Impact Factor ISSN: 1837-9664
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 17; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Special Issues
Introduction
Novel therapies targeting the...
Antibodies and antibody...
Ongoing clinical trials with...
Small molecule HER/EGFR...
Anti HER-3 agents
Targeting DNA repair pathways:...
mTOR/PI3K
Other therapeutic targets
PI3K inhibitors
Insulin-like growth factor...
Heat Shock Protein 90 (HSP 90)
Histone Deacetylase Inhibitors
Concluding Remarks
References

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(2):117-132. doi:10.7150/jca.4925 This issue Cite
Review
Treating Breast Cancer in the 21st Century: Emerging Biological Therapies
Gabriel Tinoco1*, Sean Warsch2*, Stefan Glück3 ![]() , Kiran Avancha4, Alberto J. Montero3
, Kiran Avancha4, Alberto J. Montero3
1. Department of Medicine, Division of Hospital Medicine, University of Miami Miller School of Medicine, Miami, FL, USA.
2. Department of Internal Medicine, University of Miami Miller School of Medicine, Miami, FL, USA.
3. Department of Medicine, Division of Hematology/Oncology, Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, Miami, FL, USA.
4. Office of Research, University of Miami Miller School of Medicine, Miami, FL, USA.
* Both authors contributed equally to this paper, and serve as first co-authors.
Received 2012-11-1; Accepted 2012-11-29; Published 2013-1-11
Abstract
For many years, the medical treatment of breast cancer was reliant solely on cytotoxic chemotherapy. However, over the past twenty years, treatment has evolved to a more target-directed approach. We now employ tailored therapy based on the presence or absence of receptors for estrogen, progesterone, and human epidermal growth factor 2 (HER2). We expect this trend to continue, as agents that use novel approaches to target HER2, as well as targeting different portions of the HER signaling pathway, are in various stages of development. Notably, pertuzumab, a humanized monoclonal antibody that binds to a different domain of the extracellular portion of the HER2 receptor than trastuzumab, was recently approved for use, as was lapatinib, a small-molecule tyrosine kinase inhibitor. Patients with triple negative breast cancer, particularly those with the BRCA mutation, have more limited treatment options and carry a worse prognosis than those who are hormone receptor positive. However, recent data has shown that PARP inhibitors may have significant anti-tumor effect in those with this subtype of breast cancer. Novel agents that inhibit mTOR, PI3K, the insulin-like growth factor, heat shock protein 90, and histone deacetylase have shown promise in phase I-III trials and offer exciting new possibilities for the treatment of this often fatal disease. As we are presented with an ever increasing number of treatment options, the timing and combinations of therapeutic agents used becomes ever more complex in the age of personalized care, but we are hopeful that ultimately this will lead to improved patient outcomes.
Keywords: breast cancer, chemotherapy, novel therapeutics, biologics, HER2, PARP inhibitors.
Introduction
Breast cancer is the most common cancer in women worldwide (1,2,3). In 2011, an estimated 230,000 women were diagnosed with breast cancer in the U.S. alone, with an estimated 40,000 deaths, making it the second most common cause of cancer related death in women (4). While only approximately 5% of all newly diagnosed breast cancer patients in the U.S. present with metastatic disease at the time of initial diagnosis, up to one third of patients with early stage disease will subsequently develop metastasis (3). Over the past two decades, breast cancer mortality rates have declined by approximately 30%, with corresponding improvements in 5-year overall survival rates to 90% (4). Despite these advances, metastatic breast cancer (MBC) remains incurable, with an estimated 5-year overall survival rate of only 23% (5). However, more recent data from the SEER-Medicare database painted a more pessimistic picture, reporting median overall survival of Medicare patients with MBC to be only 22 months (6).
The biological underpinnings of breast cancer, and the major pathways involved in tumor progression and metastases, are still incompletely understood. Breast cancer traditionally has been classified into three different subtypes based on the presence or absence of three receptors found on cancer cells (7). Hormone receptor (HR) positive breast cancers express estrogen and/or progesterone receptors (ER/PR), and constitute approximately 60% of all breast cancer cases (8). The oncogene human epidermal growth factor receptor 2 (HER-2/neu) is over-expressed in approximately 20% of all breast cancer cases; while approximately 20% of breast cancer cases are negative for the expression of ER,PR, and HER-2/neu, also known as triple negative breast cancer (TNBC) (9,10).
More sophisticated genomic microarray analyses have also corroborated the presence of several distinct intrinsic molecular subtypes of breast cancer: luminal A, luminal B, basal, normal breast-like, and HER-2 like subsets (11,12). Subsequent to this initial research, the claudin low subtype was also recognized as yet another distinct molecular subtype. In about 70% of cases, the molecular breast cancer subtypes correlate with the expression of ER, PR, and HER-2 (13). Research is currently focusing on the clinical utility of molecular profiling that in the near future may replace traditional immunohistochemical staining, as initial evidence suggests that molecular profiling may be more accurate in predicting prognosis (13-14) .
Patients with hormone receptor positive tumors typically receive endocrine therapy (e.g. selective estrogen-receptor response modulators [SERM] and aromatase inhibitors [AI]) as one of many options of their treatment. Moreover, patients with HER-2/neu overexpressing tumors typically receive anti-HER/2 targeted therapy in combination with cytotoxic chemotherapeutic agents. Patients with triple negative breast cancer (TNBC) do not have these targeted treatment options, with cytotoxic chemotherapy being the primary modality (15, 16). In this article, we will review the most promising novel biologic agents under clinical development over the last 5 years in women with MBC. We will focus primarily on agents that target the HER-2 receptor family, poly ADP ribose polymerase (PARP) inhibitors, the mTOR pathway, insulin-like growth factor receptors, heat shock protein 90, and histone deacetylase (HDAC) inhibitors.
Novel therapies targeting the HER signaling pathway
The human epidermal growth factor receptor 2 (HER2) is a cell membrane tyrosine kinase receptor member of the epidermal growth factor receptor (EGFR) family (2, 17) that is over-expressed in approximately 15 - 25% of primary human breast cancers, and is associated with poor clinical outcomes and aggressive tumor progression (18- 21). The first commercially available HER 2 targeting agent was the monoclonal antibody trastuzumab (Herceptin®) (22, 23). The humanized monoclonal antibody trastuzumab binds to an extracellular segment of the HER2/neu receptor, leading to inhibition of the proliferation of human tumor cells that overexpress HER 2 (22). The exact mechanism of action is not fully understood. The use of trastuzumab has proven to improve survival for patients with breast cancer overexpressing HER 2; as adjuvant therapy for patients with early stage disease (24) and in combination with chemotherapy or as monotherapy for patients with metastatic disease (9, 25-36).
Antibodies and antibody conjugates
Trastuzumab established a new treatment paradigm for HER2-positive breast cancer. It is currently the standard first line treatment for patients with HER2-positive MBC in combination with chemotherapy or as a single agent (26, 30, 37-40). Lapatinib is a reversible small-molecule tyrosine kinase inhibitor that also targets HER2 by interfering with downstream signaling through the HER-2 pathway; it binds to the ATP-binding pocket of the EGFR/HER2 protein kinase domain, preventing self-phosphorylation and subsequent activation (41, 42). Lapatinib is currently approved as first-line therapy in combination with letrozole for patients with MBC who overexpress HER2 and are estrogen receptor (ER) positive (42- 44), as well as for the treatment of HER2- positive MBC in combination with capecitabine for patients who progressed on prior therapy including a taxane, an anthracycline, and trastuzumab (45, 46).
Despite the beneficial impact of HER-2 targeted therapy, numerous patients still develop recurrences or progression of the disease due to trastuzumab resistance (43). In patients with HER-2 amplified breast cancer, approximately 70% may have primary resistance to trastuzumab (26, 47). In addition, an important number of patients who achieve initial response to the treatment tend to develop secondary trastuzumab resistance (47, 48). Nevertheless, the continuous use of trastuzumab has shown to be beneficial, even in patients who develop MBC after its use as adjuvant therapy or whose disease progresses while receiving this medication (20, 49-51).
The antibody-drug conjugate (ADC) trastuzumab-DM1 (Genentech) combines, via a specially designed linker, trastuzumab with a fungal toxin DM1 (Emtansine), a potent cytotoxic- microtubulin disruptive chemotherapeutic agent derivate of maytansine (52, 53). This allows trastuzumab to be used in the targeted delivery of the potent chemotherapy drug DM1 (2).
Using trastuzumab mainly as a carrier, this ADC was designed to target the delivery of DM1 into the HER2 overexpressing cells via endocytosis (53), wherein DM1 is subsequently detached from trasuzumab and released intracellularly, causing inhibition of microtubule assembly and posterior apoptosis (54-57), which leads to the recently described mitotic catastrophe (58). Trastuzumab-DM1 has been shown to have a mild dosage-related toxicity profile (59) (transient thrombocytopenia, elevated transaminases, fatigue, and anemia are the most common reactions), with low inter-individual variability in its pharmacokinetic parameters (60) and a maximal tolerated dose of 3.6 mg/kg every 3 weeks (61).
The efficacy of this ADC has been demonstrated in both in vitro and in vivo models of trastuzumab resistant breast cancer (53). More recently it has been proposed that trastuzumab-DM1 is able to circumvent the cross-resistance phenomenon observed with the use of lapatinib and trastuzumab (58). Preliminary efficacy data of a phase Ib/II study of trastuzumab-DM1given with Pertuzumab in HER2-positive, trastuzumab pre-treated patients showed partial responses (PR) among 23 patients (62).
In a recent phase II study involving patients with HER2-positive MBC who had progressed on earlier treatment with a HER2-directed agents plus chemotherapy (n=112), patients were schedule to receive trastuzumab-DM1 at a dose 3.6 mg/kg every 3 weeks. An overall response rate (ORR) of 25.9% was reported with a median PFS of 4.6 months. The median duration of response was not reached due to a low number of events (63). Early results from another phase II study comparing trastuzumab plus docetaxel to T-DM1 in first-line HER2-positive MBC indicated comparable response rates of 41 and 48%, respectively, without docetaxel related toxicities (64). A recent update indicated that PFS was significantly longer with T-DM1 versus trastuzumab/docetaxel (52, 65). Currently there are two prospective randomized phase 3 trials designed to evaluate the efficacy of T-DM1 in the management of MBC compared to the current standard of care. First, MARIANNE is a three arm trial that compared trastuzumab plus a taxane to T-DM1 combined with a placebo or pertuzumab (66). This study met enrollment goals in April 2012. The second trial is the TH3RESA trial, where TDM1will be compared to treatment of physician's choice as third line therapy in women previously having received taxanes, trastuzumab, and capecitabine/lapatinib, with or without prior anthracyclines (67).
