Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(1):66-83. doi:10.7150/jca.5112 This issue Cite
Review
The Tumor Microenvironment Contribution to Development, Growth, Invasion and Metastasis of Head and Neck Squamous Cell Carcinomas
Sittichai Koontongkaew1,2 ![]()
1. Oral Biology Unit, Faculty of Dentistry, Thammasat University, Klong Luang, Prathumtani 12121, Thailand
2. Medicinal Herb Research Unit, Thammasat University, Thailand.
Received 2012-11-16; Accepted 2012-12-20; Published 2013-1-1
Abstract
Head and neck squamous cell carcinoma (HNSCC) is a complex tissue that contains tumor cells and the surrounding stroma, which is populated by different types of mesenchymal cells and the extracellular matrix (ECM). Collectively, they are referred to as the tumor microenvironment (TME). Recent studies have shown that TME has a more profound influence on the growth and metastasis of HNSCC than was previously appreciated. Because carcinoma-associated fibroblasts (CAFs) are frequently observed in the stroma of the tumor, this review focuses on the potential role of tumor-CAFs interactions in progression of HNSCC. Tumor-CAFs crosstalk enhances the production of growth factors, cytokines, chemokines, matrix metalloproteinases (MMPs), and inflammatory mediators, which eventually facilitates tumor growth. In fact, factors and cells that do not support tumor growth are usually down regulated or mitigated in TME. Therefore TME may determine the fate of the tumors at the site of invasion and metastasis. For tumor cells that survive at these sites, stromal activation may serve to establish a supportive tumor stroma, fostering the outgrowth of the metastatic cells. The concept of tumor-stromal interactions and microenvironmental niche has profound consequences in tumor growth and metastasis and therefore, it's understanding will open up new strategies for the diagnosis, prognosis and therapy of HNSCC.
Keywords: Head and neck cancer, Cancer associated fibroblasts (CAFs), Matrix metalloproteinases (MMPs), Cycloxygenase-2 (COX-2), CXCR4, CCL12.
Introduction
Head and neck squamous cell carcinoma (HNSCC) occurs in the oral cavity, oropharynx, larynx or hypopharynx. It is the sixth leading cancer by incidence worldwide [1]. At present the most important risk factors identified are tobacco use and alcohol consumption. The two factors seem to have a synergistic effect. In addition, a subgroup of HNSCC, particularly those of the oropharynx, is caused by infection with high-risk types of human pailloma virus (HPV) [2].
The most knowledge on the pathogenesis of HNSCC has been acquired from the studies of oral cancer, likely because oral cancer is the most commonly diagnosed HNSCC. Additionally, oral premalignant lesions are the most frequently diagnosed pathology. Oral leukoplakias are visible precursor lesions that are macroscopically recognizable [3, 4]. However, there are several studies indicating that many precursor changes in the oral mucosa are not clinically visible. In 1953, the term of 'field cancerization' was proposed to explain the high propensity of HNSCC to develop local recurrences after treatment, and the high likelihood that multiple independent tumors can develop in the head and neck region [5].
In general, cancers including HNSCC arise from the accumulation of genetic and epigenetic changes and abnormalities in cancer-associated signaling pathways, causing the acquisition of cancer-related phenotypes that have previously been summarized by Hanahan and Weinberg [6]. This includes limitless replicative potential of tumors, self-sufficiency in growth signals, insensitivity to anti-growth signals, ability to evade apoptosis, increased angiogenesis, and invasion and metastasis. However, cancers are complex tissues. They contain tumor cells and surrounding stroma, which is constructed by various types of mesenchymal cells and the extracellular matrix (ECM). Collectively, this tissue is referred to as the tumor microenvironment (TME). Therefore, the tumor cell-centric view of cancer does not take into account the context in which malignant cells subsist. In fact, as the cancer progresses, the surrounding microenvironment also co-evolves into an activated state through continuous tumor-stromal interactions. For these reasons, six hallmarks of cancer delineated by Hanahan and Weinberg are provided by various stromal components [7].
This review discusses recent insights into the tumor-stromal crosstalk in the pathogenesis of HNSCC. The roles of cancer- associated fibroblasts in tumor growth and invasion is addressed. Additionally, chemokines, cytokines, and inflammatory signal pathways that have been implicated in tumor-stromal crosstalk are briefly reviewed. Finally, the important role of matrix metalloproteinases (MMPs) in tumor-stromal interactions is also discussed.
