Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(2):96-103. doi:10.7150/jca.5470 This issue Cite
Mini-Review
Crosstalk between Wnt Signaling and RNA Processing in Colorectal Cancer
Michael Bordonaro ![]()
Department of Basic Sciences, The Commonwealth Medical College, 525 Pine Street, Scranton, PA 18509, USA.
Received 2012-10-31; Accepted 2012-11-21; Published 2013-1-5
Abstract
RNA processing involves a variety of processes affecting gene expression, including the removal of introns through RNA splicing, as well as 3' end processing (cleavage and polyadenylation). Alternative RNA processing is fundamentally important for gene regulation, and aberrant processing is associated with the initiation and progression of cancer. Deregulated Wnt signaling, which is the initiating event in the development of most cases of human colorectal cancer (CRC), has been linked to modified RNA processing, which may contribute to Wnt-mediated colonic carcinogenesis. Crosstalk between Wnt signaling and alternative RNA splicing with relevance to CRC includes effects on the expression of Rac1b, an alternatively spliced gene associated with tumorigenesis, which exhibits alternative RNA splicing that is influenced by Wnt activity. In addition, Tcf4, a crucial component of Wnt signaling, also exhibits alternative splicing, which is likely involved in colonic tumorigenesis. Modulation of 3' end formation, including of the Wnt target gene COX-2, also can influence the neoplastic process, with implications for CRC. While many human genes are dependent on introns and splicing for normal levels of gene expression, naturally intronless genes exist with a unique metabolism that allows for intron-independent gene expression. Effects of Wnt activity on the RNA metabolism of the intronless Wnt-target gene c-jun is a likely contributor to cancer development. Further, butyrate, a breakdown product of dietary fiber and a histone deacetylase inhibitor, upregulates Wnt activity in CRC cells, and also modulates RNA processing; therefore, the interplay between Wnt activity, the modulation of this activity by butyrate, and differential RNA metabolism in colonic cells can significantly influence tumorigenesis. Determining the role played by altered RNA processing in Wnt-mediated neoplasia may lead to novel interventions aimed at restoring normal RNA metabolism for therapeutic benefit. Therefore, this minireview presents a brief overview of several aspects of RNA processing of relevance to cancer, which potentially influence, or are influenced by, Wnt signaling activity.
Keywords: colon cancer, butyrate, Wnt activity, RNA processing, splicing, polyadenylation.
Introduction
The protective action of dietary fiber against colorectal cancer (CRC) has been attributed to the fermentation of fiber by anaerobic bacteria in the colon, producing butyrate (1), an inhibitor of histone deacetylases (HDACi), which produces cell cycle arrest, differentiation, and/or apoptosis of CRC cells (2-4). Butyrate upregulates canonical Wnt transcriptional activity in CRC cells (5-7). Wnt activity derives from the accumulation of beta-catenin, which becomes associated with DNA-binding Tcf factors; the resulting beta-catenin-Tcf complexes stimulate transcriptional activity from target genes (8). The constitutive activation of Wnt signaling, primarily due to mutations in the APC and beta-catenin genes, promotes tumorigenesis in the colon (8). Wnt signaling also plays a role in RNA metabolism that may have consequences for tumorigenesis (9,10). For example, the Wnt factor beta-catenin has been shown to influence various steps of RNA metabolism in CRC cells, modulating the levels of gene products relevant to colonic tumorigenesis, including COX-2 and cyclin D1 (10).
Since Wnt activity can influence RNA metabolism (10) and butyrate can influence Wnt signaling (5-7), the findings that butyrate can directly modulate RNA metabolism is provocative. For example, butyrate inhibits the splicing of RNA from the Wnt-target gene c-myc, important for many forms of cancer including CRC (11), and also alters alternative splicing of the survival motor neuron (SMN) gene, resulting in enhanced SMN levels in vitro and in vivo (12). In addition, butyrate influences the relative levels of calcitonin and calcitonin gene-related peptide mRNAs (13), the ratio of which is controlled by alternative polyadenylation. This latter finding suggests that butyrate modulates 3' end processing as well as splicing. These findings indicate that Wnt signaling, and the activation of this signaling by butyrate, influences RNA metabolism, and that altered RNA metabolism modifies patterns of gene expression related to tumorigenesis. Further, Wnt signaling itself can be differentially modulated by the products of alternative RNA processing, suggesting the possibility of bidirectional crosstalk between Wnt activity and RNA metabolism.