Results of the EMILIA study, an open-label, randomized phase 3 trial comparing T-DM1 to capecitabine plus lapatinib (XL) as second line therapy in women with MBC previously treated with anthracyclines, taxanes, and trastuzumab, were recently reported (68). Patients who had received T-DM1 had significantly longer median progression free survival (9.6 vs 6.4 months, HR=0.650 p <0.0001 ), with a trend towards longer median overall survival time ([1-year : T-DM1 84.7% (80.76-88.55%) versus XL 77.0% (72.40-81.50%), 2-year: T-DM1 65.4% (58.65-72.15%) versus XL 47.5% (39.20-55.89%)]. The median overall survival was not reached in the T-DM1 arm and was 23.3 months in the capecitabine plus lapatinib arm. T-DM1 was beneficial for patients in different sub-groups, including those with visceral metastases and positive ER/PR status. T-DM1 was also well tolerated; the most common grade 3 adverse events for T-DM1: thrombocytopenia (12.9% vs 0.2%), increased AST (4.3% vs 0.8%), and increased ALT (2.9% vs 1.4%), and for XL: diarrhea (20.7% vs 1.6%), palmar plantar erythrodysesthesia (16.4% vs 0) and vomiting (4.5% vs 0.8%) (68).
Pertuzumab is a humanized monoclonal antibody that binds to the HER2 receptor, binding to a different domain of the extracellular portion of the HER2 receptor than trastuzumab, and blocks HER2- dimerization (43, 69). This agent has been actively investigated in combination with trastuzumab, aiming to explore the theoretical advantage of using two HER2 targeted agents (43, 70) for more complete blockade of the HER-2 signaling pathway (37). The phase III trial (CLEOPATRA) showed that the addition of pertuzumab to trastuzumab plus docetaxel, when used as first-line treatment for HER2-positive metastatic breast cancer, significantly prolonged median PFS by 6.1 months (HR, 0.62; 95% CI, 0.51-0.75; P<0.001), with no increase in cardiac toxicity (71). These data led to the approval by the FDA on June 8, 2012 of the compound as Perjeta® in combination with docetaxel and trastuzumab as first line therapy for HER2+ MBC.
The trial that led to pertuzumab's approval was a double-blind, placebo controlled, multicenter, phase III trial that enrolled 808 patients with HER2-positive MBC for first-line therapy. The patients were randomly assigned into two groups: 406 to the placebo plus trastuzumab plus docetaxel group (control group) and 402 to pertuzumab plus trastuzumab plus docetaxel group (pertuzumab group). The primary end point was independently assessed progression-free survival, and secondary end points included overall survival and safety (71). The median progression-free survival was 12.4 months in the control group vs. 18.5 months in the pertuzumab group. The median investigator-assessed progression-free survival was 12.4 months in the control group vs.18.5 months in the pertuzumab group (HR, 0.65; 95% CI,0.54 -0.78; P<0.001) (71).
Data for OS are not yet mature; the interim analysis of overall survival was performed after 165 events had occurred (43% of the pre-specified total number of events for the final analysis) and is expected to be complete in 2013 (71). With the exception of cardiotoxicity, all other adverse events (of any grade), including diarrhea, rash, mucosal inflammation, febrile neutropenia, and dry skin, were reported more frequently in the pertuzumab group (71). Data so far available suggest that pertuzumab and trastuzumab in combination is more efficacious in the treatment of HER-2 positive breast cancer than the use of a single anti-HER 2 agent in both MBC and preoperatively, as neoadjuvant therapy (43).
The NeoSphere trial assessed the effect of pertuzumab and trastuzumab plus chemotherapy in treatment-naive women with locally advanced, inflammatory or early stage HER2-positive breast cancer (72).
The patients were assigned to one of four groups: group A received trastuzumab and docetaxel, group B received pertuzumab, trastuzumab and docetaxel, group C received pertuzumab and trastuzumab, and group D received pertuzumab and docetaxel.
Patients given pertuzumab and trastuzumab plus docetaxel (group B) had a significantly improved pathological complete response rate (49 of 107 patients; 45.8% [95% CI 36.1—55.7]) compared with those given trastuzumab plus docetaxel (group A) (31 of 107; 29.0% [20.6—38.5]; p=0.0141), without substantial differences in tolerability (72). The most common adverse events of grade 3 or higher was neutropenia (61 of 107 women in group A, 48 of 107 in group B, one of 108 in group C, and 52 of 94 in group D) and febrile neutropenia (eight, nine, none, and seven, respectively) (72).
Ongoing clinical trials with other HER2 targeted agents
MM-302 is a liposomal encapsulation of doxorubicin attached to antibodies that target HER2-overexpressing cancer cells, attempting to limit off target toxicity to non-malignant cells (73). Currently an ongoing phase I study is attempting to establish the safety and pharmacokinetics of this agent in patients with HER-2 positive MBC (74).
Ertumaxomab (Rexomun; Fresenius Biotech, Hamburg, DE) is a hybrid trifunctional monoclonal antibody that targets the CD3 antigen on T cells and the HER-2 expressed on the tumor cells (75).
This compound forms a HER-2-ertumaxomab-CD3 complex, leading to the aggregation and activation of T cells, macrophages, dendritic cells, and natural killer cells, which results in the phagocytosis and death of the tumor cells (75). A phase II study was terminated due to a change in development plan from the sponsor, not due to safety concerns (76). Further clinical studies are required to determine the clinical relevance of this medication.
MM-111 is a novel antibody fusion protein that targets the HER 2/HER 3 heterodimer, and blocks the ligand binding to HER 2-3 heterodimers, thereby inhibiting downstream signaling (77). The fusion protein is conformed by two human single-chain variable fragment (scFv) antibodies linked to a modified human serum albumin (78). An ongoing phase I/II trial is evaluating the safety and tolerability of MM-111 in patients with HER 2 MBC (77).
Novel HER-2 monoclonal antibodies, drug conjugates, or antibody fusion proteins under clinical development in breast cancer.
| Drug Name | Description | Phase | References |
|---|---|---|---|
| TDM-1 | Trastuzumab-DM1 conjugate | I-III | 53-55,58,60-64,66-68 |
| MM-302 | Nanotherapeutic encapsulation of doxorubicin with attached antibodies | I | 73,74 |
| Ertumaxomab | Murine monoclonal antibody with two antigen-recognition sites(CD3 & HER-2/neu) | I | 75,76 |
| Pertuzumab | Recombinant humanized monoclonal antibody targeting Subdomain II of (HER2) | II-III | 69,71,72 |
| MM-111 | Novel antibody fusion protein that targets HER 2/HER 3 heterodimer | I-II | 77,78 |
Small molecule HER/EGFR inhibitors other than lapatinib
Afatinib (BIBW 2992) is an irreversible small molecule inhibitor of the ErbB-receptor family, targeting the intracellular tyrosine kinase of the HER-2 molecule (79-81). In initial phase I studies, diarrhea, vomiting, nausea, fatigue and rash were the most common adverse events. Results of an open-label phase II study in advanced HER2-positive breast cancer patients after failure of trastuzumab were recently published, showing clinical benefit in 53% of the patients enrolled (79). Additional phase II studies showed acceptable tolerance and moderate activity of afatinib in ER-positive, HER2-negative breast cancer patients (82-84).
Neratinib (HKI-272) is a small molecule irreversible pan-HER receptor tyrosine kinase inhibitor (activity against HER1, HER2, and HER4) (85). It does not have activity against HER3, which is relevant because HER3 does not have an intracellular domain with tyrosine kinase activity. Early phase studies established the dose limiting toxicity to be diarrhea.
TAK-285 is a novel oral small-molecule dual HER-2/EGFR tyrosine kinase. The initial phase I study in primarily Japanese patients with advanced cancers demonstrated good tolerance of TAK-285, with elevation in aminotransferases and hyporexia being the most prominent grade 3 dose-limiting toxicities (86). In the U.S., a multicenter phase I study designed to determine the pharmacokinetics in a more diverse population of patients with advanced cancer was recently completed, and the results are forthcoming (87).
ARRY-380 is an orally active, small molecule, reversible-selective inhibitor of the HER-2 tyrosine kinase receptor with antitumor activity in HER2-positive breast cancer in vitro and in vivo (88). These findings led to a phase I trial which found ARRY-380 to be well tolerated, with rash and diarrhea as the most frequent adverse effects, along with promising signs of clinical activity, especially in pretreated patients with HER2+ MBC (89).
In an open-label, phase II study Burstein et al, showed the clinical activity of neratinib in patients with advanced HER2+ MBC with and without prior trastuzumab treatment: 16-week PFS was 78% for patients with no prior treatment with trastuzumab (n = 64) and 59% for those who had previously received trastuzumab (n = 63), with the median PFS 39.6 and 22.3 weeks, respectively. The most significant toxicities were diarrhea, vomiting, nausea, and fatigue (90). Neratinib has been included as one of the arms of the ongoing I-SPY 2 trial (91).
Anti HER-3 agents
MM-121 is a fully human monoclonal antibody that binds to HER3 and blocks the binding of its ligand, leading to inhibition of HER3 signaling. Schoeberl et al, provided data suggesting that MM-121 can be an effective therapeutic agent for cancers with ligand dependent HER3 activation (93). Currently Moyo et al. are conducting a phase II study, with MM-121 plus exemestane vs. exemestane alone in ER+ and/or PR + and HER2 negative MBC patients who have progressed on prior anti-estrogen therapy (94).
Novel targeted anti-HER/EGFR therapies.
| Drug Name | Description | Phase | References |
|---|---|---|---|
| Afatinib [BIBW 2992] | Orally active, small molecule irreversible pan-HER TKI | I-II | 79-84 |
| Neratinib | Orally available, irreversible TKI of HER-2 | I-II-III | 85,90-92 |
| TAK-285 | Novel, orally active, dual HER2/EGFR inhibitor | I | 86,87 |
| ARRY-380 | Orally active, reversible and selective ErbB-2 inhibitor | I-II | 88,89 |
| MM-121 | Fully human monoclonal antibody ErbB3 (Her3) | I-II | 93,94 |
Novel drugs targeting HER-3.
| Drug Name | Description | Phase | References |
|---|---|---|---|
| MM-121 | Fully human monoclonal antibody ErbB3 (Her3) | I-II | 93,94 |
Therapies targeting HER signaling
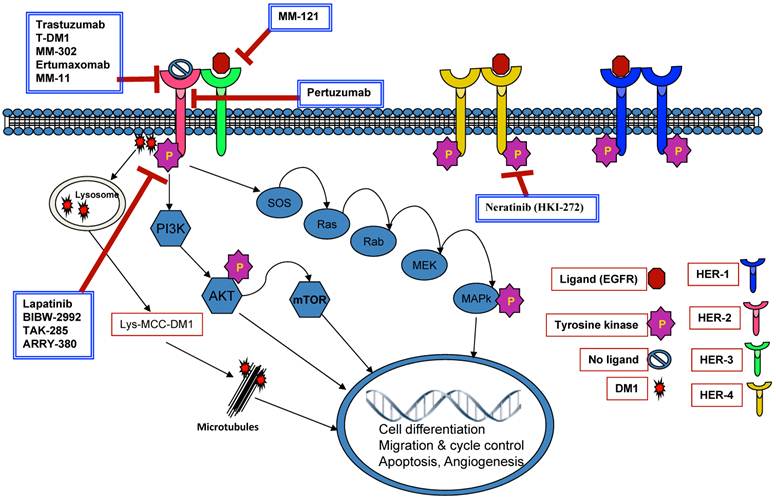
Targeting DNA repair pathways: PARP1 Inhibitors
The poly (adenosine diphosphate [ADP])-ribose) polymerases (PARPs) are a family of enzymes involved in DNA repair, gene transcription, chromatin architecture, and apoptosis in normal human cells (95-97). The most abundant is PARP1, a key player in single-stranded DNA base-excision repair (95-100). PARP inhibition leads to the accumulation of single strand DNA breaks and subsequent double strand breaks at replication forks that ultimately induce apoptosis and cell death (100-102).