Molecular genetics of head and neck cancer
Genetic and epigenetic alterations in HNSCC may lead to protein changes including decreased or increased expression. The accumulation of these alterations in oncogenes and tumor suppressors can lead to the development of HNSCC. Critically altered pathways in HNSCC include cyclinD1, p53, retinoblastoma (Rb), epidermal growth factor receptor (EGFR), signal transducer and activator of transcription 3 (STAT3) and vascular endothelial growth factor receptor (VEGFR), among other important molecules that may serve as therapeutic targets (Figure 1) [8, 9].
Cell cycle deregulation in HNSCC. Normally, the cell cylcle is regulated by complexes of cyclins and cyclin-dependent kinases (CDKs). pRb binds to and inactivated E2F transcription factor, which induces in the expression of S phase genes. The CDK2-cyclin E complex phosphorylated Rb and causing release of E2F. In response to a mitogenic signal such as MAPK activation, the cyclin D1-CDK4/6 complex is activated. Activation of receptor tyrosine kinase (RTK) by mitogens can signal through ERK/MAPK and PI3 K pathways. AKT is a major downstream of PI3K. AKT prevents apoptosis by acting through different pathways. It could inactivate the apoptotic protein (BAD). The p16 and p 27 proteins are the inhibitors of cyclin D1-CDK4/6 and CDK2-cyclin E complexes, respectively. The p53 is another key protein, involving in the response of DNA damage. Deregulation of cell cycle-regulating proteins was observed in HNSCC. E6 and E7 proteins from human papillomavirus (HPV) could inactivate p53 and pRb functions, respectively. Activation of cyclin D1 and the RTK commonly occur in HNSCC.
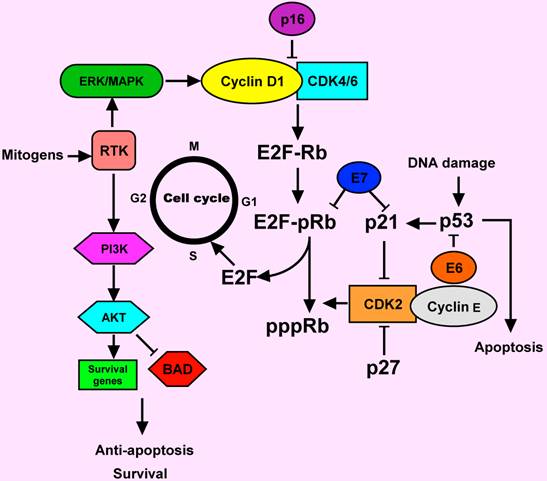
Cyclin D1 is a proto-oncogene encoding a positive regulator of G1 phase progression through the cell cycle that regulates the initiation of DNA synthesis. Over-expression of the cyclin D1 gene has been reported in 30-65% of HNSCC, suggesting that cyclin D1 gene amplification and subsequent the cyclin D1 protein over-expression are early events during HNSCC development [10-12]. However, whilst it seems likely that cyclin D1 up-regulation does play a role in the development of at least a subset of HNSCC, there may be other genes in the pathway controlling G1/S transition that may also be altered in the process of HNSCC development.
EGFR is a member of a membrane-bound receptor tyrosine kinase (RTK) family, which is composed of erbB1, erbB2, erbB3, and erbB4 [13, 14]. The known natural ligands of EGFR are EGF and transforming growth factor alpha (TGF-α). After binding to one of its ligands, EGFR forms a dimer, leading to autophosphorylation and activation of intracellular signaling events, including activation of mitogen-activated protein kinases (MAPKs), AKT, mammalian target of rapamycin (mTOR), signal transducer and activator of transcription (STAT), Janus kinase (Jak), phosphoinositide 3-kinase (PI3K), and protein kinase C (PKC) pathways. These signaling pathways, in turn, result in a multiplecellular functions, including cell proliferation and survival, invasion, metastasis, and angiogenesis [15-17].
Expression of EGFR can be deregulated in many cancers, including HNSCC. Over-expression of the EGFR ligands is observed frequently in HNSCC. This finding is associated with the outcome of poor treatment. Several studies have shown that EGFR over-expression is an independent prognostic marker that correlates with increased tumor size, decreased radiation sensitivity, and increased risk of recurrence [16, 18-20].
