Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(6):481-490. doi:10.7150/jca.6583 This issue Cite
Research Paper
CBP Activity Mediates Effects of the Histone Deacetylase Inhibitor Butyrate on WNT Activity and Apoptosis in Colon Cancer Cells
Darina L Lazarova1, Christopher Chiaro1, Terrence Wong1, Eric Drago1, Anthony Rainey1,2, Shannon O'Malley1,3, Michael Bordonaro1 ![]()
1. Department of Basic Sciences, The Commonwealth Medical College, 525 Pine Street, Scranton, PA 18509, USA
2. Marywood University, 2300 Adams Avenue, Scranton, PA 18509
3. The Pennsylvania State University, 201 Old Main, University Park, PA 16802
Received 2013-4-29; Accepted 2013-6-13; Published 2013-7-15
Abstract
Mutations in the WNT/beta-catenin pathway are responsible for initiating the majority of colorectal cancers (CRCs). We have previously shown that hyperactivation of this signaling by histone deacetylase inhibitors (HDACis) such as butyrate, a fermentation product of dietary fiber, promotes CRC cell apoptosis. The extent of association between beta-catenin and the transcriptional coactivator CREB-binding protein (CBP) influences WNT/catenin signaling and, therefore, colonic cell physiology. CBP functions as a histone acetylase (HAT); therefore, we hypothesized that the modulation of WNT/catenin activity by CBP modifies the ability of the HDACi butyrate to hyperinduce WNT signaling and apoptosis in CRC cells. Our findings indicate that CBP affects the hyperinduction of WNT activity by butyrate. ICG-001, which specifically blocks association between CBP and beta-catenin, abrogates the butyrate-triggered increase in the number of CRC cells with high levels of WNT/catenin signaling. Combination treatment of CRC cells with ICG-001 and butyrate results in cell type-specific effects on apoptosis. Further, both butyrate and ICG-001 repress CRC cell proliferation, with additive effects in suppressing cell growth. Our study strongly suggests that ICG-001-like agents would be effective against butyrate/HDACi-resistant CRC cells. Therefore, ICG-001-like agents may represent an important therapeutic option for CRCs that exhibit low-fold hyperactivation of WNT activity and apoptosis in the presence of HDACis. The findings generated from this study may lead to approaches that utilize modulation of CBP activity to facilitate CRC therapeutic or chemopreventive strategies.
Keywords: CBP, ICG-001, colorectal cancer, WNT, butyrate, fiber.
Introduction
Colorectal cancers (CRCs) are amenable to a high degree of prevention through diet (1-8). The protective effect of dietary fiber has been attributed to its fermentation product, butyrate, a histone deacetylase inhibitor (HDACi) (9,10). HDACis induce cell cycle arrest, differentiation, and/or apoptosis of CRC cells (11-16). In CRC cells with mutations in the WNT/beta-catenin (WNT/catenin) signaling pathway, butyrate hyper-activates WNT/catenin activity, and this hyperinduction is causally linked to the apoptosis of the CRC cells exposed to the agent (1,2,17).
In the absence of activating mutations, WNT/catenin signaling is initiated by the binding of WNT ligands to their cell surface receptors, resulting in inhibition of the proteosomal degradation of beta-catenin (18). Transcriptionally active (dephosphorylated) beta-catenin accumulates, interacts with LEF/TCF DNA-binding proteins (18-27), and the resulting complexes drive transcription from genes with LEF/TCF binding sites. This WNT/catenin transcriptional activity can be measured with WNT-sensitive reporter constructs (25,26). Constitutively active WNT/catenin signaling, supported by mutations in the adenomatous polyposis coli (APC) and beta-catenin genes (25-27), promotes colonic cell proliferation and tumorigenesis; whereas, relatively high and relatively low levels of WNT/catenin activity lead to CRC cell apoptosis (1,2,17,28,29). Therefore, WNT/catenin activity can be viewed as a gradient, within which absence of detectable WNT signaling results in terminal differentiation and apoptosis, relatively low levels maintain controlled self-renewal (as in stem cells), moderate levels promote uncontrolled cell proliferation, and relatively high levels of signaling lead to enhanced apoptosis.
The association between beta-catenin and the transcriptional coactivators CREB-binding protein (CBP) and p300 influences WNT signaling (30-35). CBP is a histone acetyl transferase (HAT) that has both positive and negative effects on WNT/catenin signaling in vertebrate cell lines (30-39). The interaction between CBP and WNT signaling can be dissected utilizing the small molecule inhibitor ICG-001, which binds to CBP but not to p300 (30). Treatment of CRC cells with ICG-001 suppresses the association between CBP and beta-catenin without blocking p300/beta-catenin association, and ICG-001 was shown to downregulate basal WNT/catenin reporter activity in CRC cells (30). ICG-001 inhibited CRC cell proliferation, and in SW480 CRC cells, ICG-001 increased caspase activity, indicative of enhanced apoptosis (30). The expression of survivin (an anti-apoptotic gene) is enhanced by CBP-WNT/catenin activity, an effect which is blocked by ICG-001 (33). In addition, a water soluble version of ICG-001 reduced the formation of intestinal neoplasms in the APCMin mouse model of CRC, demonstrating preliminary in vivo efficacy (30). An ICG-001 analog has advanced to stage one clinical trials (40), emphasizing the clinical relevance of studying the mechanisms of the agent and the interactions between CBP and WNT/catenin activity. These interactions likely affect the function of the HDACi butyrate on WNT/catenin activity and physiology of CRC cells.
HDACis, including butyrate, hyperinduce WNT/catenin activity and apoptosis in CRC cells (1,2,17,28,29). Both HDACis and HATs result in net acetylation of cellular proteins. Blocking CBP-mediated WNT/catenin signaling may suppress or enhance the effects of butyrate. Since treatment of CRCs with ICG-001 and its analogs affects the colon, where dietary fiber-derived butyrate is present at high levels (up to 10 mM), analyzing how ICG-001 and butyrate interact to influence the physiology of CRC cells is important.