In normal cells, these breaks are repaired via the homologous recombination double-stranded DNA repair pathway. The tumor-suppressor proteins BRCA1 and BRCA2 are key components of the DNA repair pathway in humans (95,100). Normal cells, with intact homologous recombination, are able to tolerate the PARP inhibitors. However, in tumor cells lacking homologous recombination (i.e. BRCA1/2), PARP inhibition leads to cell death (101,102). This phenomenon has been described as synthetic lethality.
Farmer et al, demonstrated in vitro that PARP inhibitors are active only in the presence of BRCA mutations (103). The majority of woman with BRCA1 mutations develop triple negative breast cancer (102,103). This fact highlights the importance of the study of PARP inhibitors because currently there are no biological or targeted therapies approved for the treatment of BRCA-associated TNBC, which is an aggressive subtype of breast cancer with a poor prognosis after relapse (102). Iniparib, olaparib, and veliparib are the most investigated PARP inhibiting agents to date.
Until recently, iniparib was considered to be an irreversible inhibitor of PARP 1. However, Liu et al. showed that the primary mechanism of action of iniparib was not via the inhibition of PARP activity. Apparently, the cysteine-containing proteins in tumor cells are modified non-selectively by iniparib (104). O'Shaughnessy et al. conducted a phase II randomized study to compare the efficacy and safety of gemcitabine and carboplatin with or without iniparib. One hundred and twenty three patients with triple-negative MBC were included. The addition of iniparib prolonged the median progression-free survival from 3.6 months to 5.9 months (HR 0.59; P = 0.01) and the median overall survival from 7.7 months to 12.3 months (HR 0.57; P= 0.01), with no significant difference in the toxicity (neutropenia and thrombocytopenia being the most common grade 3 or 4 toxicities) between the two groups (105). Based on these results, a phase III trial was subsequently conducted in order to evaluate the overall survival and progression-free survival. Its primary endpoint was not met (106).
Olaparib (AZD2281- KU-0059436) is an orally active PARP inhibitor that induces apoptosis and cell death in homozygous BRCA-deficient cells (107). Fong et al. conducted a phase I study of olaparib focusing on BRCA1 and BRCA2 mutation carrying breast cancer patients, establishing the maximum tolerated dose and toxic profile of this agent. Olaparib was well tolerated, with grade 1-2 nausea, fatigue and vomiting being the most commonly observed adverse reactions (95). In the same study, investigators observed high rates of tumor response only in the BRCA1 or BRCA2 carriers (95). These promising results strongly suggest that BRCA mutation is necessary for the clinical activity of this agent in patients with MBC (100).
A phase II trial in patients with BRCA1 or BRCA2 mutation-associated, chemotherapy-refractory MBC evaluated two different dose schedules of olaparib: olaparib 400 mg once daily (cohort 1) vs. 100 mg twice daily (cohort 2). Overall radiographic response rates were 41% (11 of 27) for the first cohort and 22% (6 of 27). The most common grade 3-4 adverse reactions in both groups were nausea and fatigue (107). Gelmon et al. recently published results of a phase II study of olaparib in patients with advanced high-grade serous and/or undifferentiated ovarian carcinoma or TNBC. However, no confirmed objective responses were reported in the breast cancer group (108).
Veliparib is another potent PARP1 inhibitor (109). Donawho et al., using a breast xenograft model, were able to show the potentiating effect of veliparib when combined with cisplatin, carboplatin, and cyclophosphamide in tumor regression (110). Results of a phase I trial conducted by Kummar et al. established the maximum tolerated dose of veliparib at 10 mg orally twice a day, with neutropenia and thrombocytopenia as the most relevant dose limiting toxicities (111). A phase II study showed a synergistic activity for veliparib combined with temozolomide in breast cancer patients. The progression-free survival was 5.5 months in the BRCA-mutated group versus 1.8 months for patients without a BRCA mutation, findings that suggests that veliparib is only clinically active when the BRCA mutation is present (112).
Rucaparib (AG014699) demonstrates synergy with temozolamide in preclinical solid tumor models (113). A phase I trial of rucaparib combined with temozolomide in advanced solid tumors was conducted. This study established that doses up to 12 mg/m2 AG-014699 and 200 mg/m2 TMZ were safe and able to inhibit PARP in peripheral blood lymphocytes and tumor. No dose-limiting toxicity was observed. The adverse events were grade 1/2, except 1 case each of grade 3 infection, fatigue, low phosphate and lymphopenia, in a total of 27 patients. (114). Phase 2 trials are currently ongoing (115).
MK-4827 is an oral PARP-1/2 inhibitor. A recent study recruited 39 patients with multiple malignancies (11 of those were BRCA mutation carriers). PARP inhibition in peripheral blood mononuclear cells was observed at doses of > 110 mg daily, achieving antitumor activity in both BRCA mutated and not mutated tumors. Grade 3 fatigue, pneumonitis and anorexia were the most common dose limiting toxicity factors found (116). Currently a phase I study aims to determine the MTD and evaluate the degree of inhibition of PARP activity in patients with advanced solid tumors (including breast cancer) or hematologic malignancies (117).
PARP inhibitors in clinical development.
| Drug Name | Description | Phase | Target Population | Ref |
|---|---|---|---|---|
| Iniparib | Intravenous small-molecule prodrug inhibitor of PARP-1 | I-III | triple-negative MBC | 104-106,118 |
| Olaparib [AZD2281] | Oral small molecule inhibitor of PARP | I-II | BRCA1 or BRCA2 mutation | 95,107-108 |
| Veliparib [ABT-888] | Oral PARP -1 and -2 inhibitor with chemosensitizing and antitumor activities | 0-II | BRCA1 or BRCA2 mutation | 109-112 |
| Rucaparib [AG-014699] | Oral tricyclic indole PARP1 inhibitor with potential chemosensitizing, radiosensitizing, and antineoplastic activities | I | BRCA1 or BRCA2 mutation | 113-115 |
| MK-4827 | Oral PARP 1/2 inhibitor | I | BRCA1 or BRCA2 mutation | 116,117 |
mTOR/PI3K
The mammalian target of rapamycin (mTOR) is a kinase that is part of the PI3K-related kinase family (119). mTOR plays a key role in cell cycle progression, and is inhibited by the antibiotic rapamycin (120). Over-activation of PI3K and mTOR has been observed in many cancers (121). As such, rapamycin, along with several rapamycin analogues (rapalogs), have been studied for the treatment of a variety of different cancers. The mTOR inhibitors temsirolimus and everolimus are currently approved for the treatment of renal cell carcinoma (122,123), and mTOR inhibitors have shown promise in a number of other types of cancers, including breast cancer.
Studies have shown that activation of the mTOR/PI3K pathway promotes anti-estrogen resistance (124). Pre-clinical models have shown that letrozole and everolimus both inhibit estrogen-induced breast cancer proliferation, and that these two agents in combination act synergistically to augment their anti-tumor activity even further (125). This cross-talk in activity has led researchers to combine an mTOR inhibitor and an aromatase inhibitor in the treatment of ER positive breast cancer. Everolimus has been the most widely studied mTOR inhibitor in breast cancer. The effect of everolimus added to an aromatase inhibitor was studied in a randomized phase II trial of 272 postmenopausal patients with operable ER positive breast cancer (126). Patients received 4 months of neoadjuvant letrozole combined with either everolimus or placebo. Response rate, determined by clinical palpation, favored the everolimus group 68.1% to 59.1%. In the everolimus group, 22.6% of the patients experienced a grade 3 or 4 adverse event, compared to 3.8% in the placebo group. A dose reduction or interruption in treatment due to an AE occurred in over half of the patients in the everolimus group. The most common grade 3 or 4 AEs were pneumonitis, pneumonia, and stomatitis. All three cases of pneumonitis resolved shortly after discontinuing everolimus.
Two schedules of everolimus as first or second line therapy in metastatic breast cancer were compared in a phase II trial of 49 patients (127). Patients were randomized to receive either 10mg daily or 70mg weekly doses of everolimus. Twelve percent of the patients in the daily therapy group responded, versus zero in the weekly therapy group. Pneumonitis occurred more frequently than expected; 11 of 33 patients in the daily group and 2 of 16 patients in the weekly group experienced grade two (symptomatic) or higher pneumonitis. Of note, most patients in this study had previously received radiation therapy. The most common adverse events include fatigue, rash, cytopenias, and elevated liver enzymes. Serious (grade 3 or 4) AEs include fatigue, infection, pneumonitis, cytopenias, and hyperglycemia, which lead to 27% of patients in the daily schedule and 13% on the weekly schedule to discontinue everolimus. Five of the 11 patients who withdrew from the study as a result of adverse toxicities did so due to pneumonitis.
BOLERO-2, a landmark phase 3 randomized placebo controlled trial, evaluated exemestane with or without everolimus in a 2:1 ratio in 724 postmenopausal patients with hormone receptor positive breast cancer who recurred or progressed while receiving a non-steroidal aromatase inhibitor (letrozole or anastrozole) and had advanced disease (128). Patients were allowed to have received other anticancer endocrine therapies and no more than one prior chemotherapy regimen. Patients were stratified, based on the presence of visceral metastasis and prior sensitivity to endocrine therapy, and received daily oral everolimus (10mg) or placebo combined with exemestane (25mg daily). Sensitivity to endocrine therapy was defined as at least 24 months before recurrence in patients receiving endocrine therapy as adjuvant therapy and 24 weeks of at least stable disease in patients with advanced breast cancer. PFS was the primary endpoint, with OS, ORR, and clinical benefit rate, among others, as secondary endpoints. Patients continued treatment until they developed disease progression, unacceptable toxicity, or withdrew from the study. 724 women at 189 centers in 24 countries were enrolled, with 485 receiving everolimus. The median age of the enrolled patients was 62, with 56% having visceral disease, and 76% with bony metastasis. 36% of patients had metastatic disease involving at least 3 sites, with lung and liver being the most commonly affected organs. All patients were ER positive and HER2 negative; 72% were PR positive. Every patient had received prior AI therapy with either letrozole or anastrozole, 48% had previously received tamoxifen, 16% had previously received fulvestrant, and 68% prior chemotherapy.