Effects of butyrate and Wnt activity on the expression of RNA processing factors and on RNA splicing in CRC cells
As part of our study of differential gene expression affecting colonic tumorigenesis, a full human genome microarray analysis (Genus Biosystems) was performed on RNA derived from HCT-116 CRC cells, to determine genes modulated by a physiologically relevant concentration (5 mM) of butyrate. The expression of a number of genes involved with RNA metabolism was found to be influenced by butyrate. For example, butyrate significantly (P < 0.001) decreased by 2.4-fold the expression of the splicing factor gene SRp20; this latter action by butyrate may mediate its therapeutic activity, since SRp20 has been linked to CRC tumorigenicity (14). Expression of the intronless gene c-jun, which can promote colonic tumorigenesis (see below), was upregulated by butyrate 9.5-fold (P < 0.001). Furthermore, the expression of other genes important in RNA metabolism was up- or downregulated by butyrate treatment of HCT-116 cells. Genes whose expression was upregulated include the nuclear RNA export factor NXF3, while a number of genes exhibited downregulation of expression, including genes whose products influence 3' end processing (CSTF3, several other CPSF and CSTF genes, and PABPN1), as well as genes whose products influence RNA stability (hnRNP D) and splicing (SRSF10 and PTBP1). The modulation, by butyrate, of the expression of CSTF3, SRSF10, PTBP1, and PABPN1 is also influenced by Wnt activity, as the degree of that modulation was decreased in HCT-116 cells expressing an inducible form of dominant-negative Tcf4 (DN-Tcf4), which is a potent inhibitor of canonical Wnt signaling (8).
To evaluate effects of Wnt activity on overall intron splicing efficiency in CRC cells, we utilized the luciferase-beta galactosidase double reporter system from the Eperon laboratory (15) which is based on a tropomyosin TMP3 intron. Two plasmids, pTN23 and pTN24, were used, which differ in the structure of the TMP3 intron sequences. With efficient intron splicing, coding sequences from both the luciferase and beta-galactosidase cassettes are fully included in the final transcript and both reporter activities are detected. However, in the absence of splicing, only beta-galactosidase reporter activity is observed. Thus, the ratio of luciferase to beta-galactosidase reporter activity is indicative of splicing efficiency.
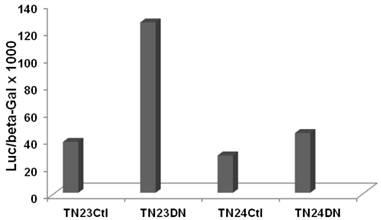
SW620 CRC cells exhibit a relatively high basal level of canonical Wnt signaling that is strongly inhibited by DN-Tcf4 (5-7 and refs. therein). We performed a pilot experiment by cotransfecting the pTN23 or pTN24 vectors with a control vector or with the DN-Tcf4 expression vector. Inhibition of Wnt activity in SW620 cells enhanced splicing efficiency by more than three-fold with the pTN23 vector and more than 1.5-fold with the pTN24 vector (Fig. 1). This demonstrates Wnt activity-specific effects on intron splicing in a well characterized CRC cell line, with the general trend of increased intron splicing with repressed Wnt activity.
Wnt activity influences splicing efficiency in SW620 CRC cells. Splicing constructs pTN23 (TN23) or pTN24 (TN24) were transfected into SW620 cells with pcDNA3 (Ctl) or DN-Tcf4 (DN). Luciferase and beta-galactosidase activities were assayed with the Dual Light kit (Applied Biosystems) and the relative activity of luciferase vs. beta-galactosidase, indicative of splicing efficiency, is shown.
Based on these preliminary findings, a review of the literature was conducted to evaluate evidence for physiologically relevant cross-talk between Wnt activity and RNA processing that is (a) of relevance to CRC, and (b) potentially modifiable by butyrate/HDACis, and thus possibly amenable to preventive and/or therapeutic approaches against CRC.
Review of Literature and Discussion
Intron splicing
Most eukaryotic genes contain introns and undergo mRNA splicing. At least 60% of human genes exhibit alternative splicing, and microarray methodologies have been developed to analyze the patterns of pre-mRNA splicing in different cells and under varied conditions (16-18). Tumor-specific splicing variants have been identified and some of these variants influence Wnt activity (19-22). Wnt activity in turn modulates RNA processing, particularly RNA splicing (10,22). For example, the splicing factor SRp20 is a Wnt target gene; SRp20 upregulates CRC cell proliferation and has been linked to tumorigenesis (14).
It is known that variants of Tcf/Lef factors such as Tcf1 and Tcf4, which bind to beta-catenin and drive Wnt transcriptional activity, can be generated by alternative splicing (23). Further, Lee et al. (10) demonstrated that an RNA aptamer that interferes with the interaction between beta-catenin and Tcf4 in CRC cells not only repressed Wnt activity but also regulated the alternative splicing of RNA from an adenovirus E1A minigene. Conversely, alternative splicing influences Wnt activity; thus, while the full length form of sulfatase 1 (SULF1A) stimulates Wnt signaling, a shorter SULF isoform (SULF1B) produced by alternative splicing inhibits Wnt activity (19). Thus, cross-talk between RNA splicing and Wnt signaling activity is bidirectional, and is associated with colonic neoplasia.