In the present study, we demonstrate that the high levels of WNT/catenin activity induced by the HDACi butyrate are repressed by ICG-001, thus implicating CBP in WNT/catenin hyperactivation. Consistent with previous reports (1,2,17,28,29), apoptosis is influenced by the modulation of butyrate-induced WNT signaling; however, cotreatment with butyrate and ICG-001 has cell type-specific effects on CRC cell apoptosis. In conclusion, the modulation of WNT/catenin activity by butyrate, a fermentation product of dietary fiber, is dependent upon CBP, and the combination of butyrate and ICG-001-like agents promotes or interferes with apoptosis in a cell type-specific manner. Since synthetic HDACis that are currently in clinic mimic the effects of butyrate on WNT signaling and apoptosis (2), it is likely that the effects of a combination of clinically available HDACis and ICG-001 also depend upon the CRC cell phenotype, and its underlying mutations. Therefore, altering the levels of CBP-WNT activity in the context of diet-derived butyrate or other HDACis may lead to novel preventive and/or therapeutic approaches against CRC.
Materials and Methods
Reagents
Sodium butyrate was obtained from Sigma, ICG-001 was provided by Dr. Michael Kahn (USC, Los Angeles, CA); ICG-001 was also obtained from Selleckchem.
Cell lines, Plasmids, siRNA, Transfection, reagent treatment
Cell lines were obtained from the ATCC and cultured as described previously (1,2). The TOPFlash and FOPFlash vectors were described previously (1,2,25,26). HCT-116 and SW620 cells were transfected with the TOPFlash (wild-type) and FOPFlash (mutant) luciferase reporter vectors along with the normalization vector pRLTK (Promega) using Lipofectamine 2000 as previously described (1,2). Cells were treated with 5 mM sodium butyrate or mock-treated for 17.5 hr along with small molecule inhibitor ICG-001 (30), or DMSO vehicle at the indicated for each experiment concentrations. Cells were lysed and assayed for reporter expression by the Dual Luciferase system (Promega) as previously described (1,2).
Apoptosis
Apoptotic analyses were performed using the caspase 3/7 Caspase-Glo luciferase system (Promega) (30). 20,000 HCT-116 or SW620 cells/well in a 96 well plate were plated and the next day treated with 75 μM or 100 μM ICG-001, 5 mM sodium butyrate, both agents, or mock treated 24 hr, a time point which we have found yields optimal butyrate-induced apoptosis subsequent to WNT hyperactivation (1,2). The next day caspase 3/7 activity was measured with the Caspase Glo kit (Promega) according to manufacturer's instructions. Statistical analyses of caspase data was performed on background corrected raw data.
Coimmunoprecipitation
For CBP/p300-beta-catenin coimmunoprecipitations, HCT-116 or SW620 cells were incubated overnight (17.5 hr) with or without ICG-001, and nuclei were isolated with the Nuclei EZ Prep kit (Sigma). Nuclear pellets were lysed in SDS-containing buffer (1,2), and 100 μg nuclear protein extract was diluted to 1 mL in coimmunoprecipitation (Co-IP) buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% NP-40, and Sigma protease inhibitor cocktail). Two μg of either CBP (sc-369, Santa Cruz Biotechnology) or p300 (sc-584, Santa Cruz Biotechnology) antibody were added to the protein samples, and incubated overnight at 4oC with rotation. Fifty μL of a 50% protein A/G-agarose bead slurry (equilibrated in Co-IP buffer) were added, and after 1h-incubation at 4oC, the beads were washed three times (0.5 ml per wash) with Co-IP buffer and extracted with 30 μL of Laemmli buffer. Western blotting was performed, and total beta-catenin was detected using a mouse monoclonal antibody (sc-53483, Santa Cruz Biotechnology).
Western blot
Western blotting was performed as previously described (1,2), using the antibodies described above, as well antibodies for p21 (sc-6246, Santa Cruz Biotechnology), survivin (SC-17779, Santa Cruz Biotechnology), and Actin (A-5441, Sigma).
Proliferation assays and cell cycle analysis
Proliferation assays were performed with the QuickCell Proliferation kit (Biovision), according to manufacturer's instructions. Statistical analyses of proliferation data was performed on background corrected raw data. Cell cycle analysis was performed with propidium iodide treatment; for flow cytometry, the propidium iodide was excited at 488 nm, and its fluorescence collected between 620 and 650 nm.
Analysis of high/low Wnt cell fractions
HCT-116 cells were stably transfected with TOP-EGFP and FOP-EGFP (1), selected and maintained in G418. Flow cytometry analyses of cells into WNT/catenin-positive and WNT/catenin-negative fractions based on expression of enhanced green fluorescent protein, driven by WNT/beta-catenin dependent promoter has been described previously (1). Untransfected HCT-116 cells were used as EGFP-negative controls to set gating.
Statistics
Student's T-test was utilized to determine statistical significance (P < 0.05).
Results
Levels of CBP in mock- and butyrate-treated CRC cells
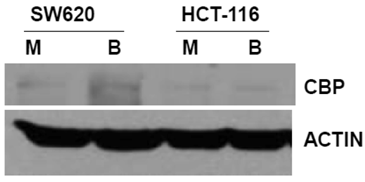
The primary objective of this study was to evaluate how ICG-001 abrogation of CBP/beta-catenin association modulates the hyperactivation of WNT/catenin activity and apoptosis in CRC cells exposed to butyrate. Therefore, we first established the steady-state levels of CBP in two commonly used CRC cell lines in the presence or absence of 5 mM butyrate, a physiologically relevant concentration which induces apoptosis in butyrate-sensitive CRC cells (1,2,17, 28,29) (Fig.1). We analyzed SW620 cells (APC mutant) and HCT-116 cells (beta-catenin mutant) cells. Immunoblot analyses revealed that the two cell lines express CBP at variable levels.