At the first formal interim analysis, which occurred after 359 events (approximately 60% of the PFS events), the median PFS was 6.9 months in patients receiving the combination of exemestane plus everolimus, versus 2.8 months for exemestane alone (HR for progression or death 0.43, 95% CI 0.35-0.54). Updated data was recently reported at the 2011 San Antonio Breast Cancer Symposium. After 457 events, the median PFS by central assessment favored the group that received the combination of exemestane/everolimus vs. exemestane alone (11.0 vs.4.1 months; HR 0.36, 95% CI 0.28-0.45) (129). ORR and clinical benefit rate favored the everolimus-receiving group, 12.0% vs.1.3% and 50.5% vs.25.5%, respectively. 22.6% of the patients in the exemestane-only group died, compared to 17.3% in the everolimus- exemestane group. Serious adverse events attributed to the study treatment occurred in 11% of patients receiving everolimus vs. only 1% of patients in the placebo arm. Discontinuation of therapy as a result of AEs occurred more frequently in the everolimus arm, 19% to 4%. Grade 3 or 4 AEs that occurred more frequently in the everolimus plus exemestane arm included: stomatitis/mucositis (8% vs.1%), anemia (7%vs. 1%), hyperglycemia (5% vs. <1%), dyspnea (4% vs. 1%), fatigue (4% vs. 1%), and pneumonitis (3% vs. 0%). The drug Everolimus, a kinase inhibitor, was recently approved (July 20, 2012) by the FDA for the treatment of postmenopausal women with locally advanced, unresectable or metastatic hormone receptor-positive, HER2-negative breast cancer (advanced HR+ BC) in combination with exemestane after failure of treatment with letrozole or anastrozole.
Other therapeutic targets
Multiple factors lead to resistance to anti-HER2 agents, including increased signaling via upstream growth factor receptors, such as those in the EGFR and IGFR1 families, PTEN mutations, and changes in the HER2 receptor. However, the major mechanism of trastuzumab resistance appears to be increased activation/signaling of PI3K/Akt (130). Preclinical models have shown that the combination of trastuzumab with an mTOR inhibitor act synergistically to inhibit tumor proliferation, and the addition of trastuzumab to an mTOR inhibitor reduces the activity of the PI3K, MAPK, and HER3 signaling pathways (131). As such, several studies examining the addition of mTOR inhibitors to anti-HER2 therapy as a means of overcoming developed resistance have been undertaken. The combination of everolimus and trastuzumab, with or without chemotherapy, has been explored in several phase I and II clinical trials (132 -135). The ORR of this combination therapy was approximately 20% in all 4 trials. Toxicities were tolerable, with the most common grade 3-4 AE being neutropenia. BOLERO-1 is a randomized, double-blind phase III RCT that will compare trastuzumab and paclitaxel, with or without everolimus, as first line therapy in women with locally advanced or metastatic breast cancer (136). In BOLERO-3, a double-blind, randomized phase III RCT, the addition of everolimus to vinorelbine and trastuzumab is being compared to vinorelbine and trastuzumab alone in women with HER2 positive, locally advanced or metastatic breast cancer (137). Both of these trials are currently enrolling patients.
Much less data is available in MBC with the mTOR inhibitor temsirolimus. To date, there have been two reported trials involving temsirolimus, one phase II trial (138) and one phase I (139). Of the 31 patients in the phase II trial, none had an objective response to temsirolimus.
PI3K inhibitors
Phosphatidylinositol 3-kinases (PI3K) are a family of enzymes involved in multiple important cellular functions including proliferation, cell growth, differentiation, motility, and survival (140). Aberrant activation of PI3K has been implicated in different cancers. The gene that encodes the p110α catalytic subunit of PI3K (PIK3CA) is the most commonly mutated gene in breast cancer (141,142). PI3K promotes estrogen receptor activity, and mutations of PI3K can mediate resistance to endocrine therapy (143). Clinical trials with PI3K inhibitors in breast cancer patients are still in early phases of development. There have been 2 phase I/II trials reported involving SAR245408 or SAR245409, pan-inhibitors of PI3K (144,145). GDC-0941, a pan-inhibitor of PI3K, has been studied in two phase I trials (146,147). A third trial involving GDC-0941 in combination with fulvestrant is also currently ongoing (148).
Insulin-like growth factor inhibitors
Insulin-like growth factors I and II (IGF-I and IGF-II) play an integral role in the growth and development of somatic tissue, including bone and skeletal muscle (148). IGFs bind to IGF-receptors (IGF-R), helping regulate cellular functioning (149). Expression of both IGF and IGF-R increase during fetal development and in several types of cancers, including breast cancer (150), making the IGF-R a promising target for antibody directed therapy. An anticipated complication of therapy that targets IGF-R is hyperglycemia, as targets to the IGF-R may also bind to the insulin receptor (151). Aside from directly leading to the inhibition of tumor growth, IGF-R targeted therapy may also play a role in treating cancer by reversing resistance to endocrine, cytotoxic, or other therapies that may develop during the course of treatment.
Monoclonal antibodies targeting the IGF receptor have been the subject of many phase I and II clinical trials. One phase III trial involving figitumumab, a monoclonal antibody (MOAB) that targets/inhibits IGF-1R, combined with chemotherapy in the treatment of non-small cell lung cancer, has been reported (151). The study was terminated early due to lack of clinical benefit. Serious adverse events occurring more often in the figitumumab arm included dehydration, hyperglycemia, and hemoptysis.
Five agents that target the IGF-R pathway have been or are being studied in the treatment of breast cancer. Ganitumab (AMG 479), figitumumab (CP-751,871), dalotuzumab (MK-0646, h7C10), and cixutumumab (IMCA12) are MOABs that target IGF-1R, while BMS-754807 is an IGF-1R/insulin receptor kinase inhibitor. The effect of ganitumab combined with hormonal therapy was studied in a randomized phase II double blind trial of 156 patients with ER and/or PR positive metastatic or locally advanced breast cancer who had received prior anti-estrogen therapy (152). In a 2:1 allotment, patients received either ganitumab or placebo, combined with exemestane or fulvestrant, per investigator's choice. The addition of ganitumab to exemestane or fulvestrant did not appear to improve median PFS or ORR. Grade 3-4 SAE occurred in 42% of patients receiving ganitumab, the most common being: hyperglycemia (6%), neutropenia (6%), thrombocytopenia (4%), asthenia (4%), and transaminitis (4%). A second trial involving ganitumab in breast cancer is currently ongoing (85).
In a randomized phase II trial, 205 patients with stage IIIB - IV, hormone receptor positive breast cancer received figitumumab plus exemestane or exemestane alone as first-line therapy (153). There was a non-significant trend toward improved median PFS with figitumumab. The most common grade 3-4 SAE associated with figitumumab included: transient hyperglycemia, the development of diabetes mellitus, weight loss, elevated GGT, and fatigue. Grade 3-4 hyperglycemia occurred in 12% of patients. Dalotuzumab (MK-0646, h7C10) will be combined with ridaforolimus, an mTOR inhibitor, in a phase II trial that will explore the efficacy and safety of these two agents in the treatment of ER positive breast cancer that progressed on aromatase inhibitors (154). There are currently three trials involving cixutumumab (IMCA12) in the treatment of breast cancer in various phases of development (155,156). One of these trials is studying the combination of cixutumumab and temsirolimus, an mTOR inhibitor (158). BMS-754807, the sole TKI that targets the IGF-R that has been studied in breast cancer, will be compared to BMS-754807 plus letrozole in a phase II trial involving patients with locally advanced or metastatic ER positive breast cancer (158).
mTOR, PI3K, and Insulin-like growth factor inhibitors in clinical development.
| Drug Name | Description | Phase | Target Population | References |
|---|---|---|---|---|
| Everolimus | Inhibits the mTORC1 protein | I-III | ER or PR positive (126, 128) HER2 positive (132-137) | 126-128,132-137 |
| Temsirolimus | Inhibits the mTORC1 protein | I-III | ER, PR, or HER2 positive (138) TNBC or HER2 positive (139) | 138,139 |
| BEZ235 | Dual inhibitor of PI3K and mTOR | I | ER positive | 158 |
| GDC-0941 | PI3K inhibitor | HER2 positive (146) | 146,147 | |
| SAR245408 | PI3K inhibitor | I/II | HER2 positive | 144 |
| SAR245408 or SAR245409 | PI3K inhibitor | I/II | ER or PR positive | 145 |
| Ganitumab (AMG 479) | MOAB that targets IGF-1R | II | ER or PR positive | 152 |
| Figitumumab (CP-751,871) | MOAB that targets IGF-1R | II | ER or PR positive | 153 |
| Dalotuzumab (MK-0646, h7C10) | MOAB that targets IGF-1R | II | ER positive | 154 |
| Cixutumumab (IMCA12) | MOAB that targets IGF-1R | II | HER2 positive (155) ER or PR positive (156) ER positive (158) | 155,156,158 |
| BMS-754807 | IGF-1R/insulin receptor kinase inhibitor | II | ER positive | 158 |
Heat Shock Protein 90 (HSP 90)
HSP 90 is a molecular chaperone protein which assists in the folding and stabilization of proteins vital to cell survival (160). It assists in the stability and function of many proteins associated with cancer cell propagation, including estrogen receptors, HER2, EGFR, VEGFR, BCR-ABL, AKT, FLT3, MET, BRAF, and CRAF, among others, making it an ideal target for cancer treatment strategies. Breast cancers that express higher levels of HSP-90 are associated with a higher nuclear grade, larger tumors, increased lymph node involvement, increased expression of HER2 and ER, and more aggressive clinical features, e.g. decreased survival (161). Several different HSP-90 inhibitors have recently been studied in phase II trials. Tanespimycin (17-AAG) was combined with trastuzumab in 31 patients with advanced, HER-2 positive breast cancer who had progressed on trastuzumab (162).The ORR was 22%, median PFS was 6 months, and median OS was 17 months. There were no grade 4 AEs. Grade 3 AEs that occurred in more than 5% of the patients were transaminitis (10%), headache (7%), and fatigue (7%). Five patients withdrew from the study, one each for: fatigue, decreased ejection fracture (which also had occurred when she received trastuzumab previously), depression, transaminitis, and an atypical reaction to therapy (tremor plus unresponsiveness to verbal stimuli). Tanespimycin was added to trastuzumab in 29 patients with HER2 positive MBC who progressed after receiving trastuzumab (163). Of the 21 evaluated patients, five had a PR. The most common AEs were fatigue (39%), diarrhea (33%), dizziness (24%), and headache (19%). No grade 3 or 4 AEs occurred in more than 5% of the patients.