Importantly, Wnt activity is influenced not only by the expression of the splicing factor SRp20 (14,22) but also by that of ASF/SF2 (22,24). Thus, changes in the relative levels of SRp20 and ASF/SF2 results in altered RNA splicing and differential expression of Rac1b, a Rac1 variant overexpressed in certain colon tumors that contributes to cell survival (14,22), and which can enhance Wnt signaling (25). SRp20 enhances skipping of the Rac1 alternative exon 3b in CRC cells, while ASF/SF2 promotes exon 3b inclusion; thus, ASF/SF2 increases and SRp20 decreases Rac1b expression. Increased levels of Wnt activity enhance expression of SRp20, which in turn results in repressed expression of the Rac1b variant (22). Rac1b is more highly expressed in CRC cells with lower levels of Wnt activity, promoting survival of these low-Wnt activity cells (20). Conversely, the expression of ASF/SF2 is blocked by phosphatidylinositol 3-kinase (PI3-K) signaling; therefore, formation of Rac1b is ultimately controlled by levels of Wnt and PI3-K signaling (22,24).
CRC cells characterized by low levels of Wnt activity and PI3-K signaling utilize Rac1b to promote survival; however, CRC cell types with higher levels of Wnt and PI3-K signaling exhibit low or no expression of Rac1b and utilize alternative signaling pathways to promote cell survival (20). These findings are consistent with our observations that very high levels of Wnt activity induced by HDACis promote apoptosis of CRC cells, since lower levels of Wnt signaling, associated with increased expression of Rac1b, promote cell survival (5-7). Further, Rac1b itself upregulates Wnt activity when overexpressed in HCT-116 cells, a cell line that normally expresses relatively low levels of Rac1b (25). This upregulation of Wnt activity by Rac1b occurs upstream of beta-catenin (24,25); this is similar to the activation of Wnt activity by HDACis at the plasma membrane, resulting in enhanced levels of active beta-catenin (5-7). Therefore, one intriguing possibility is that upregulation of Wnt activity by HDACis may be partially mediated by the action of Rac1b.
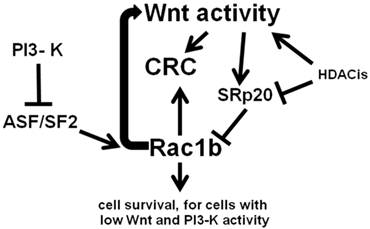
Remarkably, Rac1b-negative SW480 CRC cells can be made to endogenously express levels of Rac1b similar to that of the Rac1b-positive HT-29 CRC cell line, through a combination of ASF/SF2 overexpression and siRNA knockdown of SRp20 (22). This suggests plasticity of Rac1b phenotypes, perhaps extending to downstream effects of Rac1b expression. Manipulation of Rac1b levels likely can induce Rac1b-mediated cell survival pathways, including induction of moderate levels of Wnt signaling activity. However, since increased Wnt activity downregulates Rac1b expression via SRp20, a negative feedback loop would likely occur. Thus, low levels of Wnt activity promotes increased expression of Rac1b, which simulates Wnt activity; this increase in Wnt signaling would in turn downregulate levels of Rac1b through alternative RNA splicing. Our previous findings (5-7) are consistent with the hypothesis that moderate levels of Wnt activity drive CRC cell proliferation and survival, while Wnt hyperactivation promotes CRC cell death. Thus, the negative Wnt-Rac1b feedback loop may serve to keep levels of Wnt activity in the pro-proliferative range and below levels that would promote CRC cell death. The overall relationship between Wnt signaling, Rac1b, and CRC, with respect to alternative RNA splicing, is outlined in Fig. 2.
Wnt activity and Rac1b expression in CRC cells. Wnt activity promotes expression of SRp20 which blocks expression of Rac1b through control of alternative splicing. In contrast, the splicing factor ASF/SF2, the expression of which is repressed by PI3-K signaling, enhances expression of Rac1b through alternative splicing. Rac1b itself not only promotes cell survival, but enhances Wnt signaling, forming a negative feedback loop that controls Rac1b levels through Wnt-mediated SRp20 expression. Histone deacetylase inhibitors (HDACis) influence these processes by affecting both Wnt activity and the expression of factors such as SRp20. Arrows represent known or potential positive interactions; blocked lines represent known or potential repressive interactions.
The development of other forms of cancer is also influenced by splicing factors whose expression is controlled by cancer-related cell signaling pathways. For example, the levels of ASF/SF2 as well as of hnRNP A1 are increased in lung neoplasia; this altered expression is associated with cancer-related changes in the alternative splicing of CD44, which is involved in cell-cell and cell-matrix interactions (26). Thus, the effects of SRp20 and ASF/SF2 on Rac1b may be part of a broader modulation of RNA processing in tumorigenesis, responsible for altering the levels of protein variants in the neoplastic cell (22,26).
Consistent with a significant role for RNA metabolism in mediating the effects of cell signaling pathways in normal and neoplastic cells, analysis of beta-catenin-containing nuclear complexes showed the presence of a variety of proteins that bind RNA and/or influence RNA metabolism, such as hnRNP A2/B1, hnRNP M, hnRNP G, hnRNP A1, and hnRNP K, among others (27). In addition, beta-catenin interacts with fusion (FUS)/translocated in liposarcoma (FUS/TLS), a RNA binding protein that influences mRNA transcription and splicing (27). Overexpression of FUS/TLS repressed Wnt activity in CRC cells and repressed CRC cell clonogenic growth; in addition, FUS/TLS levels are increased in CRC and may serve as negative feedback, limiting the effects of deregulated Wnt activity (27). This interaction further links RNA metabolism to Wnt activity, which in turn influences CRC cell growth. Thus, FUS/TLS and similar factors may integrate Wnt-mediated RNA transcription with effects on RNA processing, to exert more fine-grained control of the expression of genes associated with tumorigenesis.