Expression of CBP in CRC cell lines. The indicated CRC cell lines were left untreated or treated overnight (17.5 hr) with 5 mM butyrate, total protein was isolated, and analyzed with western blot, with anti-CBP antibody. Actin was used as a loading control. Mock treated (M), butyrate treated (B). Representative data are shown.
Butyrate-mediated hyperactivation of WNT/catenin signaling, induction of apoptosis, and repression of cell proliferation are influenced by ICG-001
The effects of butyrate on WNT signaling and apoptosis may be mediated differently by the association of CBP with beta-catenin. First, we measured WNT/catenin transcriptional activity in cells treated with ICG-001, an inhibitor of the CBP/beta-catenin association (30), in the presence or absence of butyrate.
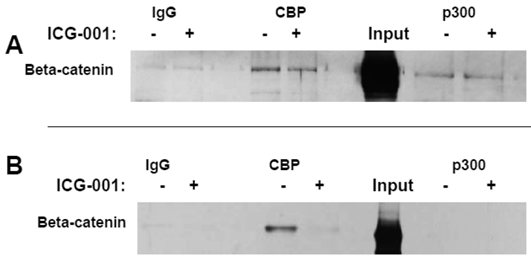
Titration experiments demonstrated that 17.5 hr treatment with 75 μM or 100 μM ICG-001 results in optimal inhibition of WNT signaling in HCT-116 cells or SW620 cells, respectively (data not shown). The concentrations of ICG-001 required for efficient knockdown of WNT/catenin activity in our study were somewhat higher than previously reported (30). Therefore, we performed coimmunoprecipitation analyses to evaluate CBP/beta-catenin and p300/beta-catenin association in HCT-116 and SW620 cells treated with 75 μM or 100 μM ICG-001. These experiments ascertained that these concentrations of ICG-001 disrupt CBP/beta-catenin, but not p300/beta-catenin, association, as previously reported with lower concentrations of this agent (30). As expected, treatment of HCT-116 cells with 75 μM ICG-001 decreased CBP/beta-catenin association without affecting p300/beta-catenin association (Fig. 2A). Treatment of SW620 cells with 100 μM ICG-001 also effectively abolished the CBP/beta-catenin association; however, in these cells we did not detect p300/beta-catenin association with or without treatment with ICG-001 (Fig.2B). These data confirm the ability of ICG-001 to specifically target CBP/beta-catenin association at the concentrations utilized.
Coimmunoprecipitation analysis of HCT-116 and SW620 cells treated with ICG-001. (A) Coimmunoprecipitation of HCT-116 CRC cells, performed as described in Materials and Methods, using 75 μM ICG-001. Anti-CBP or anti-p300 antibody was used for the immunoprecipitation and beta-catenin was detected with an anti-beta-catenin antibody after SDS-PAGE. (B) Coimmunoprecipitation assay, similar to that described in (A), showing activity of 100 μM ICG-001 against CBP-beta-catenin association in SW620 CRC cells. Representative coimmunoprecipitation data are shown.
HCT-116 and SW620 cells both exhibit enhanced WNT/catenin activity and apoptosis when exposed to a physiologically relevant concentration (5 mM) of butyrate (1,2). Therefore, to ascertain how CBP influences butyrate-mediated WNT hyperactivation, we treated these CRC cell lines with ICG-001 and 5 mM butyrate (Fig.3A). In agreement with previous findings (1,2), WNT/catenin activity was markedly enhanced by exposure to 5 mM butyrate (P<0.01). Treatment with ICG-001 alone reduced WNT/catenin activity compared to mock-treated HCT-116 cells (P<0.05). Butyrate/ICG-001 cotreatment resulted in low levels of WNT/catenin activity, markedly lower than that observed with butyrate alone (P < 0.01). However, butyrate/ICG-001 cotreatment resulted in a 2.9-fold enhancement of WNT/catenin activity compared to ICG-001 alone (P<0.005), demonstrating that butyrate retains the ability to upregulate WNT signaling in the presence of ICG-001.
Effects of ICG-001 on butyrate-mediated WNT signaling and apoptosis in CRC cells. (A) ICG-001 inhibits butyrate-induced WNT activity in HCT-116 cells. HCT-116 cells were transfected with TOP/FOPFlash reporter vectors and with pRLTK for normalization of transfection efficiency. After 5 hours cells were mock treated (M) or treated with 75 μM ICG-001 (I), 5 mM sodium butyrate (B) or both agents (IB) 17.5 hr. WNT signaling measured by the ratio of luciferase expression from TOPFlash (T) to FOPFlash (F) is shown (B) ICG-001 inhibits butyrate-induced WNT activity in SW620 cells. Experiment was performed as in (A). (C) Butyrate potentiates apoptosis mediated by ICG-001 in HCT-116 cells. The fold induction of caspase activity compared to mock treated control is shown for butyrate-treated (B), ICG-001 treated (I) and cells cotreated with both reagents (IB). (D) ICG-001 interferes with butyrate-induced apoptosis in SW620 cells. Experiment was performed as in (C). For (A)-(D), data are for three separate experiments; Bars, SDs; * = statistical significance.
