Actin was used as a loading control; OVCAR-3 cells served as COX-1 positive cell line control. (B) Comparison of cell viability in COX-1 null SKpcDNA and positive SKCOX-1 cells. Experiments were performed in triplicate and all data are shown as means ± SE. * Indicates a significant (p≤0.05) increase in SKCOX-1 cells compared to SKpcDNA cells when a Student's-t test was made.
Overexpression of COX-1 increases cell viability in response to EGF and aspirin blocks the response
We examined whether COX-1 contributes to EGF responsiveness by comparing the effects of EGF in SKpcDNA and SKCOX-1 cells. EGF significantly increased cell viability in SKCOX-1 cells but not in SKpcDNA cells (figure 5A and 5B). In addition, aspirin inhibited EGFR-activated cell viability only in SKCOX-1 cells (figure 5B). Furthermore, EGF activated EGFR, Erk and Akt in both SKpcDNA and SKCOX-1 cells, but aspirin inhibited EGFR, Erk and Akt activation in SKCOX-1 cells but not in SKpcDNA cells (figure 5C and 5D).
ASA blocks EGFR-activated cell viability by blocking phosphorylation of Erk and Akt in COX-1 expressing cells. Effects of aspirin on EGFR-activated cell viability in SKpcDNA (A) and SKCOX-1 cells (B). Cells were treated for 48 hours with aspirin (1 mmol/L) in the absence or presence of EGF (10 ng/ml). The cell viability assay was performed by using MTT, and values were normalized to untreated controls. Experiments were performed in triplicate and all data are shown as means ± SE. *, # Indicate a significant increase or decrease (p≤0.05), respectively, by Student's-t test. The inhibitory effect of aspirin on EGFR, Erk and Akt activation in SKpcDNA (C) and SKCOX-1 cells (D) by western blot analysis. Cells were pretreated with aspirin (1 mmol/L) for 24 hours followed by the addition of EGF (10 ng/ml) for 0-120 minutes. pEGFR, pErk and pAkt indicate phosphorylated EGFR, Erk and Akt, respectively. β-Actin was used as a loading control.
Discussion
One of the primary findings in this study is that aspirin inhibits EGFR-activated cell viability in a COX-1 dependent manner by blocking phosphorylation of Akt and Erk in COX-1 positive ovarian cancer cells. Reportedly, some authors found that ovarian cancer cells express higher levels of COX-1 than COX-2 [18, 19] while others report a similar expression of COX-1 (59.9%) and COX-2 (60.3%) in epithelial ovarian cancer cells [31]. In our studies, OVCAR-3 cells expressed COX-1 while the other three cell lines (SKOV-3, A2780 and TOV-21G) did not. Aspirin inhibited cell viability in COX-1 expressing OVCAR-3 cells (figure 1A) as observed by others using COX-1 expressing ovarian cancer cells [18-20, 22]. Since the relationship between aspirin use and the risk ovarian cancer is controversial [9, 12-17], these findings suggest that the status of COX-1 expression in tumor subtypes needs to be considered in order to judge whether or not aspirin will be effective.
As malignant ovarian cancer cells express higher levels of EGFR when compared to nonmalignant type cells [24, 25], targeting EGFR could be a useful approach to treating ovarian cancer. Because aspirin inhibited EGF-stimulated cell viability in COX-1 expressing OVCAR-3 cells (figure 2A), our findings suggest an interaction between an EGFR-activated signaling pathway and COX-1. We found that aspirin blocked EGFR downstream Erk and Akt activation (figure 2) which is consistent in part with other reports about the inhibitory effect of aspirin on tumor cell growth by inhibiting Erk [22] and by down-regulating Akt activity [31]. In addition, aspirin did not attenuate EGF-stimulated cell proliferation in COX-1 knockdown cells (figure 3), indicating the involvement of COX-1. Overexpression of COX-1 accelerated cell viability (figure 4) suggesting it too may be a potential target for the prevention and treatment of ovarian cancers expressing this enzyme [20]. Furthermore, transfecting a COX-1 null ovarian cancer cell line with COX-1 increased the level of cell viability in response to EGF (figure 5A and 5B).