Members of the STAT family are latent cytoplasmic transcription factors activated by extracellular signaling proteins, such as cytokines, growth factors, hormones and peptides. Activated STAT proteins deliver the signals by translocating into nucleus and regulating transcription of target genes involved in normal cell functions, including growth, differentiation and apoptosis. There is strong evidence that STATs, especially STAT3 and STAT5, are involved in tumorigenesis. Activation of STAT3 is known to up-regulate transcription of target genes, including cell-cycle regulators, anti-apoptotic genes, and pro-angiogenic factors, leading to uncontrolled cellular proliferation, anti-apoptotic response, and angiogenesis, all hallmarks of cancer [21, 22].
Previous studies have suggested that STATs play important roles in HNSCC development and growth. Both tumor and normal epithelia of HNSCC patients show higher levels of STAT3 expression than in epithelium derived from control subjects [23]. This result suggests that STAT3 activation seems to be an early step in HNSCC development. Furthermore, activated STAT3 is also highly expressed in poorly differentiated HNSCC, and its expression is correlated with lymph node metastasis and poor prognosis [24].
The p53 gene is one of the most commonly mutated genes in HNSCC, with mutations detected in over 50% of HNSCC malignancies [11, 25]. Inactivation of the tumor suppressor p53 leads to a lack of growth control and renders the cells incapable of responding to stress or DNA damage [26]. In HNSCC, other proteins in the p53 pathway are often deregulated causing dysfunction of the p53 pathway [27]. In addition to upstream effectors of p53, there may also be alterations in downstream molecules such as the apoptotic proteins Bcl-2 and Bax in HNSCC cell lines and tumor tissues [28-31]. However, endogenous genetic alterations are not the only disrupters of p53 function. Human papillomavirus (HPV), specifically HPV16, is a risk factor for oropharyngeal cancer [32]. E6, a viral oncoprotein of HPV16 could inactivate p53 via ubiquitination [33].
The retinoblastoma gene product, a key regulator of G1/S cell cycle progression, is normally hypophosphorylated, enabling it to form a complex with the transcription factor E2F, thereby inhibiting E2F-mediated transcription of the genes that regulate DNA synthesis [34]. Although Rb mutations are rare in HNSCC, loss of this protein expression has been observed in 66-73% of HNSCC [11, 35].
Small tumor deposits (up to 1-3 mm in diameter) can receive nutrition by diffusion. For further growth, angiogenesis is necessary [36]. Therefore, all solid tumors including HNSCC exploit methods to induce neo-angiogenesis, usually by producing angiogenic factors. There are many inducers of angiogenesis, but the important inducer is vascular endothelial growth factor (VEGF). VEGF plays a pivotal role in the regulation of normal and pathological angiogenesis. It also increases vessel permeability and enhances endothelial cell growth, proliferation, migration and differentiation. At present, six VEGF family members have been identified. These include VEFG-A, placental growth factor, VEGF-B, VEGF-C, VEGF-D, and VEGF-F [37]. Tumor angiogenesis is also one of the vital components of 'successful' malignant neoplasia. VEGF, certain integrin subunit complexes and MMPs may initiate the development of new blood vessels in cancerous tissues [38-43]. This may be caused by increased production of such factors by the tumor cells but may also be a result of the release of ECM-bound growth factors by increased ECM turnover [44, 45]. Several studies have shown that tumor angiogenesis is correlated with tumor progression and aggressiveness in human cancers, including HNSCC. Expression of VEGF was stronger in cancerous tissues of HNSCC than in normal oral epithelium [46-50].
HNSCC is characterized by local invasion and a propensity for dissemination to cervical lymph nodes. The ability of malignant cells to invade surrounding tissues is one of the major hallmarks that distinguish the cancer cells from normal cells. Cancer cell invasion and metastasis represent complex, multistep process involving cell adhesion, cytoskeletal rearrangements, cell migration and degradation of the basement membrane, intravasation, survival in the blood vessel, extravasation at a distant site, and growth of metastatic cells in the distant site, with stimulation of neo-angiogenesis [51, 52]. MMPs have long been viewed as ideal candidates for proteolytic enzymes, which enable tumor cells to permeate basement membrane defense and invade surrounding tissues. Several reports support the roles of MMPs in cell adhesion, migration, epithelial to mesenchymal transition (EMT), tumor angiogenesis, and proteolytic processing of cytokines, chemokines, growth factors and their receptors, underlying the complex nature of tumorigenesis [53-57].