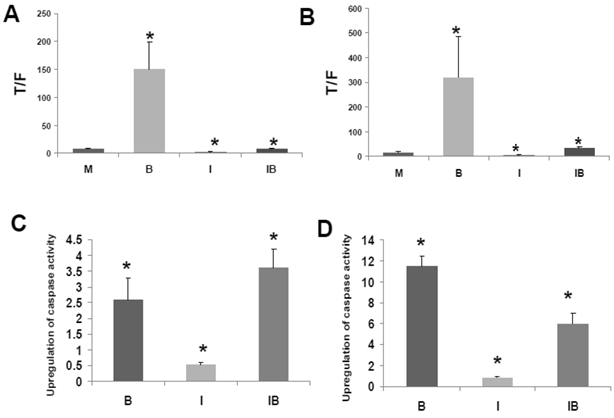
Similar to HCT-116 cells, SW620 CRC cells exhibit an upregulation of WNT/catenin activity and apoptosis in the presence of butyrate (1). ICG-001 was used to ascertain effects of CBP on the ability of butyrate to hyperactivate WNT/catenin signaling in SW620 cells (Fig. 3B). Consistent with previous findings (1,2), WNT/catenin activity in SW620 cells was enhanced by exposure to 5 mM butyrate (P<0.02). Treatment with ICG-001 alone reduced WNT/catenin activity compared to mock-treated cells (P < 0.001). Butyrate/ICG-001 cotreatment resulted in levels of WNT/catenin activity, significantly lower than what was observed with butyrate alone (P < 0.02). However, cotreatment resulted in enhanced WNT/catenin activity compared to ICG-001 alone (P < 0.001). The findings from both cell lines indicate that (a) CBP activity is required for the high levels of WNT signaling observed in butyrate-treated HCT-116 and SW620 CRC cells, and (b) although ICG-001 sharply reduces WNT/catenin signaling in these cells, this repressed activity is still upregulated by cotreatment with butyrate.
The clinical use of ICG-001-like agents against CRC (40) may be influenced by dietary factors. For example, dietary fiber is fermented to butyrate, which modulates WNT/catenin activity in a manner opposite of that of ICG-001. Conversely, ICG-001, by repressing WNT/catenin hyperactivation, may interfere with the ability of butyrate to induce apoptosis. Therefore, we measured the effects of cotreatment of ICG-001 and butyrate on apoptosis in HCT-116 (Fig. 3C) and SW620 (Fig. 3D) cells. Consistent with previous studies on ICG-001 (30), we measured activation of caspase 3/7, a downstream event in the apoptotic cascade. 75 μM ICG-001 alone did not induce caspase activity in HCT-116 cells; however, caspase activity was upregulated upon cotreatment with butyrate. The induction of apoptosis with butyrate/ICG-001 cotreatment was approximately 7-fold higher compared to ICG-001 alone (P<0.002), and markedly higher than mock treated control (P<0.03). As expected, treatment with butyrate alone induced caspase activity compared to control (P<0.02). The level of caspase activity observed with butyrate/ICG-001 cotreatment was statistically equivalent (slightly increased, not statistically) to that produced by butyrate alone. Thus, ICG-001 did not interfere with the ability of butyrate to upregulate apoptosis in HCT-116 cells. In SW620 cells, treatment with 100 μM ICG-001 did not induce caspase activity. Butyrate/ICG-001 cotreatment enhanced caspase activity 7-fold compared to ICG-001 alone (P < 0.002), and 6-fold higher than control (P<0.002). However, in SW620 cells, ICG-001 interfered with butyrate-induced apoptosis, as butyrate/ICG-001 cotreatment reduced caspase activity by approximately one-half compared to butyrate treatment alone (P<0.003).
HCT-116 and SW620 cells are common in vitro models for CRC, and SW620 cells are frequently utilized to generate xenograft mouse models. Therefore, we further analyzed the effects of butyrate/ICG-001 cotreatment in the two cell lines. Cell proliferation has been positively linked to the induction of apoptosis (41, 42 and refs. therein), and it has been reported that ICG-001 inhibits CRC cell proliferation (30). Therefore, we evaluated the effects of treatment with butyrate, ICG-001, or both agents on HCT-116 and SW620 cell growth/viability. Butyrate treatment of the HCT-116 cell line reduced the numbers of viable cells (P < 0.02) and exposure to ICG-001 led to a larger decrease in cell proliferation (P < 0.002). Cotreatment with both agents resulted in the least cell growth compared to mock treated cells (P < 0.001). Treatment with butyrate also reduced the numbers of viable SW620 cells (Fig. 4B) compared to mock treatment (P < 0.01); exposure to ICG-001 reduced cell proliferation to a greater extent, to 29% of mock treated samples (P < 0.005). Cotreatment with both butyrate and ICG-001 resulted in the lowest percentage of viable cells, only 8.8% of that observed with mock treatment (P < 0.001). SW620 cells were more sensitive to the effects of ICG-01 on cell proliferation/viability than HCT-116 cells.
Possible relationship between CRC cell proliferation and apoptosis. (A) HCT-116 CRC cells were mock treated (M) or treated with 5 mM butyrate (B), 75 μM ICG-001 (I) or both agents (BI) for 24 hr. Samples were assayed with the QuickCell Proliferation kit (Biovision) according to manufacturer's instructions. (B) SW620 cells were assayed for proliferation as in (A), except that the ICG-001 concentration was 100 μM. (C) Western blot data showing expression of p21 and survivin in HCT-116 (H) and SW620 (S) CRC cells mock treated (M) or treated with 5 mM butyrate (B) , 75 (HCT-116) or 100 mM (SW620) ICG-001 (I), or both agents (BI) for 17.5 hr. For (A)-(B), data are from three separate experiments; Bars, SDs. * = statistical significance.