Although further studies are required to clarify the relationship between EGFR signaling pathways and COX-1 in ovarian cancer, here we show that aspirin blocked EGF-stimulated Akt and Erk activation in COX-1 stable transfected cells (figure 5C and 5D). This fact indicates a COX-1-dependent effect of aspirin rather than a direct effect of aspirin on EGFR-activated signaling. As aspirin potentiated the effects of a histone deacetylase inhibitor in COX-1 positive ovarian cancer cells [23], it may also potentiate the efficacy of EGFR inhibitors. Although sorafenib, a tyrosine kinase inhibitor, has become the first-line therapy for advanced hepatocellular carcinoma, it also promotes the invasiveness and metastasis of hepatocellular carcinoma [32]. Interestingly, aspirin minimizes the pro-metastasis effect of sorafenib in hepatocellular carcinoma, resulting in improved survival in a mouse model [33]. Therefore, while aspirin cannot be the primary medication to treat cancer, when COX-1 is involved, aspirin could be used as a supportive medication to enhance EGFR based therapy.
In conclusion, aspirin inhibits EGFR-activated cell viability by blocking Akt and Erk activation in a COX-1 dependent manner, probably leading to potentiate the efficacy of chemotherapy treatments particularly in COX-1 positive ovarian cancer subsets.
Acknowledgements
The authors are grateful to Dr. Andrew Godwin (University of Kansas, Kansas City, KS) for A2780 human ovarian cancer cell line and Dr. Sudhansu K. Dey (Cincinnati Children's Hospital Medical Center, Cincinnati, OH) for COX-1 expression vector. We thank Dr. Diana Marver (Meharry Medical College, Nashville, TN) for her editorial suggestions. This research was supported by NIH grants U54CA091408 (DK & DS), G12RR003032 (DK & DS) and SC1AI089073 (DS).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chobanian N, Dietrich CS. Ovarian cancer. Surg Clin North Am. 2008;88:285-99
2. Sudo T. Molecular-targeted therapies for ovarian cancer: prospects for the future. Int J Clin Oncol. 2012;17:424-9
3. Macciò A, Madeddu C. Inflammation and ovarian cancer. Cytokine. 2012;58:133-47
4. Pejovic T, Nezhat F. Missing link: inflammation and ovarian cancer. Lancet Oncol. 2011;12:833-4
5. Mitchell JA, Akarasereenont P, Thiemermann C. et al. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci USA. 1993;90:11693-7
6. Zhou Y, Boudreau DM, Freedman AN. Trends in the use of aspirin and nonsteroidal anti-inflammatory drugs in the general U.S. population. Pharmacoepidemiol Drug Saf. 2013; doi: 10.1002/pds.3463. [Epub ahead of print].
7. Shebl FM, Sakoda LC, Black A. et al. Aspirin but not ibuprofen use is associated with reduced risk of prostate cancer: a PLCO study. Br J Cancer. 2012;107:207-14
8. Veitonmäki T, Tammela TL, Auvinen A. et al. Use of aspirin, but not other non-steroidal anti-inflammatory drugs is associated with decreased prostate cancer risk at the population level. Eur J Cancer. 2013;49:938-45
9. Bosetti C, Rosato V, Gallus S. et al. Aspirin and cancer risk: a quantitative review to 2011. Ann Oncol. 2012;23:1403-15
10. Johnson CC, Hayes RB, Schoen RE. et al. Non-steroidal anti-inflammatory drug use and colorectal polyps in the Prostate, Lung, Colorectal, And Ovarian Cancer Screening Trial. Am J Gastroenterol. 2010;105:2646-55
11. Neill AS, Nagle CM, Protani MM. et al. Aspirin, nonsteroidal anti-inflammatory drugs, paracetamol and risk of endometrial cancer: A case-control study, systematic review and meta-analysis. Int J Cancer. 2013;132:1146-55
12. Murphy MA, Trabert B, Yang HP. et al. Non-steroidal anti-inflammatory drug use and ovarian cancer risk: findings from the NIH-AARP Diet and Health Study and systematic review. Cancer Causes Control. 2012;23:1839-52
13. Ni X, Ma J, Zhao Y. et al. Meta-analysis on the association between non-steroidal anti-inflammatory drug use and ovarian cancer. Br J Clin Pharmacol. 2013;75:26-35