Retaspimycin (IPI-504) was combined with trastuzumab in 26 patients with advanced, HER-2 positive breast cancer who had progressed on trastuzumab (164). Among the 20 evaluable patients, there was 1 PR and SD in 14. The only grade 3-4 toxicities were grade 3 transaminits in a patient with liver metastasis, grade 3 vomiting, and grade 3 hypokalemia due to grade 1 diarrhea. Ganetespib, an HSP-90 inhibitor with broader activity then tanespimycin, was studied as single-agent therapy in an unselected cohort of patients with locally advanced or metastatic breast cancer (165). Data on 14 patients has been reported thus far. Of the 10 evaluable patients, there is 1 PR, 1 minor response, and 2 SD. Grade 3 abdominal pain and diarrhea requiring dose reduction and asymptomatic and reversible elevation in serum amylase occurred in one patient each.
Histone Deacetylase Inhibitors
The transcriptional factor hypoxia-inducible factor-1 alpha (HIF-1 α) regulates multiple cellular signaling pathways (166). HIF-1 α promotes angiogenesis by increasing the expression of VEGF, and HIF-1 α-induced over-expression of VEGF has been identified in various malignancies (165). Histone deacetylase inhibitors have been shown to indirectly (167) and directly (168) negatively regulate HIF-1 α, offering promising options for the treatment of tumors, which proliferate via stimulation from VEGF.
The use of valproic acid as an HDAC inhibitor was tested in a phase I/II clinical trial conducted by Munster et al (176). In this study, 44 patients with advanced solid malignancies were enrolled in the phase I portion, with 15 women with MBC enrolled into the cohort expansion. Breast cancer patients in the cohort expansion group received 120 mg/kg/day of valproic acid followed by FEC100 (epirubicin 100 mg/m2 with 5-fluorouracil 500 mg/m2 and cyclophosphamide 500 mg/m2). Partial responses were seen in 9 of 41 (22%) patients in the phase 1 portion. Objective responses were seen in 9 of 14 (64%) evaluable patients at the dose expansion cohort, after a median number of 6 cycles of therapy. The most common observed toxicities were valproic acid-associated somnolence and epirubicin-induced myelosuppression (169).
Three other HDAC inhibitors have been clinically evaluated to date in breast cancer patients: vorinostat, entinostat, and panobinostat. Initial studies used an HDAC inhibitor as an adjunct to AI therapy in patients who had progressed on an AI. Vorinostat was combined with tamoxifen in 19 patients with MBC who progressed on prior AI therapy (170). Significant toxicities included two venous thromboembolic events and grade 3 fatigue in 3 patients. The most common grade 2 toxicities included: fatigue, nausea/vomiting, anorexia, bleeding, and myelosuppression. Of the 17 patients evaluated for efficacy, 1 had a CR, 3 had PR, and 1 had SD for 12 months. A phase II trial involving 27 patients who were progressing on AI therapy received entinostat while continuing their same AI therapy (171). One patient had a PR and one had SD > 6 months. The most common grade 3-4 toxicities were: fatigue, dyspnea, diarrhea, and lethargy. A phase II trial of 114 patients with ER positive metastatic breast cancer progressing on an AI in which patients will be randomized, in a double-blind fashion, to continue their AI with or without the addition of entinosat, is currently enrolling patients (172). Panobinostat and capecitabine, with or without lapatinib, was studied in a phase I trial involving 15 patients (173). There was one DLT of grade IV thrombocytopenia, as well as 2 cases of grade 3 thrombocytopenia. Grade 3 anemia, dehydration, fatigue, peripheral edema, and hand-foot syndrome each occurred in one patient.
HSP-90 and HDAC inhibitors in clinical development.
| Drug Name | Description | Phase | Target Population | References |
|---|---|---|---|---|
| Tanespimycin (17-AAG) | HSP-90 inhibitor | II | HER2 positive | 162, 163 |
| Retaspimycin (IPI-504) | HSP-90 inhibitor | II | HER2 positive | 164 |
| Ganetespib | HSP-90 inhibitor | II | advanced or MBC | 165 |
| Vorinostat | HDAC inhibitors | II | ER positive | 170 |
| Entinostat | HDAC inhibitors | II | ER positive | 171, 172 |
| Panobinostat | HDAC inhibitors | I | advanced or MBC | 173 |
Concluding Remarks
The paradigm for treating breast cancer has changed rather dramatically over the last decade. Several targeted therapies, e.g. trastuzumab and anti-estrogen therapies, have greatly improved patient outcomes. A much more detailed understanding of the underlying biology that drives malignant progression and metastases has yielded other novel targets including PI3K/mTOR and PARP, which are currently being tested clinically. One of the challenges, particularly in treating metastatic breast cancer, is that even with improved systemic therapies, the disease remains incurable. As more drugs that target specific pathways are developed, tumors develop means to evade these agents, particularly when there is redundancy in most biologic processes. The robustness, evolvability, modularity, redundancy, diversity, system control, tolerance, and plasticity are hallmarks of network pathways (174) which will further lead to difficulties of individual compounds to be successfully used against cancer growth for longer periods of time. One potential strategy is to combine multiple drugs that hit biologically important pathways at different places. Such a combinatorial strategy (175) will definitely increase efficacy, but we must choose wisely which combinations to pursue in future clinical trials. We believe that more thorough preclinical testing may help us make more informed decisions on which combinations should be brought forward into the clinic, especially when it is unlikely that we will be able to empirically test every possible combination. The next decade will hopefully bring new treatment paradigms that will continue to build on progress made over preceding two decades, and further improve clinical outcomes and survival rates for patients with breast cancer.
For HER2 positive MBC, given the availability of several HER2 targeted agents and the anticipated approval of newer agents as detailed above, in the future the clinician may be challenged as to how to sequence these therapies in clinical practice. An evidence-based approach should guide therapeutic decision making in an era where economic considerations are also paramount. Certainly, this question will be the subject of future studies. However, if one uses the paradigm of choosing highly efficacious therapies which have the least toxicities, this can be a guiding principle upon which treatments are chosen.
The negative reputation of HER2 positive metastatic breast cancer is changing with the approval of several new and well tolerated HER2 targeted treatments. In some aspects, this disease may be considered a “double-edged sword” due to the aggressive tumor biology, but at the same time beneficial due to the availability of a biomarker that can become a target for successful therapy. This in turn gives clinicians the availability of several less toxic targeted therapies which can drastically change the natural history of the disease. In fact it speaks to how far we have come in treating breast cancer as not just one disease, as our treatments will become “personalized” to specific subtypes of the disease. We can tailor our therapy to the presence of functional genes: molecular profiling will become much more used in the near future and more such targeted compounds may become reality. Much work is of course still needed to unfold the complex personalized networks of tumor proliferation and resistance mechanisms (176).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. GLOBOCAN 2008 v1.2, Cancer Incidence and Mortality Worldwide: IARC CancerBase No.10 [Internet]. Lyon, France: International Agency for Research on Cancer. 2010
2. Mathew J, Perez E.A. Trastuzumab emtansine in human epidermal growth factor receptor 2-positive breast cancer: a review. Curr Opin Oncol. 2011;23:594-600
3. Jemal A, Siegel R, Xu J. et al. Cancer statistics. CA Cancer J Clin. 2010;60:277-300
4. Siegel R, Ward E. et al. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61(4):212-36
5. American Cancer Society. Cancer Facts and Figures, 2011. Atlanta, GA: American Cancer Society. 2011
6. Rugo H, Taylor D, Sanon M, Clements K, Balu S, Faria C, Teitelbaum A. Survival in US Women Following an Indication of Metastatic Breast Cancer Diagnosis and Chemotherapy Initiation: A SEER-Medicare Analysis, (P1-08-07). Poster Presentation. San Antonio Breast Cancer Symposium, San Antonio, Texas. 2012
7. Carlson RW, Allred DC, Anderson BO, Burstein HJ. et al. Breast cancer. Clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2009Feb;7(2):122-92
8. Buzdar AU. Role of biologic therapy and chemotherapy in hormone receptor- and HER2-positive breast cancer. Ann Oncol. 2009Jun;20(6):993-9
9. Slamon DJ, Leyland-Jones B, Shak S. et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783-792
10. Anders CK, Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer. 2009Jun;9(Suppl 2):S73-81
11. Perou CM, Sørlie T, Eisen MB. Molecular portraits of human breast tumours. Nature. 2000Aug17;406(6797):747-52
12. Sotiriou C, Pusztai L. Gene-expression signatures in breast cancer. N Engl J Med. 2009;360(8):790-800
13. Buyse M, Loi S, van't Veer L, Viale G. et al. Validation and clinical utility of a 70-gene prognostic signature for women with node-negative breast cancer. J Natl Cancer Inst. 2006Sep6;98(17):1183-92
14. Glück S, Yip AY, Ng EL. Can we replace the microscope with microarrays for diagnosis, prognosis and treatment of early breast cancer? Expert Opin Ther Targets. 2012(Suppl 1):S17-22
15. Hernandez-Aya LF, Chavez-Macgregor M, Lei X. et al. Nodal status and clinical outcomes in a large cohort of patients with triple-negative breast cancer. J Clin Oncol. 2011Jul1;29(19):2628-34
16. Ismail-Khan R, Bui MM. A review of triple-negative breast cancer. Cancer Control. 2010Jul;17(3):173-6
17. Dawood S, Broglio K, Buzdar AU. et al. Prognosis of women with metastatic breast cancer by HER2 status and trastuzumab treatment: an institutional based review. J Clin Oncol. 2010;28:92-98
18. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177-182
19. Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A. et al. Studies of the HER-2/neu protooncogene in human breast and ovarian cancer. Science. 1989;244:707-712
20. Olson EM. et al. Responses to subsequent anti-HER2 therapy after treatment with trastuzumab-DM1 in women with HER2-positive metastatic breast cancer. Ann Oncol. 2012;23(1):93-97
21. Yarden Y, Sliwkowski MX. Untangling the ErbB signaling network. Nat Rev Mol Cell Biol. 2001;2:127-137
22. Package Insert Herceptin (Trastuzumab) Genetech, Inc. http://www.accessdata.fda.gov/drugsatfda_docs/label/2000/trasgen020900LB.htm
23. Leyland-Jones B. Trastuzumab: hopes and realities. Lancet Oncol. 2002;3(3):137-44
24. Yin W. et al. Trastuzumab in the adjuvant treatment of HER2-positive early breast cancer patients: a meta-analysis of published randomized controlled trials. PLoS One. 2011;6(6):e21030
25. Baselga J, Tripathy D, Mendelsohn J. et al. Phase II study of weekly intravenous recombinant humanized antip185HER2 monoclonal antibody in patients with HER2/neu-overexpressing metastatic breast cancer. J Clin Oncol. 1996;14:737-744
26. Vogel CL, Cobleigh MA, Tripathy D. et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:719-726
27. Burstein HJ, Kuter I, Campos SM. et al. Clinical activity of trastuzumab and vinorelbine in women with HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2001;19:2722-2730
28. Montemurro F, Choa G, Faggiuolo R. et al. A phase II study of three-weekly docetaxel and weekly trastuzumab in HER2-overexpressing advanced breast cancer. Oncology. 2004;66:38-45
29. Jahanzeb M, Mortimer JE, Yunus F. et al. Phase II trial of weekly vinorelbine and trastuzumab as first-line therapy in patients with HER2(þ) metastatic breast cancer. Oncologist. 2002;7:410-417
30. Cobleigh MA, Vogel CL, Tripathy D. et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17:2639-2648
31. Romond EH, Perez EA, Bryant J. et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353:1673-1684
32. Esteva FJ, Valero V, Booser D. et al. Phase II study of weekly docetaxel and trastuzumab for patients with HER-2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:1800-1808
33. Valero V, Forbes J, Pegram MD. et al. Multicenter phase III randomized trial comparing docetaxel and trastuzumab with docetaxel, carboplatin, and trastuzumab as first-line chemotherapy for patients with HER2-gene-amplified metastatic breast cancer (BCIRG 007 study): two highly active therapeutic regimens. J Clin Oncol. 2011;29:149-156
34. Andersson M, Lidbrink E, Bjerre K. et al. Phase III randomized study comparing docetaxel plus trastuzumab with vinorelbine plus trastuzumab as first-line therapy of metastatic or locally advanced human epidermal growth factor receptor 2-positive breast cancer: the HERNATA study. J Clin Oncol. 2011;29:264-271
35. Perez EA, Suman VJ, Davidson NE. et al. Results of Chemotherapy Alone, with Sequential or Concurrent Addition of 52 Weeks of Trastuzumab in the NCCTG N9831 HER2-Positive Adjuvant Breast Cancer Trial [Abstract #80]. Cancer Res. 2010;70:5640
36. Perez EA, Romond EH, Suman VJ. et al. Original report: 4-year follow-up of trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer: Joint analysis of data from NCCTG N9831 and NSAB B-31. J Clin Oncol. 2011;29:3366-3373
37. Saini K. et al. Beyond trastuzumab: New treatment options for HER2-positive breast cancer. The Breast. 2011;20:S20-S27
38. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology; Breast Cancer Version 2.2011. http://www.nccn.org/professionals/physician_gls/PDF/breast.pdf
39. Baselga J. Clinical trials of Herceptin(trastuzumab). Eur J Cancer. 2001;37:S18-24
40. Burstein HJ, Keshaviah A, Baron AD. et al. Trastuzumab plus vinorelbine or taxane chemotherapy for HER2-overexpressing metastatic breast cancer: the trastuzumab and vinorelbine or taxane study. Cancer. 2007;110:965-972
41. Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M, Untch M, Rusnak DW, Spehar G, Mullin RJ, Keith BR, Gilmer TM, Berger M, Podratz KC, Slamon DJ. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66:1630-1639
42. Tykerb [package insert] (2011). http://us.gsk.com/products/assets/us_tykerb.pdf
43. Ahn ER, Vogel CL. Dual HER2-targeted approaches in HER2-positive breast cancer. Breast Cancer Res Treat; DOI: 10.1007/s10549-011-1781-y.