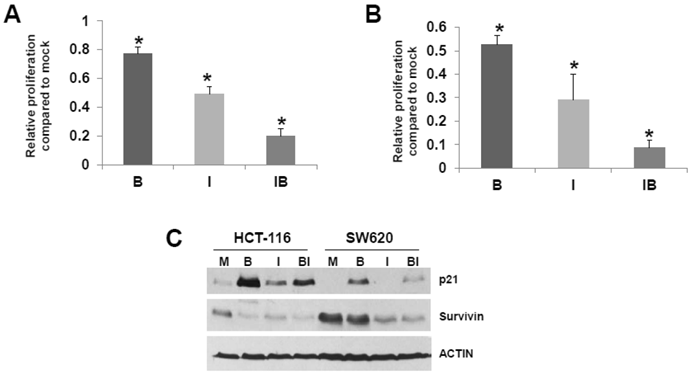
Cell cycle progression can influence apoptosis (41, 42 and refs. therein) Butyrate-treated SW620 cells demonstrate a marked G1 arrest which is abrogated by cotreatment with ICG-001, while HCT-116 cells exhibit moderate G2 arrest which is partially downregulated by ICG-001 (data not shown). We next determined how treatment of HCT-116 and SW620 cells with butyrate and/or ICG-001 modulates the expression of p21, which induces cell cycle arrest (Fig. 4C). Butyrate is known to upregulate p21 expression; as expected we detect increased steady state levels of p21 in butyrate-treated HCT-116 and SW620 cells. ICG-001 treatment downregulates expression of p21, particularly in SW620 cells, which is consistent with the effect of this agent in partially reversing SW620 cell G1 arrest. Levels of p21 are markedly lower in SW620 cells treated with both agents compared to HCT-116 cells. We also assessed effects of butyrate/ICG-001 cotreatment on expression of survivin (Fig. 4C), an anti-apoptotic factor downregulated by ICG-001 in certain CRC cell lines (33). As expected, ICG-001 downregulates survivin levels in both CRC cell lines; in contrast, butyrate markedly downregulates survivin expression in HCT-116, but not SW620, cells. Therefore, butyrate and ICG-001 can cooperate in the suppression of CRC cell growth, but combinations of these agents have different cell-type specific effects on CRC cell apoptosis.
Effects of ICG-001 on the WNT/catenin signaling heterogeneity in CRC cell lines
CRC cell populations are heterogeneous in terms of WNT signaling levels. The existence of high and low WNT/catenin activity fractions in CRC cell populations in vitro is a well-documented phenomenon, and the relative proportions of each fraction is altered by physiologically relevant concentrations of butyrate (1,2). The modulation of WNT/catenin signaling heterogeneity by butyrate is likely of physiological and clinical relevance, since tumor cell fractions with relatively higher WNT/catenin activity are thought to represent a more stem cell-like and tumorigenic phenotype (43). Considering the potential clinical application of ICG-001 and its analogs (40), we decided to evaluate how exposure of CRC cells with ICG-001 (a) alters the distribution of high and low WNT/catenin activity cell fractions, and (b) modifies the ability of butyrate to affect these fractions. We posited that ICG-001, which significantly represses WNT/catenin hyperactivation by butyrate in reporter assays, will suppress the number of CRC cells with relatively high (detectable) WNT/catenin activity.
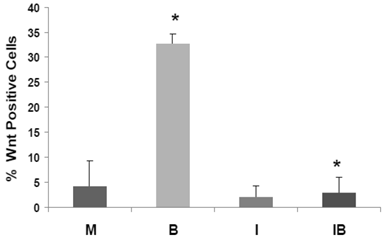
We have developed HCT-116 CRC cell lines stably transfected with TOP-EGFP (in which WNT sensitive promoter drives the expression of EGFP) or FOP-EGFP (in which WNT-non responsive promoter drives the expression of EGFP). Flow cytometry analyses of EGFP expression levels therefore determine the percentage of high-WNT activity “WNT-positive” cells: these are cells in which WNT activity-driven EGFP expression exceeds that of EGFP driven by non-WNT (background) activity (1). In our analyses, mock- and butyrate-induced levels of WNT-positive HCT-116 cells were similar to that previously reported, and butyrate sharply increased the percentage of WNT-positive cells (P < 0.002). Importantly, the butyrate-induced increase of WNT-positive HCT-116 cells was completely eliminated by cotreatment with ICG-001 (P<0.001). Therefore, in HCT-116 cells exposed to physiological levels of butyrate, ICG-001 eliminates the hyperinduction of WNT transcriptional activity (Fig. 5), and the increase in the number of WNT/catenin-positive cells.
ICG-001 represses the high-WNT signaling fraction of CRC cells induced by butyrate. HCT-116 cells stably transfected with TOP-EGFP and FOP-EGFP (1), were mock treated (M) or treated with 5 mM butyrate (B), 75 μM ICG-001 (I) or both agents (IB) 17hr. Data are from three separate experiments; Bars, SDs. * = statistical significance.
Discussion
Our findings demonstrate that the upregulation of WNT/catenin signaling by butyrate is dependent upon CBP-mediated WNT/catenin activity. ICG-001, an agent that disrupts the association between CBP and beta-catenin (Fig. 2) (30), markedly inhibits butyrate-induced WNT/catenin activity. The role of CBP in buffering WNT/catenin activity suggests that modulation of CBP activity can upregulate or downregulate WNT signaling to affect the cellular phenotype (e.g., proliferation, apoptosis or differentiation). ICG-001 has in vivo efficacy in mouse models of CRC (30); further, this agent and similar CBP-WNT inhibitors are entering clinical trial for colon and other cancers (40). Therefore, since ICG-001 has significant therapeutic potential, we evaluated how the agent influences two physiological consequences of the WNT/catenin signaling hyperactivation by butyrate: (a) changes in the number of WNT-positive cells in CRC cell populations, and (b) induction of CRC cell apoptosis.
CRC cell populations in vitro exhibit heterogeneity in terms of WNT/beta-catenin levels, and the proportion of WNT-positive cells is increased after exposure to physiologically relevant concentrations of butyrate (1). In this study, we demonstrate that the WNT-positive (high-WNT) subpopulation of HCT-116 cells is reduced by ICG-001, and the butyrate-induced increase in WNT-positive cells is completely abrogated by ICG-001 treatment (Fig. 5). This is significant, since cells with WNT activity are linked to a more stem cell-like and tumorigenic phenotype (43). Our findings suggest the possibility of using modulation of CBP-mediated WNT activity to transition CRC cells to less tumorigenic types, with positive consequences for CRC therapy.