A microenvironmental view of cancer
Cancer has been long viewed as a disease consisting of transformed cells acquiring cell autonomous hyperproliferative, invasive and limitless survival capacities. Accordingly, therapeutic anticancer strategies have been focused on and limited to targeting the tumor cell itself. Emerging evidence indicates that to effectively control cancer, we need to consider tumorigenesis and tumor progression not as a cell autonomous, cancer cell-centered condition, but rather as a disease involving complex heterotypic multicellular interactions within a newly formed tissue, the cancerous tissue. In fact a solid tumor is a tissue disease and a systemic disease rather than a cell disease. Hence, the concept of TME as an integrated and essential part of the cancer tissue was proposed. Recent evidence emerging from the study of TME is forcing the cancer research community to revise basic concepts of cancer biology [58].
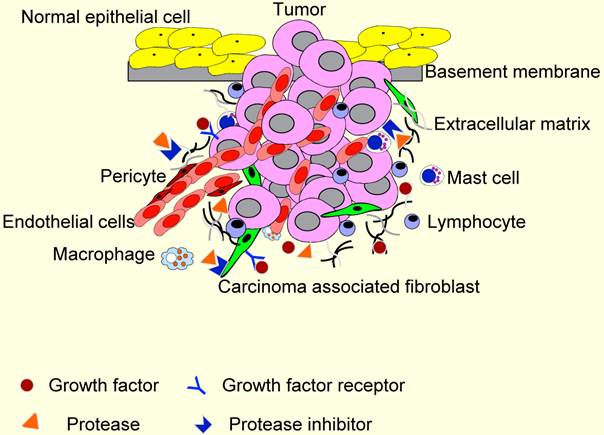
TME contains many distinct cell types, including fibroblasts, carcinoma-associated fibroblasts (CAFs), myofibroblasts, smooth muscle cells, endothelial cells and their precursors, pericytes, neutrophils, eosinophils, basophils, mast cells, T and B lymphocytes, natural kill cells, and antigen presenting cells (APC) such as macrophages and dendritic cells (Figure 2). Numerous data have demonstrated a role for these individual components, in particular CAFs, macrophages and endothelial cells, in promoting tumor growth and progression. While most cellular components of the immune system are capable of rejecting tumors, basically, they are enslaved by cancer cells to promote tumor growth and invasion. For these reasons, knowledge and control of TME is becoming as essential as the knowledge and control of the cancer cells for better understanding of cancer biology and for devising novel therapeutic approaches [59, 60].
The tumor microenvironment (TME). TME comprises different stromal cells in addition to tumor cells. These include vascular or lymphatic endothelial cells, supporting pericytes, fibroblasts, and both innate and adaptive infiltrating immune cells. Moreover, TME contains non-cellular components, including extracellular matrixes, growth factors, proteases, protease inhibitors and other signaling molecules that play important roles in stromal reactions in TME.
As mentioned above, six acquired, hallmark capabilities of cancer are thought to be required for tumorigenesis. The orders by which these hallmark capabilities are acquired vary across cancer types. However, recently several studies suggested the essential role of tumor stroma in acquisition of hallmark capabilities. The stroma provides support with growth factors and cytokines and promotes angiogenesis, tissue invasion, and metastasis. In addition, it has become evident that the stroma provides a chemoresistant capability to the tumor, preventing chemotherapeutics from reaching their targets [7, 59, 60].
The first evidence that non-cancerous tissue elements might affect tumor formation and growth came from the field of inflammation. A link between inflammation and cancer has been recognized since 1863 when Rudolf Virchow, demonstrated the presence of leukocytes in tumor tissues. Based on his observation he proposed the hypothesis that cancer originates at sites of chronic inflammation [61]. The presence of leukocytes in tumors was subsequently interpreted as an aborted attempt of the immune system to reject the tumor. However, this observation remained largely neglected for over a century until it was demonstrated that innate immune cells, in particular phagocytes, play an active role in promoting the tumorigenesis. In addition to leukocyte infiltration, angiogenesis is now being recognized as another stromal reaction promoting cancer progression. Therefore, chronic inflammatory and neovascularization are critical, if not essential, for cancer progression [62-64].