44. Johnston S, Pippen JJr, Pivot X, Lichinitser M, Sadeghi S, Dieras V, Gomez HL, Romieu G, Manikhas A, Kennedy MJ, Press MF, Maltzman J, Florance A, O'Rourke L, Oliva C, Stein S, Pegram M. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol. 2009;27:5538-5546
45. Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, Crown J, Chan A, Kaufman B, Skarlos D, Campone M, Davidson N, Berger M, Oliva C, Rubin SD, Stein S, Cameron D. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733- 2743
46. Blackwell KL, Burstein HJ, Storniolo AM, Rugo H, Sledge G, Koehler M, Ellis C, Casey M, Vukelja S, Bischoff J, Baselga J, O'Shaughnessy J. Randomized study of lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J Clin Oncol. 2010;28:1124-1130
47. Fang L. et al. Targeted therapy in breast cancer: what's new? Swiss Med Wkly. 2011;141:w13231
48. Nahta R, Esteva FJ. Trastuzumab: triumphs and tribulations. Oncogene. 2007;26(25):3637-43
49. Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3:269-280
50. Bartsch R, Wenzel C, Gampenrieder SP. et al. Trastuzumab and gemcitabine as salvage therapy in heavily pre-treated patients with metastatic breast cancer. Cancer Chemother Pharmacol. 2008;62:903-910
51. Von Minckwitz G, du BA, Schmidt M. et al. Trastuzumab beyond progression in human epidermal growth factor receptor 2-positive advanced breast cancer: a German Breast Group 26/Breast International Group 03-05 study. J Clin Oncol. 2009;27:1999-2006
52. EA Perez. et al. Current and Emerging Targeted Therapies for Metastatic Breast Cancer. Cancer. 2011
53. Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blattler WA, Lambert JM, Chari RV, Lutz RJ, Wong WL, Jacobson FS, Koeppen H, Schwall RH, Kenkare-Mitra SR, Spencer SD, Sliwkowski MX. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68:9280-9290
54. Remillard S, Rebhun LI, Howie GA, Kupchan SM. Antimitotic activity of the potent tumour inhibitor maytansine. Science. 1975;189:1002-1005
55. Oroudjev E, Lopus M, Wilson L, Audette C, Provenzano C, Erickson H, Kovtun Y, Chari R, Jordan MA. Maytansinoid-antibody conjugates induce mitotic arrest by suppressing microtubule dynamic instability. Mol Cancer Ther. 2010;9:2700-2713
56. Chari RV. Targeted cancer therapy: conferring specificity to cytotoxic drugs. Acc Chem Res. 2008;41:98-107
57. Kovtun YV, Goldmacher VS. Cell killing by antibody-drug conjugates. Cancer Lett. 2007;255:232-240
58. Barok et al. Trastuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in vivo. Breast Cancer Research. 2011;13:R46
59. LoRusso P. et al. Trastuzumab Emtansine: A Unique Antibody-Drug Conjugate in Development for Human Epidermal Growth Factor Receptor 2-Positive Cancer. Clin Cancer Res. 2011;17:6437-6447
60. Gupta M. et al. Clinical Implications of pathophysiological and demographic covariates on the population pharmacokinetics of trastuzumab emtansine, a HER2-targeted antibody-drug conjugate, in patients with HER2-positive metastatic breast cancer. J Clin Pharmacol. 2012May;52(5):691-703
61. Krop IE, Beeram M, Modi S, Jones SF, Holden SN, Yu W. et al. Phase I study of trastuzumab-DM1, a HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol. 2010;28:2698-704
62. Miller K, Gianni L, Andre F. et al. A phase Ib/II trial of trastuzumab-DM1 (T-DM1) with Pertuzumab for women with HER2-positive, locally advanced or metastatic breast cancer (BC) who were previously treated with trastuzumab (T) [abstract]. J Clin Oncol. 2010 28
63. Burris HA III, Rugo HS, Vukelja SJ. et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J Clin Oncol. 2011;29:398-405
64. Perez EA, Dirix L, Kocsis J. et al. Efficacy and safety of trastuzumab- DM1 versus trastuzumab plus docetaxel in HER2-positive metastatic breast cancer patients with no prior chemotherapy for metastatic disease: preliminary results of a randomized, multicenter, open-label phase 2 study (TDM4450G) [abstract]. Ann Oncol. 2010;21:viii2
65. Roche announces positive phase II results for trastuzumab emtansine (T-DM1) in HER2-positive metastatic breast cancer. Media release, April 7, 2011 Roche. http://www.roche.com/media/media_releases/med-cor-2011-04-07b.htm
66. A Study of Trastuzumab-Emtasine (T-DM1) Plus Pertuzumab/Pertuzumab Placebo Versus Trastuzumab [Herceptin] Plus a Taxane in Patients With Metastatic Breast Cancer (MARIANNE). NCT01120184. http://clinicaltrials.gov
67. A Phase III Randomized, Multicenter, Two-Arm, Open-Label Trial to Evaluate the Efficacy of Trastuzumab Emtansine Compared With Treatment of Physician's Choice in Patients With HER2 Positive Metastatic Breast Cancer Who Have Received at Least Two Prior Regimens of HER2 Directed Therapy. NCT01419197. http://clinicaltrials.gov
68. Blackwell KL, Miles D, Gianni L. et al. Primary results from EMILIA, a phase III study of trastuzumab emtansine (T-DM1) versus capecitabine (X) and lapatinib (L) in HER2-positive locally advanced or metastatic breast cancer (MBC) previously treated with trastuzumab (T) and a taxane. 2012 ASCO Annual Meeting. J Clin Oncol. 2012;30:abstrLBA1
69. Baselga J, Gelmon KA, Verma S. et al. Phase II trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J Clin Oncol. 2010;28:1138-1144
70. Baselga J. et al. Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist. 2011;16(Suppl 1):12-19
71. Baselga J, Cortés J, Sung-Bae K. et al. Pertuzumab plus Trastuzumab plus Docetaxel for Metastatic Breast Cancer. N Engl J Med. 2012;366:109-119
72. Gianni L, Pienkowski T, Im YH. et al. Neoadjuvant pertuzumab (P) and trastuzumab (H): antitumor and safety analysis of a randomized phase II study ('NeoSphere'). San Antonio: Thirty-Third Annual San Antonio Breast Cancer Symposium; December 8-12. 2010: Abstract.