The effects of ICG-001 on butyrate-induced CRC cell apoptosis were cell type-dependent (Fig. 3). The levels of caspase activity in HCT-116 cells cotreated with both butyrate and ICG-001 are equal to or greater than that observed with butyrate alone. In SW620 cells, fold-induction of caspase activity was higher in butyrate/ICG-001- than in ICG-001-treated cells (Fig. 3B,D). However, caspase activity in SW620 cells exposed to butyrate/ICG-001 was significantly lower than that observed in butyrate-treated cells (Fig. 3D). ICG-001, known to suppress CRC proliferation (30), affects proliferation of SW620 cells to a greater extent compared to HCT-116 cells (Fig. 4A,B). Since cell proliferation, but not growth arrest, positively associates with the induction of apoptosis (44-47), the more potent cytostatic effect of ICG-001 on SW620 cells may explain the interference of the agent with butyrate-induced apoptosis.
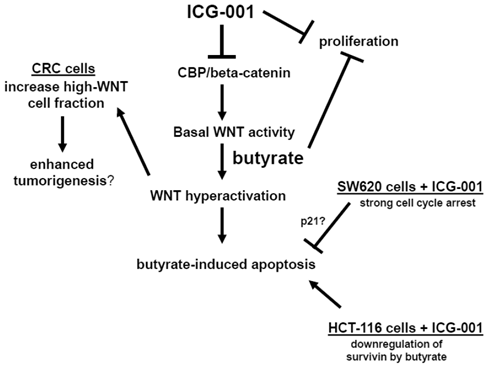
Therefore, in some CRC cell lines, ICG-001 may interfere with butyrate-induced changes in cell cycle, and thus, block the induction of apoptosis. We have observed significant differences in cell cycle progression between HCT-116 and SW620 cells (data not shown). Butyrate induces G2 arrest in HCT-116 cells, however, in SW620 cells butyrate primarily results in G1 arrest, and the level of G1 arrest is reduced by ICG-001 treatment. ICG-001 also interferes with the ability of butyrate to induce expression of p21 (Fig. 4C), which promotes cell cycle arrest, and this effect of ICG-001 is particularly pronounced in SW620 cells. Therefore, cell cycle variations between CRC cell lines exposed to ICG-001/butyrate may contribute to the observed differences in apoptosis. HCT-116 and SW620 cells also differ in patterns of expression of the anti-apoptotic factor survivin (Fig. 4C). Consistent with previous reports (33), ICG-001 downregulates survivin expression in both CRC cell lines. In contrast, butyrate markedly downregulates survivin expression in HCT-116, but not in SW620, cells. These differences in survivin expression may also contribute to differential effects of butyrate/ICG-001 exposure on apoptosis in HCT-116 and SW620 cells. Our hypothesis of how CBP/beta-catenin mediates the effects of ICG-001 and butyrate on CRC cells is outlined in Fig. 6.
Central role of CBP/beta-catenin in the effects of butyrate on CRC cells. (Center). The association between CBP and beta-catenin, which is blocked by ICG-001, contributes to basal WNT/catenin activity and is required for butyrate-induced WNT hyperactivation. The hyperactivation of WNT/catenin activity mediated by butyrate enhances apoptosis. (Left) Butyrate-mediated WNT hyperactivation increases the proportion of cells exhibiting high levels of WNT/catenin activity, which may influence tumorigenesis. ICG-001 represses WNT hyperactivation and decreases the proportion of high-WNT activity cells. (Right Top). Both ICG-001 and butyrate repress CRC cell proliferation. (Right Bottom). Treatment of SW620 CRC cells with ICG-001 induces a strong cell cycle arrest, which may interfere with butyrate-mediated apoptosis. This negative interaction between ICG-001 and butyrate may involve altered expression of factors such as p21. In HCT-116 cells, butyrate downregulates expression of the anti-apoptotic factor survivin, and in this cell line, ICG-001 does not interfere with butyrate-induced apoptosis. Arrows indicate positive interactions (activation); blocked lines indicate negative interactions (repression).
Our findings indicate that the combination of butyrate and ICG-001-like agents is effective in suppressing CRC cell growth (Fig. 4), which is of therapeutic utility. However, apoptosis is generally preferable to cytostatis for therapy; therefore, butyrate/ICG-001 cotreatment should be avoided in CRCs in which ICG-001-like agents and butyrate counteract each other's effect on apoptosis. Since butyrate is produced by dietary fiber, the effects of a fiber-rich diet on the anti-tumor activity of ICG-001-like agents should be investigated through animal studies. We speculate that a subset of CRC patients should be treated with ICG-001-like agents in fasting state, or at least on a diet that does not include fiber. In addition, butyrate-sensitive CRCs may benefit from alternating exposure to HDACis (WNT-hyperactivation therapeutic mode) and ICG-001-like agents (WNT-suppressive therapeutic mode) (48).
Thus, the findings of our study have chemopreventive and therapeutic significance. Increased understanding of CBP-mediated WNT signaling will also improve the utility of agents (e.g., HDACis), the activity of which is modulated by CBP. Our findings also suggest that butyrate derived from dietary fiber interferes with the apoptotic action of ICG-001-like agents, and vice versa; therefore, these interactions should be taken into account in future clinical trials. A follow-up study will examine the role of the HAT factor p300 in mediating butyrate-induced WNT hyperactivation and its downstream physiological consequences.