14. Setiawan VW, Matsuno RK, Lurie G. et al. Use of nonsteroidal anti-inflammatory drugs and risk of ovarian and endometrial cancer: The Multiethnic Cohort. Cancer Epidemiol Biomarkers Prev. 2012;21:1441-9
15. Ammundsen HB, Faber MT, Jensen A. et al. Use of analgesic drugs and risk of ovarian cancer: results from a Danish case-control study. Acta Obstet Gynecol Scand. 2012;91:1094-102
16. Lo-Ciganic WH, Zgibor JC, Bunker CH. et al. Aspirin, nonaspirin nonsteroidal anti-inflammatory drugs, or acetaminophen and risk of ovarian cancer. Epidemiology. 2012;23:311-9
17. Prizment AE, Folsom AR, Anderson KE. Nonsteroidal anti-inflammatory drugs and risk for ovarian and endometrial cancers in the Iowa Women's Health Study. Cancer Epidemiol Biomarkers Prev. 2010;19:435-42
18. Gupta RA, Tejada LV, Tong BJ. et al. Cyclooxygenase-1 is overexpressed and promotes angiogenic growth factor production in ovarian cancer. Cancer Res. 2003;63:906-11
19. Daikoku T, Tranguch S, Trofimova IN. et al. Cyclooxygenase-1 is overexpressed in multiple genetically engineered mouse models of epithelial ovarian cancer. Cancer Res. 2006;66:2527-31
20. Daikoku T, Wang D, Tranguch S. et al. Cyclooxygenase-1 is a potential target for prevention and treatment of ovarian epithelial cancer. Cancer Res. 2005;65:3735-44
21. Li W, Ji ZL, Zhuo GC. et al. Effects of a selective cyclooxygenase-1 inhibitor in SKOV-3 ovarian carcinoma xenograft-bearing mice. Med Oncol. 2010;27:98-104
22. Daikoku T, Tranguch S, Chakrabarty A. et al. Extracellular signal-regulated kinase is a target of cyclooxygenase-1-peroxisome proliferator-activated receptor-delta signaling in epithelial ovarian cancer. Cancer Res. 2007;67:5285-92
23. Son DS, Wilson AJ, Parl AK. et al. The effects of the histone deacetylase inhibitor romidepsin (FK228) are enhanced by aspirin (ASA) in COX-1 positive ovarian cancer cells through augmentation of p21. Cancer Biol Ther. 2010;9:928-35
24. Normanno N, De Luca A, Bianco C. et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2-16
25. Landen CN Jr, Birrer MJ, Sood AK. Early events in the pathogenesis of epithelial ovarian cancer. J Clin Oncol. 2008;26:995-1005
26. Palayekar MJ, Herzog TJ. The emerging role of epidermal growth factor receptor inhibitors in ovarian cancer. Int J Gynecol Cancer. 2008;18:879-90
27. Gui T, Shen K. The epidermal growth factor receptor as a therapeutic target in epithelial ovarian cancer. Cancer Epidemiol. 2012;36:490-6
28. Wilken JA, Badri T, Cross S. et al. EGFR/HER-targeted therapeutics in ovarian cancer. Future Med Chem. 2012;4:447-69
29. Nordt SP, Clark RF, Castillo EM. et al. Comparison of three aspirin formulations in human volunteers. West J Emerg Med. 2011;12:381-5
30. Siwak DR, Carey M, Hennessy BT. et al. Targeting the epidermal growth factor receptor in epithelial ovarian cancer: current knowledge and future challenges. J Oncol. 2010;2010:568938
31. Uddin S, Ahmed M, Hussain A. et al. Cyclooxygenase-2 inhibition inhibits PI3K/AKT kinase activity in epithelial ovarian cancer. Int J Cancer. 2010;126:382-94
32. Zhang W, Sun HC, Wang WQ. et al. Sorafenib down-regulates expression of HTATIP2 to promote invasiveness and metastasis of orthotopic hepatocellular carcinoma tumors in mice. Gastroenterology. 2012;143:1641-9
33. Lu L, Sun HC, Zhang W. et al. Aspirin minimized the pro- metastasis effect of Sorafenib and improved survival by up-regulating HTATIP2 in hepatocellular carcinoma. PLoS One. 2013;8:e65023
Author contact
![]() Corresponding author: DS Son, Department of Biochemistry and Cancer Biology, Meharry Medical College, 1005 Dr DB Todd Jr Blvd, Nashville, TN 37208, USA, Tel: +1-615-327-6501, Fax: +1-615-327-6042, Email: dsonedu.
Corresponding author: DS Son, Department of Biochemistry and Cancer Biology, Meharry Medical College, 1005 Dr DB Todd Jr Blvd, Nashville, TN 37208, USA, Tel: +1-615-327-6501, Fax: +1-615-327-6042, Email: dsonedu.