73. Geretti E. et al. Abstract C90: HER2-targeted liposomal doxorubicin MM-302 has a favorable cardio safety profile in preclinical models. Molecular Cancer Therapeutics. 2011 10(11)
74. A Phase 1, Multi-Center, Open-Label, Dose-Escalation, Safety, and Pharmacokinetic Clinical Study of Intravenously Administered MM-302 in Patients With Advanced Breast Cancer. NCT01304797. http://clinicaltrials.gov
75. Kiewe P, Thiel E. Phase I Trial of the Trifunctional Anti-HER2 × Anti-CD3 Antibody Ertumaxomab in Metastatic Breast Cancer. Clin Cancer Res. 2006May15;12(10):3085-91
76. Phase II Study Of The Trifunctional Bispecific Anti-HER-2/Neu x Anti-CD3 Antibody Ertumaxomab In Patients With HER-2/Neu Overexpressing (3+ Or 2+/FISH+) Metastatic Breast Cancer Progressing After Trastuzumab Treatment. NCT00522457. http://clinicaltrials.gov
77. Denlinger CS. et al. A phase I/II and pharmacologic study of MM-111 in patients with advanced, refractory HER2-positive (HER2+) cancers. J Clin Oncol. 2010;28:abstrTPS169
78. McDonagh CF. et al. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Mol Cancer Ther. 2012
79. Yap TA, Vidal L, Adam J. et al. Phase I trial of the irreversible EGFR and HER2 kinase inhibitor BIBW 2992 in patients with advanced solid tumors. J Clin Oncol. 2010;28:3965-72
80. Stopfer P, Marzin K. et al. Afatinib pharmacokinetics and metabolism after oral administration to healthy male volunteers. Cancer Chemother Pharmacol. 2011
81. Hickish T, Wheatley D, Lin N. et al. Use of BIBW 2992, a novel irreversible EGFR/HER2 tyrosine kinase inhibitor (TKI), to treat patients with HER2-positive metastatic breast cancer after failure of treatment with trastuzumab. J Clin Oncol. 2009;27(15S):Abstract1023
82. Gunzer K. et al. Addition of BIBW 2992, an irreversible inhibitor of EGFR/HER1 and HER2, to letrozole in estrogen receptor (ER)-positive metastatic breast cancer (mBC) progressing on letrozole monotherapy. Cancer Res. 2009;69(24 Suppl):Abstract4098
83. Schuler MH. et al. BIBW 2992, a novel irreversible EGFR/HER1 and HER2 tyrosine kinase inhibitor, for the treatment of patients with HER2-negative metastatic breast cancer after failure of no more than two prior chemotherapies. J Clin Oncol. 2010;28:abstr1065
84. Harbeck N, Schmidt M, Harter P. et al. BIBW 2992, a novel irreversible EGFR/HER1 and HER2 tyrosine kinase inhibitor for the treatment of patients with HER2-negative metastatic breast cancer after failure of no more than two prior chemotherapies. Cancer Res. 2009;69(24 Suppl):Abstract5062
85. Wong KK, Fracasso PM, Bukowski RM. et al. A phase I study with neratinib (HKI- 272), an Irreversible pan ErbB receptor tyrosine kinase inhibitor, in patients with solid tumors. Clin Cancer Res. 2009;15:2552-8
86. Doi T. Phase I first-in-human study of TAK-285, a novel investigational dual HER2/EGFR inhibitor, in cancer patients et al. British Journal of Cancer. 2012;106:666- 672
87. A multicenter, open-label, non comparative phase I clinical and pharmacokinetic study of oral TAK-285 in patients with advanced cancer. NCT00535522. http://clinicaltrials.gov
88. Lee P. et al. in Vivo Activity of ARRY-380, a Potent, Small Molecule Inhibitor of ErbB2 in Combination with Trastuzumab, Docetaxel or Bevacizumab. Cancer Research. 2009 69(24)
89. Moulder SL. et al. A phase I study to assess the safety, tolerability, and PK of ARRY-380, an oral HER2 inhibitor. Poster ASCO Breast Cancer Symposium. 2010
90. Burstein HJ, Sun Y, Dirix LY. et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Oncol. 2010;28:1301-7
91. I-SPY 2 Trial (Investigation of Serial Studies to Predict Your therapeutic Response With Imaging And moLecular Analysis 2). NCT01042379. http://clinicaltrials.gov
92. Gajria D, King TA, Pannu H. et al. Combined inhibition of mTORC1 with temsirolimus and HER2 with neratinib: a phase I study in patients with metastatic HER2-amplified breast cancer. Abstract ASCO Annual Meeting. 2011
93. Schoeberl B. et al. An ErbB3 Antibody, MM-121, Is Active in Cancers with Ligand-Dependent Activation. Cancer Res. 2010;70(6):2485-2494
94. Moyo V. A. A randomized, double-blind phase II trial of exemestane with or without MM-121 in postmenopausal women with locally advanced or metastatic estrogen receptor-positive (ER+) and/or progesterone receptor-positive (PR+), HER2-negative breast cancer. J Clin Oncol 29. 2011(suppl):abstrTPS112
95. Fong PC, Boss DS, Yap TA. et al. Inhibition of poly(ADPribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123-34
96. Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39:8-24
97. Amir E, Seruga B, Serrano R, Ocana A. Targeting DNA repair in breast cancer: a clinical and translational update. Cancer Treat Rev. 2010;36:557-565
98. Amé J-C. Spenlehauer C, de Murcia G. The PARP superfamily. BioEssays. 2004;26:88293
99. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nature Rev Cancer. 2008;8:193-204
100. Burstein H.J. Novel Agents and Future Directions for Refractory Breast Cancer. Semin Oncol. 2011;38(Suppl 2):S17-S24
101. Farmer H, McCabe N, Lord CJ. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917-21
102. Carey L.A. et al. PARP and Cancer — If It's Broke, Don't Fix It. N Engl J Med. 2011;364:277-279
103. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938-48
104. Liu X. et al. Iniparib non-selectively modifies cysteine-containing proteins in tumor cells and is not a bona fide PARP inhibitor. Clin Cancer Res. 2011 [Epub ahead of print]
105. O'Shaughnessy J. et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med. 2011;364(3):205-14
106. A phase 3, multi-center study of gemcitabine/carboplatin, with or without BSI-201, in patients with ER-, PR-, and Her2-negative metastatic breast cancer. NCT00938652. http://www.clinicaltrials.gov
107. Tutt A, Robson M, Garber JE. et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of concept trial. Lancet. 2010;376:235-244
108. Gelmon KA. et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011Sep;12(9):852-61
109. Yang SX, Kummar S, Steinberg SM, Murgo AJ, Gutierrez M. et al. Immunohistochemical detection of poly(ADP-ribose) polymerase inhibition by ABT-888 in patients with refractory solid tumor lymphomas. Cancer Biol Ther. 2009;8:2004-9
110. Donawho CK, Luo Y, Penning TD. et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13:2728-37
111. Kummar S. et al. Phase I Study of ABT-888, a PARP Inhibitor, in Combination with Topotecan Hydrochloride in Adults with Refractory Solid Tumors and Lymphomas. Cancer Res. 2011Sep1;71(17):5626-34
112. Isakoff SJ, Overmoyer B, Tung NM. et al. A phase II trial of the PARP inhibitor veliparib (ABT888) and temozolomide for metastatic breast cancer [abstract 1019]. J Clin Oncol. 2009;28(15S):118s
113. Ali M. et al. Vasoactivity of AG014699, a clinically active small molecule inhibitor of poly(ADP-ribose) polymerase: a contributory factor to chemopotentiation in vivo? Clin Cancer Res. 2009;15(19):6106-12
114. Plummer R. et al. First in human phase I trial of the PARP inhibitor AG-014699 with temozolomide (TMZ) in patients (pts) with advanced solid tumors. Journal of Clinical Oncology. 2005;23(16S):3065
115. A Phase I/II, Open-Label, Safety, Pharmacokinetic, and Preliminary Efficacy Study of Oral Rucaparib in Patients With BRCA Mutation Breast Cancer or Other Solid Tumor. NCT01482715. http://clinicaltrials.gov
116. Sandhu S. K, et al. First-in-human trial of a poly(ADP-ribose) polymerase (PARP) inhibitor MK-4827 in advanced cancer patients (pts) with antitumor activity in BRCA-deficient and sporadic ovarian cancers. J Clin Oncol. 2010;28:15s
117. A Phase I Study of MK-4827 in Patients With Advanced Solid Tumors or Hematologic Malignancies. NCT00749502. http://clinicaltrials.gov
118. Moulder S. et al. A Phase 1b Study To Assess the Safety and Tolerability of the PARP Inhibitor Iniparib (BSI-201) in Combination with Irinotecan for the Treatment of Patients with Metastatic Breast Cancer (MBC). Proc SABCS. 2010:AbstractP6 ~15~01
119. Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest. 2011Apr;121(4):1231-41
120. Vézina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo). 1975Oct;28(10):721-6
121. Chiang GG, Abraham RT. Targeting the mTOR signaling network in cancer. Trends Mol Med. 2007Oct;13(10):433-42
122. Hudes G, Carducci M. et al. Temsirolimus, Interferon Alfa, or Both for Advanced Renal Cell. N Engl J Med. 2007;356:2271-2281
123. Motzer RJ, Escudier B, Oudard S. et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372(9637):449-56
124. Stoica GE, Franke TF, Moroni M. et al. Effect of estradiol on estrogen receptor-alpha gene expression and activity can be modulated by the ErbB2/PI 3-K/Akt pathway. Oncogene. 2003;22(39):7998-8011
125. Boulay A, Rudloff J, Ye J. et al. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin Cancer Res. 2005;11(14):5319-28
126. Baselga J, Semiglazov V, van Dam P. et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol. 2009Jun1;27(16):2630-7
127. Ellard SL, Clemons M, Gelmon KA. et al. Randomized phase II study comparing two schedules of everolimus in patients with recurrent/metastatic breast cancer: NCIC Clinical Trials Group IND.163. J Clin Oncol. 2009Sep20;27(27):4536-41
128. Baselga J, Campone M. et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012Feb9;366(6):520-9
129. Hortobagyi GN, Piccart M, Rugo H, Burris H, Campone M, Noguchi S, Gnant M, Pritchard KI, Vittori L, Mukhopadhyay P, Sahmoud T, Lebwohl D, Baselga J. Everolimus for postmenopausal women with advanced breast cancer: updated results of the BOLERO-2 phase III trial (S3-7). Cancer Res. 2011:105-106s
130. Nahta R, O'Regan RM. Evolving strategies for overcoming resistance to HER2-directed therapy: targeting the PI3K/Akt/mTOR pathway. Clin Breast Cancer. 2010Nov;10(Suppl 3):S72-8
131. Miller TW, Forbes JT, Shah C, Wyatt SK. et al. Inhibition of mammalian target of rapamycin is required for optimal antitumor effect of HER2 inhibitors against HER2-overexpressing cancer cells. Clin Cancer Res. 2009Dec1;15(23):7266-76
132. Morrow PK, Wulf GM, Ensor J. et al. Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J Clin Oncol. 2011Aug10;29(23):3126-32
133. Dalenc F. et al. Everolimus in combination with weekly paclitaxel and trastuzumab in patients (pts) with HER2-overexpressing metastatic breast cancer (MBC) with prior resistance to trastuzumab and taxanes: A multicenter phase II clinical trial. J Clin Oncol. 2010;28:abstr1013
134. Jerusalem G, Fasolo A, Dieras V. et al. Phase I trial of oral mTOR inhibitor everolimus in combination with trastuzumab and vinorelbine in pre-treated patients with HER2-overexpressing metastatic breast cancer. Breast Cancer Res Treat. 2011Jan;125(2):447-55
135. Jerusalem G. et al. Maintenance with everolimus (RAD001) and trastuzumab (T) after discontinuation of chemotherapy in patients (pts) with heavily pretreated HER2-positive metastatic breast cancer (MBC): Pooled data of extension cohorts of phase Ib/II studies. J Clin Oncol. 2010;28:abstr1041
136. Everolimus in combination with trastuzumab and paclitaxel in the treatment of HER2 positive locally advanced or metastatic breast cancer (BOLERO 1). NCT00876395. http://clinicaltrials.gov
137. Daily Everolimus in Combination With Trastuzumab and Vinorelbine in HER2/Neu Positive Women With Locally Advanced or Metastatic Breast Cancer (BOLERO-3). NCT01007942. http://clinicaltrials.gov
138. Fleming GF, Ma CX, Huo D. et al. Phase II trial of temsirolimus in patients with metastatic breast cancer. Breast Cancer Res Treat. 2012
139. Gajria D, King T, Pannu H, Sakr R, Seidman AD, Modi S, Dickler M, Drullinsky P, Syldor A, Sujata P, Majid M, Norton L, Rosen N, Chandarlapaty S. Combined Inhibition of mTORC1 with Temsirolimus and HER2 with Neratinib: A Phase I/II Study in Patients with Metastatic HER2-Amplified or Triple-Negative Breast Cancer (PD09-08). Cancer Res 2011:113s.