Acknowledgements
We thank Dr. Michael Kahn for supplying ICG-001. This work was supported by NIH NCI grant 1R15CA149589-01.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lazarova DL, Bordonaro M, Carbone R, Sartorelli AC. Linear relationship between WNT activity levels and apoptosis in colorectal carcinoma cells exposed to butyrate. Internat J Cancer. 2004;110:523-31
2. Bordonaro M, Lazarova DL, Sartorelli AC. The activation of beta-catenin by WNT signaling mediates the effects of histone deacetylase inhibitors. Exp Cell Res. 2007;313:1652-66
3. Richter M, Jurek D, Wrba F, Kaserer K, Wurzer G, Karner-Hanusch J, Marian B. Cells obtained from colorectal microadenomas mirror early premalignant growth patterns in vitro. Euro J Cancer. 2002;38:1937-45
4. Kautenberger T, Beyer-Sehlmeyer G, Festag G, Haag N, Kuhler S, Kuchler A, Weise A, Marian B, Peters WHM, Liehr T, Claussen U, Pool-Zobel BL. The gut fermentation product butyrate, a chemopreventive agent, suppresses glutathione S-transferase theta (hGSTT1) and cell growth more in human colon adenoma cells (LT97) than tumor (HT29) cells. J Cancer Res Clin Oncol. 2005;131:692-700
5. Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Migrating cancer stem cells - an integrated concept of malignant tumour progression. Nature Reviews. 2005;5:744-9
6. Bingham SA, Day NE, Luben R, Ferrari P, Slimani N, Norat T, Clavel-Chapelon F, Kesse E, Nieters A, Boeing H, Tjonneland A, Overvad K, Martinzez C, Dorronsoro M, Gonzalez CA, Key TJ, Trichopolou A, Naska A, Vineis P, Tumino R, Krogh V, Bueno-de-Mesquita HB, Peeters PH, Berglund G, Hallmans G, Lund E, Skeie G, Kaaks R, Riboli E. Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC): an observational study. Lancet. 2003;361:1496-501
7. Peters U, Sinha R, Chaterjee N, Subar AF, Ziegler RG, Kuldorff M, Bresalier R, Weissfeld JL, Flood A, Schtzkin A, Hayes RB. Dietary fibre and colorectal adenoma in a colorectal cancer early detection programme. Lancet. 2003;361:1491-5
8. Bingham SA, Norat T, Moskal A, Ferrari P, Slimani N, Chavel-Chapelon F, Kesse E, Nieters A, Boeing H, Tjonneland A, Overvad K, Martinzez C, Dorronsoro M, Gonzalez CA, Ardanaz E, Navarro C, Quiros JR, Key TJ, Day NE, Trichopoulo A, Naska A, Krogh V, Tumino R, Palli D, Panico S, Vineis P, Bueno-de-Mesquita HB, Ocke MC, Peeters PHM, Berglund G, Hallmans G, Lund E, Skeie G, Kaaks R, Riboli E. Is the association with fiber from foods in colorectal cancer confounded by folate intake? Cancer Epid Biom Prev. 2005;14:1552-6
9. Bingham S. Mechanisms and experimental evidence relating dietary fibre and starch to protection against large bowel cancer. Proc Nutr Soc. 1990;49:153-71
10. Cummings JH, Pomare EW, Branch WJ, Naylor CPE, MacFarlane GT. Short chain fatty acids in human large intestine, portal, hepatic, and venous blood. Gut. 1987;28:1221-7
11. Boffa LC, Luption JR, Mariani MR, Ceppi M, Newmark HL, Scalmati A, Lipkin M. Modulation of colonic epithelial cell proliferation, histone acetylation, and luminal short chain fatty acids by variation of dietary fiber (wheat bran) in rats. Cancer Res. 1992;52:5906-12
12. Heerdt BG, Houston MA, Augenlicht LH. Potentiation by specific short-chain fatty acids of differentiation and apoptosis in human colonic carcinoma cell lines. Cancer Res. 1994;54:3288-94
13. Hague A, Elder DJ, Hicks DJ, Paraskeva G. Apoptosis in colorectal tumor cells: induction by the short chain fatty acids butyrate, propionate and acetate and by the bile salt deoxycholate. Internat J Cancer. 1995;60:400-6
14. Medina V, Young GP, Edmonds B, James R, Appleton S, Zalewski PD. Induction of caspase-3 protease activity and apoptosis by butyrate and another inhibitor of histone deacetylase: dependence on protein synthesis and synergy with a mitochondrial/cytochrome c-dependent pathway. Cancer Res. 1997;57:3697-707
15. Marks PA, Dokmanovic M. Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin Investig Drugs. 2005;12:1497-511
16. Lin HY, Chen CS, Lin SP, Weng JR, Chen CS. Targeting histone deacetylase in cancer therapy. Med Res Rev. 2006;26:397-413
17. Bordonaro M, Mariadason JM, Aslam F, Heerdt BG, Augenlicht LH. Butyrate-induced apoptotic cascade in colonic carcinoma cells: modulation of the beta-catenin-Tcf pathway and concordance with effects of sulindac and trichostatin A but not curcumin. Cell Growth Differ. 1999;10:713-20
18. Li VSW, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, Mohammed S, Heck AJR, Maurice MM, Mahmoudi T, Clevers H. WNT signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell. 2012;149:1245-56
19. Cook D, Fry M, Hughes K, Sumathipala R, Woodgett J, Dale T. Wingless inactivates glycogen synthase kinase-3 via an intracellular signaling pathway which involves a protein kinase C. EMBO J. 1996;15:4526-36
20. Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR, Munimetsu S, Polakis P. Association of the APC gene product with β-catenin. Science. 1993;262:1731-4
21. Su LK, Vogelstein B, Kinzler KW. Association of the APC tumor suppresser protein with catenins. Science. 1993;262:1734-7
22. Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular β-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA. 1995;92:3046-50
23. Behrens J, Von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of β-catenin with the transcriptional factor LEF-1. Nature. 1996;382:638-42