Example of an intronless Wnt target gene: c-jun
Genomic analyses suggest that approximately 10% of human genes are intronless (28-32). Statistical analyses demonstrated that naturally intronless genes are over-represented among genes coding for proteins with binding or signal transduction/receptor activities and under-represented among genes coding for proteins with catalytic functions (33). Signal transduction/receptor proteins are highly relevant to the signaling pathways involved in the development of cancer; therefore, these data suggest the possibility that intronless RNA metabolism is associated with tumorigenesis. Rapid-response genes, which include c-jun, are often intronless; RNA metabolism of these genes reflects the requirement to bypass splicing processes for more efficient expression and function (34).
The protein product of the intronless human c-jun gene associates with c-fos, forming the transcription factor AP-1, which participates in JNK signaling (35,36). AP-1/JNK signaling plays an important role in the development of CRC (37), and a dominant negative mutant of c-jun has been proposed as a gene therapy approach for CRC, through disruption of AP-1 activity (38). Supporting this view, c-jun is overexpressed in CRC, and JNK signaling activates intestinal tumorigenesis in mouse models of CRC (39 and refs. therein). JNK signaling stimulates progenitor cell proliferation in intestinal crypts; importantly, expression of the Wnt target genes Axin 2 and Lgr5 is upregulated by JNK activity (39). Further, expression of Tcf4, a key component of the Wnt signaling complex, is upregulated by JNK activity, and chromatin immunoprecipitation analyses demonstrated that Tcf4 is a direct c-jun target gene (39). C-jun itself is a Wnt target gene; therefore, these findings suggest that positive feedback between Wnt and JNK activity, mediated through c-jun, contributes to intestinal tumorigenesis (39). In addition, c-jun, together with the Wnt signaling factors beta-catenin and Tcf, associate with a 3' enhancer region of the Wnt-target gene c-myc, a proto-oncogene necessary for colorectal neoplasia (40,41).
With respect to RNA metabolism, while intron-containing genes often require the presence of introns for proper mRNA accumulation; naturally intronless genes contain sequence elements which can substitute for introns to allow for efficient gene expression (42 and references therein). An example of such an element is the human c-jun gene enhancer (CJE) element (42). The RNA metabolism of intronless c-jun, by influencing levels of c-jun protein, may alter signaling cross-talk (e.g., Wnt-JNK) and therefore contribute to the development of CRC (41). In addition, preliminary findings from our laboratory (unpublished data) indicate that Wnt activity can enhance the activity of sequences that confer efficient intron-independent gene expression; this suggests that Wnt activity influences c-jun expression both at the level of transcription as well as at the level of post-transcriptional modulation of intronless RNA metabolism (e.g., RNA stability). Further research into the effects of Wnt activity on intronless RNA metabolism is required to evaluate this possibility.
Importantly for CRC, c-jun expression is upregulated by butyrate both in vitro and in vivo (43,44), and our microarray data (above) show a marked upregulation of c-jun expression in butyrate-treated HCT-116 cells. Thus, while HDACi treatment is generally pro-apoptotic and hence therapeutic against cancer, enhanced Wnt activity resulting from exposure to HDACis can also increase the expression of pro-tumorigenic genes. Therefore, it is important to understand the various mechanisms whereby basal and HDACi-induced Wnt activity influences the expression of c-jun, including effects on intronless RNA metabolism.
Induction of c-jun expression by HDACis can be rapid; for example, exposure of leukemic cells to the butyric acid prodrug pivaloyloxymethyl butyrate (A-9) induces significant c-jun expression in only 15 minutes (45). Therefore, c-jun can be classified as an intronless “rapid response” gene (34). While there are “rapid response” genes that contain introns, the interplay between RNA processing and gene expression, including the kinetics of gene expression, likely differs in a gene specific manner. For example, the relative importance of the intronless condition for rapid gene expression may be influenced by factors such as promoter strength, chromosome location and surrounding chromatin structure, and/or the presence or absence of other regulatory sequences. Certain intronless genes, such as c-jun, may require the intronless state for efficient and rapid expression; this dependence on the intronless state influences the levels of the relevant gene products, with consequent effects on cell physiology, including neoplasia. Therefore, differences of RNA metabolism in intron-dependent vs. intron-independent pathways of expression may in part determine the degree to which induction of gene expression by Wnt signaling contributes to tumorigenesis. Further, a complete understanding of how cell signaling pathways influence intronless gene expression in tumorigenesis may lead to approaches to suppress expression of tumorigenic factors such as c-jun, which would enhance the efficacy of, e.g., HDACi-based anti-cancer therapeutics.