140. Dumont AG, Dumont SN, Trent JC. The favorable impact of PIK3CA mutations on survival: an analysis of 2587 patients with breast cancer. Chin J Cancer. 2012Jul;31(7):327-34
141. Miller TW, Balko JM, Arteaga CL. et al. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol. 2011Nov20;29(33):4452-61
142. Bartholomeusz C, Gonzalez-Angulo AM. Targeting the PI3K signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012Jan;16(1):121-30
143. Campbell RA, Bhat-Nakshatri P, Patel NM. et al. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem. 2001Mar30;276(13):9817-24
144. Tolaney S. et al. A Phase 1/2 Study of SAR245408 (S08) in Combination with Trastuzumab (T) or Paclitaxel (P) and T in Patients with HER2+ Metastatic Breast Cancer (MBC) Who Progressed on a Previous T-Based Regimen. (P1-17-02). Cancer Res. 2011;71:240s
145. Baselga J, Tolaney S, Hart L, Gomez P, Gartner E, DeCillis A, Ruiz-Soto R, Lager J, Burris H. A Phase 1/2 Dose-Escalation Study of SAR245408 (S08) or SAR245409 (S09) in Combination with Letrozole (L) in Subjects with Hormone Receptor-Positive and HER2-Negative (HR+/HER2-) Breast Cancer (BC) Refractory to a Nonsteroidal Aromatase Inhibitor (AI) (P1-17-09). Cancer Res. 2011:243s
146. Krop IE, Wolff AC, Winer EP, Miller KD, Park BH, Ware J, Holden S, Levy GG, Derynck M, Storniolo AM. A Phase Ib Study Evaluating Safety, Tolerability, Pharmacokinetics (PK), and Activity of the Phosphoinositide-3 Kinase (PI3K) Inhibitor GDC-0941 in Combination with Trastuzumab-MCC-DM1 (T-DM1) in Patients with Advanced HER2-Positive Breast Cancer (P6-15-02). Presentated at the San Antonio Breast Cancer Symposium, San Antonio, Texas, December 12. 2010
147. Schöffski P, De Benedictis E, Gendreau S, Gianni L, Krop IE, Levy G, Ware J, Wildiers H, Winer EP. A Phase Ib, Open-Label, Dose-Escalation Study of the Safety and Pharmacology of the PI3-Kinase Inhibitor GDC-0941 in Combination with Paclitaxel and Bevacizumab in Patients with Locally Recurrent or Metastatic Breast Cancer (PD09-04). Cancer Res. 2011:150-151s
148. Maki RG, Small is beautiful. insulin-like growth factors and their role in growth, development, and cancer. J Clin Oncol. 2010Nov20;28(33):4985-95
149. Pollak M. The insulin receptor/insulin-like growth factor receptor family as a therapeutic target in oncology. Clin Cancer Res. 2012Jan1;18(1):40-50
150. Gao J, Chang YS, Jallal B, Viner J. Targeting the insulin-like growth factor axis for the development of novel therapeutics in oncology. Cancer Res. 2012Jan1;72(1):3-12
151. Jassem J. et al. Randomized, open label, phase III trial of figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin in patients with non-small cell lung cancer (NSCLC). J Clin Oncol. 2010;28:abstr7500
152. Kaufman PA, Ferrero JM, Bourgeois H, Kennecke H, De Boer R, Jacot W, McGreivy J, Suzuki S, Loh E, Robertson J. A Randomized, Double-Blind, Placebo-Controlled, Phase 2 Study of AMG 479 With Exemestane (E) or Fulvestrant (F) in Postmenopausal Women With Hormone-Receptor Positive (HR+) Metastatic (M) or Locally Advanced (LA) Breast Cancer (BC) (S1-4). Presentated at the San Antonio Breast Cancer Symposium, San Antonio, Texas, December 9. 2010
153. Ryan PD, Neven P, Blackwell KL, Dirix LY, Barrios CH, Miller Jr WH, Fein LE, Fenton D, Benner RJ, Meech SJ, Paccagnella L, Sleight B, Yee D, Goss PE. Figitumumab Plus Exemestane Versus Exemestane as First-Line Treatment of Postmenopausal Hormone Receptor-Positive Advanced Breast Cancer: A Randomized, Open-Label Phase II Trial (P1-17-01). Cancer Res. 2011:239-240s
154. Ebbinghaus S. et al. A phase II study of ridaforolimus (RIDA) and dalotuzumab (DALO) in estrogen receptor-positive (ER+) breast cancer. J Clin Oncol. 2011;29:abstrTPS110
155. NCT00684983. Capecitabine and Lapatinib with or without Cixutumumab in treating patients with previously treated HER2-Positive stage IIIB, Stage IIIC, or Stage IV breast cancer.
156. NCT00728949. A Study for Safety and Effectiveness of IMC-A12 by Itself or Combined With Antiestrogens to Treat Breast Cancer.
157. Verheul HM, Salumbides B, Van Erp K, Hammers H, Qian DZ, Sanni T, Atadja P, Pili R. Combination strategy targeting the hypoxia inducible factor-1 alpha with mammalian target of rapamycin and histone deacetylase inhibitors. Clin Cancer Res. 2008;14(11):3589-97
158. Haluska P. et al. Phase II trial of the dual IGF-1R/IR inhibitor BMS-754807 with or without letrozole in aromatase inhibitor-resistant breast cancer. J Clin Oncol. 2011;29:abstrTPS111
159. Peyton J.D. et al. A dose-escalation study with the novel formulation of the oral pan-class I PI3K inhibitor BEZ235, solid dispersion system (SDS) sachet, in patients with advanced solid tumors (Abstract No: 3066). J Clin Oncol. 2011;29:abstr3066
160. Banerji U. Heat shock protein 90 as a drug target: some like it hot. Clin Cancer Res. 2009Jan1;15(1):9-14
161. Pick E, Kluger Y, Giltnane JM. et al. High HSP90 expression is associated with decreased survival in breast cancer. Cancer Res. 2007Apr1;67(7):2932-7
162. Modi S, Stopeck A, Linden H, Solit D. et al. HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin Cancer Res. 2011Aug1;17(15):5132-9
163. Modi S. et al. Phase II trial of the Hsp90 inhibitor tanespimycin (Tan) + trastuzumab (T) in patients (pts) with HER2-positive metastatic breast cancer (MBC). J Clin Oncol. 2008;26:abstr1027
164. Modi et al. Efficacy and safety of retaspimycin hydrochloride (IPI-504) in combination with trastuzumab in patients (pts) with pretreated, locally advanced or metastatic HER2-positive breast cancer. J Clin Oncol. 2011;29:abstr590
165. Jhaveri K, Chandarlapaty S, Lake D, Gilewski T, Drullinsky P, Sugarman S, Wasserheit-Leiblich C, Moynahan ME, D'Andrea G, Haque S, Patil S, Bauman L, Vukovic V, El-Hariry I, Hudis C, Modi S. A Phase II Trial of Ganetespib: Efficacy and Safety in Patients (pts) with Metastatic Breast Cancer (MBC) (P1-17-08). Cancer Res. 2011:242-243s
166. Quintero M, Mackenzie N, Brennan PA. Hypoxia-inducible factor 1 (HIF-1) in cancer. Eur J Surg Oncol. 2004Jun;30(5):465-8
167. Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol. 2006Mar;26(6):2019-28
168. Kim SH, Kim KW, Jeong JW. et al. Inhibition of hypoxia-induced angiogenesis by sodium butyrate, a histone deacetylase inhibitor, through hypoxia-inducible factor-1alpha suppression. Oncol Rep. 2007Apr;17(4):793-7
169. Munster P, Marchion D. et al. Clinical and Biological Effects of Valproic Acid as a Histone Deacetylase Inhibitor on Tumor and Surrogate Tissues: Phase I/II Trial of Valproic acid and Epirubicin/FEC. Clin Cancer Res. 2009;15:2488-2496
170. Munster P.N. et al. Phase II trial of vorinostat, a histone deacetylase inhibitor to restore the hormone sensitivity to the anti-estrogen tamoxifen in patients with advanced breast cancer having failed prior aromatase inhibitor therapy. J Clin Oncol. 2008;26:abstr3501
171. Wardley AM. Phase II data for entinostat, a class 1 selective histone deacetylase inhibitor, in patients whose breast cancer is progressing on aromatase inhibitor therapy. J Clin Oncol. 2010;26:abstr1052
172. Yardley DA. et al. A double-blind, randomized, placebo-controlled phase II study of the steroidal aromatase inhibitor exemestane with and without the isoform selective histone deacetylase inhibitor (HDACi) entinostat in metastatic breast cancer (MBC). J Clin Oncol. 2010;28:abstrTPS128
173. Peacock NW. A phase I study of panobinostat (LBH589) with capecitabine with or without lapatinib. J Clin Oncol. 2010;28:abstr1115
174. Mahalingam D. Challenges in developing targeted therapy for pancreatic adenocarcinoma. Expert Opin Ther Targets. 2008Nov;12(11):1389-401
175. Glück S, Arteaga CL, Osborne CK. Optimizing Chemotherapy-Free Survival for the ER/HER2-Positive Metastatic Breast Cancer Patient. Clinical Cancer Research. 2011;17(17):1-3
176. Sandoval-Sus J, Mahtani R, Glück S. HER2-Positive Metastatic Breast Cancer: A Double-Edged Sword. Breast Cancer Management. 2012 in press
Author contact
![]() Corresponding author: Stefan Glück, MD PhD FRCPC. Department of Medicine, Division of Hematology/Oncology, University of Miami Miller School of Medicine, Miami, FL, USA. 1475 NW 12 Ave, 3rd floor. Miami, FL. USA. 33136. Phone: 305-243-1542. Fax: 305-243-4047. Email: SGluckmiami.edu.
Corresponding author: Stefan Glück, MD PhD FRCPC. Department of Medicine, Division of Hematology/Oncology, University of Miami Miller School of Medicine, Miami, FL, USA. 1475 NW 12 Ave, 3rd floor. Miami, FL. USA. 33136. Phone: 305-243-1542. Fax: 305-243-4047. Email: SGluckmiami.edu.