24. Molenaar M, Van De Wetering M, Osterwegel M. XTcf-3 transcription factor mediates β-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391-9
25. Korinek V, Barker N, Morin PJ, Van Wichen D, De Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1784-7
26. Morin J, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787-90
27. Bienz M, Clevers H. Linking colorectal cancer to WNT signaling. Cell. 2000;103:311-20
28. Albuquerque C, Breukel C, van der Luijt R, Fidalgo P, Lage P, Slors FGM, Leitao CN, Fodde R, Smits R. The just-right signaling model: APC somatic mutations are selected based on a special level of activation of the beta-catenin signaling cascade. Hum Mol Genet. 2002;11:1549-60
29. Bordonaro M, Lazarova D, Sartorelli AC. Hyperinduction of WNT signaling: a new paradigm for the treatment of colorectal cancer? Oncol Res. 2008;17:1-9
30. Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, Moon RT, Teo JL, Oh SW, Kim HY, Moon SH, Ha JR, Kahn M. A small molecule inhibitor of β-catenin/CREB-binding protein transcription. Proc Natl Acad Sci USA. 2004;101:12682-7
31. McMillan M, Kahn M. Investigating WNT signaling: a chemogenomic safari. Drug Disc Today. 2005;10:1467-74
32. Teo JL, Ma H, Nguyen C, lam C, Kahn M. Specific inhibition of CBP/β-catenin interaction rescues defects in neuronal differentiation caused by presenilin-1 mutayion. Proc Natl Acad Sci. 2005;102:12171-6
33. Ma H, Nguyen C, Lee KS, Kahn M. Differential roles for the coactivators CBP and p300 on TCF/β-catenin-mediated survivin gene expression. Oncogene. 2005;24:3619-31
34. Miyabayashi T, Teo JL, Yamamoto M, McMillian M, Nguyen C, Kahn M. WNT/β-catenin/CBP signaling maintains long-term murine stem cell pluripotency. Proc Natl Acad Sci USA. 2007;104:5668-73
35. Teo JL, Kahn M. The WNT signaling pathway in cellular proliferation and differentiation: a tale of two coactivators. Adv Drug Delivery Rev. 2010;62:1149-55
36. Waltzer L, Bienz M. Drosophila CBP represses the transcription factor TCF to antagonize Wingless signaling. Nature. 1998;395:521-5
37. Hecht A, Vleminckx K, Stemmler MP, van Roy F, Kremler R. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 2000;19:1839-50
38. Li J, Sutter C, Parker DS, Blauwkamp T, Fang M, Cadigan KM. CBP/p300 are bimodal regulators of WNT signaling. EMBO J. 2007;26:2284-94
39. Levy L, Wei Y, Labalette C, Wu Y, Renard CA, Buendia MA, Neuveut C. Acetylation of beta-catenin by p300 regulates beta-catenin-Tcf4 interaction. Mol Cell Biol. 2004;24:3404-14
40. Takahashi-Yanaga F, Kahn M. Targeting WNT signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16:3153-62
41. Guo M, Hay BA. Cell proliferation and apoptosis. Curr Opin Cell Biol. 1999;11:745-52
42. Alenzi FQB. Links between apoptosis, proliferation, and cell cycle. Br J Biomed Sci. 2004;61:1-4
43. Fodde R, Brabletz T. WNT/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19:150-8
44. Yu D, Jing T, Liu B, Yao J, Tan M, McDonnell TJ, Hung MC. Overexpression of ErbB2 blocks taxol-induced apoptosis by upregulation of p21cip1, which inhibits p34cdc2 kinase. Molecular Cell. 1998;2:581-91
45. Reid S, Ritchie A, Boring L, Gosling J, Cooper S, Hangoc G, Charo IF, Broxmeyer HH. Enhanced myeloid progenitor cell cycling and apoptosis in mice lacking the chemokine receptor, CCR2. Blood. 1999;93:1524-33
46. Gartel AL, Tyner AL. The role of cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639-49
47. Sturm I, Raue B, Sclag PM, Wust P, Hildebrandt B, Riess H, Hauptmann S, Dorken B, Daniel PT. Genetic dissection of apoptosis and cell cycle control in response of colorectal cancer cells treated with preoperative radiotherapy. BMC Cancer. 2006;6:124
48. Lazarova DL, Bordonaro M. Extreme fluctuations in Wnt/beta-catenin signaling as an approach for colon cancer prevention and therapy. Advanced Studies in Biology. 2012;4:351-62
Author biography
Michael Bordonaro received his PhD degree from Fordham University and did a post-doctoral fellowship at the Montefiore Medical Center at the Albert Einstein Cancer Center. It was during this period that Dr. Bordonaro discovered that the Wnt signaling pathway can be pharmacologically modulated, and that histone deacetylase inhibitors (HDACis) upregulate Wnt signaling in colorectal cancer cells in a manner associated with enhanced rates of apoptosis of these cells. While at Montefiore, Dr. Bordonaro was awarded an American Institute for Cancer Research (AICR) Post-Doctoral Award grant for his work on HDACis and Wnt signaling. Dr. Bordonaro subsequently worked at the Yale University School of Medicine, where he continued his studies on the effects of HDACis on Wnt signaling and colonic cell physiology. In addition, at Yale, Dr. Bordonaro started work on Wnt-targeted genetic therapeutics against colorectal cancer. Dr. Bordonaro is currently an Assistant Professor at The Commonwealth Medical College, and is continuing his research on these and other topics, with grant funding from the National Cancer Institute and AICR.
![]() Corresponding author: The Commonwealth Medical College, 525 Pine Street, Scranton, PA 18509. Tel: 570-504-9646; Fax: 570-504-9636; Email: mbordonaroorg.
Corresponding author: The Commonwealth Medical College, 525 Pine Street, Scranton, PA 18509. Tel: 570-504-9646; Fax: 570-504-9636; Email: mbordonaroorg.