3' end processing, COX-2, beta-catenin, RNA stability and the 3' UTR
3' end processing is also of relevance to cancer; for example, alternative 3' end processing can enhance oncogene expression in cancer cells, and differential 3' end formation is associated with clinically relevant cancer subtypes (46). Altered gene expression in cancer, caused by alternative 3' end processing, often results from enhanced RNA stability, in some cases involving shortened 3' untranslated regions (3' UTRs) and the consequent loss of sites mediating repression by miRNAs (47).
Several genes linked to neoplasia exhibit regulation of expression through control of 3' end processing. The Wnt target cyclin D2, involved in cell cycle regulation, is regulated through modulation of 3' end processing, and other cancer related genes (e.g., N-acetyltransferase 1) also exhibit regulation of expression through alternative polyadenylation (48,49 and refs. therein). Further, alternative polyadenylation produces a version of the Wnt factor Tcf4 that inhibits Wnt activity (50), suggesting a direct link between 3' end processing, Wnt activity, and colonic neoplasia.
A classic example of a CRC-related gene whose RNA stability (51) and expression is controlled by alternative polyadenylation is the Wnt target cyclooxygenase-2 (COX-2) (52 and refs. therein). HT-29 CRC cells primarily use the COX-2 distal polyadenylation site, while HepG2 liver cancer cells use both the distal and proximal COX-2 polyadenylation sites (52). This tissue-specific difference in polyadenylation usage has practical consequences, as the longer COX-2 mRNA (distal site usage) tends to be more stable than the shorter form (proximal site usage), resulting in higher steady-state levels of the COX-2 protein that may promote colonic neoplasia. Thus, differential RNA stability, such as that resulting from alternative 3' end processing of COX-2 RNA, can influence neoplasia. Further, since our preliminary microarray data (see above) demonstrated that Wnt activity and butyrate influence the expression of factors involved in 3' end processing, levels of Wnt signaling may influence alternative polyadenylation (e.g., of COX-2 RNA).
Of particular relevance for CRC, beta-catenin influences COX-2 RNA stability (10,51); beta-catenin stabilizes COX-2 transcripts through interactions with AU-rich elements in the 3' UTR, again demonstrating cross-talk between Wnt signaling and RNA metabolism. The RNA binding protein HuR likely mediates these effects of beta-catenin; the HuR protein stabilizes transcripts correlated to the number of binding sites and level of association of HuR with the targeted RNA (50, 53, 54). RNAs containing HuR binding sites in both intronic as well as 3' UTR sequences exhibited greater stability than transcripts containing either intronic or 3' UTR sites only; these findings suggest that HuR links pre-mRNA processing with mature mRNA stability (55). Therefore, Wnt activity not only influences the levels of COX-2 at the transcriptional level, but also post-transcriptionally, at the level of RNA stability.
In addition, the association of HuR with thrombospondin-1 mRNA modulates the translation of the angiogenesis inhibitor TP1, affecting neoplastic progression via altered tumor vascularity (56). Therefore, RNA metabolism involving the 3' ends of transcripts can influence multiple points along the neoplastic continuum.
Conclusion
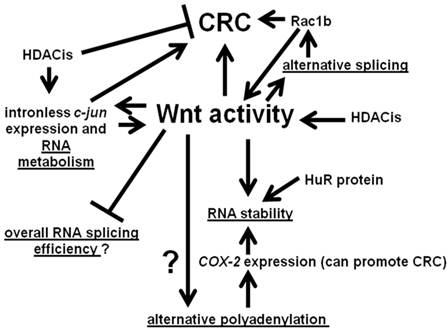
We summarize the interactions between Wnt activity and aspects of RNA processing discussed in this paper in Fig. 3. Wnt signaling interacts with various points of RNA metabolism, and the gene products so influenced themselves feed back to affect Wnt activity and, hence, influence neoplasia.
Interactions between Wnt activity and aspects of RNA metabolism. Wnt signaling can influence alternative splicing through the differential expression of RNA splicing factors. In some cases, such as Rac1b, the products of alternative processing can affect Wnt signaling. Wnt activity may also affect alternative polyadenylation. In addition, Wnt activity can influence the stability of the products of alternative polyadenylation; these effects on RNA stability may be mediated by the HuR protein. Further, our preliminary data suggest a possible role for Wnt activity in repressing general RNA splicing activity in CRC cells. Finally, Wnt activity can also promote expression of certain intronless genes, whose gene products influence Wnt activity, with possible consequences for colonic tumorigenesis. Note that the overall net effect of HDACis on CRC is believed to be inhibitory; however, at the same time, HDACis can enhance expression of potentially pro-tumorigenic factors, such as the product of the intronless c-jun gene. Arrows represent known or potential positive interactions; blocked lines represent known or potential repressive interactions. Steps involving RNA metabolism are underlined.
Analysis of cancer-specific RNA metabolism is a relatively unexplored area of research, with potentially significant implications for the prevention and treatment of CRC. RNA metabolism can be altered by Wnt signaling as well as by butyrate and other HDACis. RNA metabolism is likely a major control point mediating the changes in gene expression that result from deregulated Wnt signaling. In particular, intron-dependent expression of genes such as c-jun, and cross-talk between Wnt signaling, Rac1b expression, and mRNA processing likely play fundamental roles in colonic neoplasia. Determining how RNA processing and intron-independent gene expression influences Wnt-initiated CRC may lead to novel interventions aimed at restoring normal gene expression for therapeutic benefit.
Abbreviations
HDACi: histone acetylase inhibitor; NaB: sodium butyrate; CRC: colorectal cancer; UTR: untranslated region.
Acknowledgements
This work was supported by institutional funding from The Commonwealth Medical College.
Competing Interests
The author acknowledges that no competing interests exist.
References
1. Bingham S. Mechanisms and experimental evidence relating dietary fibre and starch to protection against large bowel cancer. Proc Nutr Soc. 1990;49:153-171
2. Boffa LC, Lupton JR, Mariani MR, Ceppi M, Newmark HL, Scalmati A, Lipkin M. Modulation of colonic epithelial cell proliferation, histone acetylation, and luminal short chain fatty acids by variation of dietary fiber (wheat bran) in rats. Cancer Res. 1992;52:5906-5912
3. Heerdt BG, Houston MA, Augenlicht LH. Potentiation by specific short-chain fatty acids of differentiation and apoptosis in human colonic carcinoma cell lines. Cancer Res. 1994;54:3288-3294
4. Medina V, Young GP, Edmonds B, James R, Appleton S, Zalewski PD. Induction of caspase-3 protease activity and apoptosis by butyrate and another inhibitor of histone deacetylase: dependence on protein synthesis and synergy with a mitochondrial/cytochrome c-dependent pathway. Cancer Res. 1997;57:3697-3707
5. Bordonaro M, Mariadason JM, Aslam F, Heerdt BG, Augenlicht LH. Butyrate-induced apoptotic cascade in colonic carcinoma cells: modulation of the beta-catenin-Tcf pathway and concordance with effects of sulindac and trichostatin A but not curcumin. Cell Growth Differ. 1999;10:713-720
6. Lazarova DL, Bordonaro M, Carbone R, Sartorelli AC. Linear relationship between WNT activity levels and apoptosis in colorectal carcinoma cells exposed to butyrate. Internat J Cancer. 2004;110:523-531
7. Bordonaro M, Lazarova DL, Sartorelli AC. The activation of beta-catenin by Wnt signaling mediates the effects of histone deacetylase inhibitors. Exp Cell Res. 2007;313:1652-1666
8. Korinek V, Barker N, Morin PJ, Van Wichen D, De Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1784-1787
9. Lee HK, Choi YS, Park YA, Jeong S. Modulation of oncogenic transcription and alternative splicing by β-catenin and an RNA aptamer in colon cancer cells. Cancer Res. 2006;66:10560-10566
10. Lee HK, Kwak HY, Hur J, Kim IA, Yang JS, Park MW, Yu J, Jeong S. Beta-catenin regulates multiple steps of RNA metabolism as revealed by the RNA aptamer in colon cancer cells. Cancer Res. 2007;67:93150-9321
11. Krupitza G, Harant H, Dittrich E, Szekeres T, Huber H, Dittrich C. Sodium butyrate inhibits c-myc splicing and interferes with signal transduction in ovarian carcinoma cells. Carcinogenesis. 1995;16:1199-1205
12. Chang J-G, Hsieh-Li H-M, Jong Y-J, Wang NM, Tsai C-H, Li H. Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci. USA. 2001;98:9808-9813
13. Nakagawa T, Nelkin BD, Baylin SB, de Bustros A. Transcriptional and posttranscriptional modulation of calcitonin gene expression by sodium n-butyrate in cultured human medullary thyroid carcinoma. Cancer Res. 1988;48:2096-2100
14. Corbo C, Orru S, Gemei M, Noto RD, Mirabelli P, Imperlini E, Ruoppolo M, Vecchio LD, Salvatore F. Protein cross-talk in CD133+ colon cancer cells indicates activation of the Wnt pathway and up-regulation of SRp20 that is potentially involved in tumorigenicity. Proteomics. 2012;12:2045-2059
15. Nasim MT, Chowdhury HM, Eperon IC. A double reporter assay for detecting changes in the ratio of spliced and unspliced mRNA in mammalian cells. Nucl Acids Res. 2002;30:e109
16. Cuperlovic-Cuif M, Belacel N, Culf AS, Ouellette RJ. Microarray analysis of alternative splicing. OMICS: A Journal of Integrative Biology. 2006;10:344-357
17. Hughes TR, Hiley SL, Saltzman AL, Babak T, Blencowe BJ. Microarray analysis of RNA processing and modification. Methods Enzymol. 2006;410:300-316
18. McKee AE, Silver PA. Systems perspectives on mRNA processing. Cell Res. 2007;17:581-590
19. Sahota AP, Dhoot GK. A novel SULF1 splice variant inhibits Wnt signaling but enhances angiogenesis opposing SULF1 activity. Expt Cell Res. 2009;315:2752-2764
20. Li Q, Liu X, Zhang M, Ye G, Qiao Q, Ling Y, Wu Y, Zhang Y, Yu L. Characterization of a novel human CDK5 splicing variant that inhibits Wnt/beta-catenin signaling. Mol Biol Rep. 2010;37:2415-2421
21. Sharma S, Lichtenstein A. Aberrant splicing of the E-cadherin transcript is a novel mechanism of gene silencing in chronic lymphocytic leukemia cells. Blood. 2009;114:4179-4185
22. Goncalves V, Matos P, Jordan P. Antagonistic SR proteins regulate alternative splicing of tumor-related Rac1b downstream of the PI3-kinase and Wnt pathways. Hum Mol Genet. 2009;18:3696-3707
23. Prokunina-Olsson L, Welch C, Hansson O, Adhikari N, Scott L, Usher N, Tong M, Sprau A, Swift A, Bonnycastle L, Erdos M, He Z, Saxena R, Harmon B, Kotova O, Hoffman E, Altshuler D, Groop L, Boehnke M, Collins F, Hall JL. Tissue-specific alternative splicing of TCF7L2. Hum Mol Genet. 2009;18:3795-3804
24. Esufali S, Charames GS, Bapat B. Suppression of nuclear Wnt signaling leads to stabilization of Rac1 isoforms. FEBS Lett. 2007;581:4850-4856
25. Esufali S, Charames GS, Pethe VV, Buongiorno P, Bapat B. Activation of tumor-specific splice variant Rac1b by disheveled promotes canonical Wnt signaling and decreased adhesion of colorectal cancer cells. Cancer Res. 2007;67:2469-2479
26. Zerbe LK, Pino I, Pio R, Cosper PF, Dwyer-Nield LD, Meyer AM, Port JD, Montuenga LM, Malkinson AM. Relative amounts of antagonistic splicing factors, hnRNP A1 and ASF/SF2, change during neoplastic lung growth: Implications for pre-mRNA processing. Mol Carcinogen. 2004;41:187-196
27. Sato S, Idogawa M, Honda K, Fujii G, Kawashima H, Takekuma K, Hoshika A, Hirohashi S, Yamada T. β-catenin interacts with FUS proto-oncogene product and regulates pre-mRNA splicing. Gastroenter. 2005;129:1225-1236
28. Sakharkar MK, Kangueane P, Petrov DA, Kolaska AS, Subbiah S. SEGE: a database on 'intronless/single exonic' genes from eukaryotes. Bioinformatics. 2002;18:1266-1267
29. Agarwal SM, Srivistava PK. Human intronless disease genes are slowly evolving. BMB Reports. 2009;42:356-360
30. Agarwal SM. Evolutionary rate variation in eukaryotic lineage specific human intronless proteins. Biochem Biophys Res Commun. 2005;337:1192-1197
31. Agarwal SM, Gupta J. Comparative analysis of human intronless proteins. Biochem Biophys Tes Commun. 2005;331:512-519
32. Lopez-Bigas N, Ouzounis CA. Genome-wide identification of genes likely to be involved in human genetic disease. Nucl Acids Res. 2004;32:3108-3114
33. Hill AE, Sorscher EJ. The non-random distribution of intronless genes across molecular function categories. FEBS Lett. 2006;580:4303-4305
34. Jeffares DC, Penkett CJ, Bahler J. Rapidly regulated genes are intron poor. Trends Genet. 2008;24:375-378
35. Bohmann D, Bos TJ, Admon A, Nishimura T, Vogt PK, Tjian R. Human proto-oncogene c-jun encodes a DNA binding protein with structural and functional properties of transcription factor AP-1. Science. 1987;238:1386-1392
36. Rahmsdorf HJ. Jun: transcription factor and oncoprotein. J Mol Med. 1996;74:725-747
37. Ashida R, Tominaga K, Sasaki E, Watanabe T, Fujiwara Y, Oshitani N, Higuchi K, Mitsuyama S, Iwao H. Arakawa T. AP-1 and colorectal cancer. Inflammopharm. 2005;13:113-125
38. Suto R, Tominaga K, Mizuguchi H, Sasaki E, Higuchi K, Kim S, Iwao H, Arakawa T. Dominant-negative mutant of c-jun gene transfer: a novel therapeutic strategy for colorectal cancer. Gene Ther. 2004;11:187-193
39. Sancho R, Nateri AS, de Vinuesa AG, Aguilera C, Nye E, Spencer-Dene B, Behrens A. JNK signalling modulates intestinal homeostasis and tumourigenesis in mice. EMBO J. 2009;28:1843-1854
40. Yochum GS, Cleland R, Goodman RH. A genome-wide screen for beta-catenin binding sites identifies a downstream enhancer element that controls c-Myc gene expression. Mol Cell Biol. 2008;28:7368-7379
41. Lee CW, Lee SH, Lee SW, Ban JO, Lee SY, Yoo HS, Jung JK, Moon DC, Oh KW, Hung JT. 2-hydroxycinnamaldehyde inhibits SW620 colon cancer cell growth through AP-1 inactivation. J Pharmacol Sci. 2007;104:19-28
42. Guang S, Mertz JE. Pre-mRNA processing enhancer (PPE) elements from intronless genes play additional roles in mRNA biogenesis than do ones from intron-containing genes. Nucl Acids Res. 2005;33:2215-2226
43. Narjano JR, Mellstrom B, Auwerx J, Mollinedo F, Sassone-Corsi P. Unusual c-fos induction upon chromaffin PC12 differentiation by sodium butyrate: loss of fos autoregulatory function. Nucl Acids Res. 1990;18:3605-3610
44. Velasquez OC, Zhou D, Seto RW, Jabbar A, Choi J, Lederer HM, Rombeau JL. In vivo crypt surface hyperproliferation is decreased by butyrate and increased by deoxycholate in normal rat colon: associated in vivo effects on c-fos and c-jun expression. J Parent Ent Nutr. 1996;20:4243-4250
45. Rabizadeh E, Shaklai M, Nudelman A, Eisenbach L, Rephaeli A. Rapid alteration of c-myc and c-jun expression in leukemic cells induced to differentiate by a butyric acid prodrug. FEBS Lett. 1993;328:225-229
46. Singh P, Alley TL, Wright SM, Kamdar S, Schott W, Wilpan RY, Mills KD, Graber JH. Global changes in processing of mRNA 3' untranslated regions characterize clinically distinct cancer subtypes. Cancer Res. 2009;69:9422-9430
47. Mayr C, Bartel DP. Widespread shortening of 3' UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673-684
48. Bell DA, Badawi AF, Lang NP, Ilett KF, Kadlubar FF, Hirvonen A. Polymorphism in the N-acetyltransferase 1 (NAT1) polyadenylation signal: association of NAT1*10 allele with higher N-acetylation activity in bladder and colon tissue. Cancer Res. 1995;55:5226-5229
49. Bell DA, Stephens EA, Castranio T, Umbach DM, Watson M, Deakin M, Elder J, Hendrickse C, Duncan H, Strange RC. Polyadenylation polymorphism in the acetyltransferase 1 gene (NAT1) increases risk of colorectal cancer. Cancer Res. 1995;55:3537-3542
50. Locke JM, Da Silva Xavier G, Rutter GA, Harries LW. An alternative polyadenylation signal in TCF7L2 generates isoforms that inhibit T cell factor/lymphoid enhancer factor (TCF/LEF)-dependent target genes. Diabetologia. 2011;54:3078-3082
51. Lee HK, Jeong S. β-catenin stabilizes cyclooxygenase-2 mRNA by interacting with AU-rich elements of 3' UTR. Nucl Acids Res. 2006;34:5705-5714
52. Hall-Pogar T, Zhang H, Tian B, Lutz CS. Alternative polyadenylation of cyclooxygenase-2. Nucl Acids Res. 2005;33:2565-2579
53. Keene JD. The global dynamics of RNA stability orchestrates responses to cellular activation. BMC Biol. 2010;8:95
54. Keene JD. Minireview: global regulation and dynamics of ribonucleic acid. Endocrinology. 2010;151:1391-1397
55. Mukherjee N, Cordoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano Jr. M, Tuschl T, Ohler U, Keene JD. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell. 2011;43:327-339
56. Mazan-Mamczarz K, Hagner PR, Corl S, Srikantan S, Wood WH, Becker KG, Gorospe M, Keene J, Levenson AS, Gartenhaus RB. Post-transcriptional gene regulation by HuR promotes a more tumorigenic phenotype. Oncogene. 2008;27:6151-6163
57. Kim I, Kwak H, Lee HK, Hyun S, Jeong S. β-catenin recognizes a specific RNA motif in the cyclooxygenase-2 mRNA 3'-UTR and interacts with HuR in colon cancer cells. Nucl Acids Res. 2012;40:6863-6872
Author biography
Michael Bordonaro received his PhD degree from Fordham University and did a post-doctoral fellowship at the Montefiore Medical Center at the Albert Einstein Cancer Center. It was during this period that Dr. Bordonaro discovered that the Wnt signaling pathway can be pharmacologically modulated, and that histone deacetylase inhibitors (HDACis) upregulate Wnt signaling in colorectal cancer cells in a manner associated with enhanced rates of apoptosis of these cells. While at Montefiore, Dr. Bordonaro was awarded an American Institute for Cancer Research (AICR) Post-Doctoral Award grant for his work on HDACis and Wnt signaling. Dr. Bordonaro subsequently worked at the Yale University School of Medicine, where he continued his studies on the effects of HDACis on Wnt signaling and colonic cell physiology. In addition, at Yale, Dr. Bordonaro started work on Wnt-targeted genetic therapeutics against colorectal cancer. Dr. Bordonaro is currently an Assistant Professor at The Commonwealth Medical College, and is continuing his research on these and other topics, with grant funding from the National Cancer Institute and AICR.
![]() Corresponding author: The Commonwealth Medical College, 525 Pine Street, Scranton, PA 18509, Tel: 570-504-9646, Fax: 570-504-9636. Email: mbordonaroorg.
Corresponding author: The Commonwealth Medical College, 525 Pine Street, Scranton, PA 18509, Tel: 570-504-9646, Fax: 570-504-9636. Email: mbordonaroorg.