The cellular microenvironment of HNSCC
Studies have demonstrated a higher incidence of malignancy in acquired or iatrogenic immunodeficient hosts, which suggest an important role for immunosurveillance in HNSCC [65-68]. In those with a competent immune system, this is an additional barrier to malignancy, which must be overcome and which shapes both the tumor and its microenvironment [69]. Although effective antitumor immune responses likely involve many components of the immune system, T-cells continue to be considered as the critical immune cells involved in antitumor immunity. T lymphocytes are considered an essential component of antitumor immunity, with CD8+ T cells serving as cytotoxic effector cells and CD4+ Th1 cells serving to 'help' and enhance the magnitude and duration of the antitumor responses. However, CD4+ Th2 cells and CD4+ T regulatory cells are capable of suppressing effective CD8+ antitumor responses. In fact, several investigators have found dysfunctional circulating and tumor-infiltrating T cells in HNSCC patients, with functional assays identifying multiple defects in T-cell activation and effector function, suggesting that the tumor has successfully suppressed an otherwise robust lymphocytic response [70-72].
Patterns of tumor-related leukocyte infiltration varies between primary tumors and metastatic lymph nodes in HNSCC with a local decrease in the number of CD8+ T-cells and increase in CD20+ B-cells being the most relevant findings. This indicated that suppression of local cellular immunity might be a mechanism by which tumor cells evade host immunity [73]. Patients with tumors expressing HPV16 had an increased frequency of CD8+ T cells specific for peptides derived from the oncogenic HPV E7 proteins, compared with those patients with tumor negative for HPV or normal controls [74, 75]. In addition to modulating immune cells in its vicinity, HNSCC cells actively recruit and trigger the production of tumor growth promoting interleukin (IL)-6 from CD34+ myeloid progenitor cells [76]. CD34+ progenitor cells differentiate into a variety of cell lineages including endothelial cells involved in angiogenesis [77]. Th17-T helper cells are characterized by the high levels of secreted pro-inflammatory cytokine IL-17 [78]. HNSCC and draining lymph nodes were infiltrated with Th-17 cells that are recruited by the tumor cells. Interestingly, Th17 cells reduce HNSCC cell proliferation while increasing angiogenesis [79].
Mast cells are white blood cells that directly associate with endothelial cells stimulating vascular tube formation. As HNSCC progresses, there is an increase in mast cell numbers which is correlated with angiogenesis suggesting a role in angiogenesis [80-82].
In neoplasms, tumor associated macrophages (TAMs) also represent a major component of the infiltrating leukocytes. The presence of these cells can be beneficial for the growth of the tumor and sometimes they may cause the death of cancer cells. TAMs are present at higher levels in HNSCC and modulate angiogenesis during tumor progression [83, 84]. Primary HNSCC tumor with high TAM infiltration is a strong predictor of lymph node metastasis, extracellular capsular spread and advanced HNSCC stages [78]. In addition, expression of macrophage inflammatory protein-3α was shown to promote oral cancer cell migration and invasion [85]. TAMs accumulate near blood vessels that are associated with tumor cells owing to the local secretion of colony-stimulating factor 1 (CSF-1) by tumor cells [86]. TAMs secrete EGF, which attracts tumor cells to the vessels by chemotaxis. Several studies have suggested the involvement of TAMs in angiogenesis and tumor progression of HNSCC. A close association was found between TAM count and integrity of vascular structure in oral squamous cell carcinoma. In addition, a significant correlation between TAM count and lymph node involvement was observed in this tumor type [87].
Hypoxia-inducible factor (HIF)-1α expression and TAMs can change cancer cell behavior resulting in more invasive and aggressive behavior. It has been suggested that the presence of tumor cell-lined vessel, HIF-1α and the high rate of TAMs could be the potential marker for the prognosis of patients with oral squamous cell carcinoma [88]. The impact of TAMs on tumor aggressiveness was previously studied in a series of oral cavity or oropharyngeal squamous cell carcinomas. In that study, the authors demonstrated a correlation between the aggressive behavior of HNSCC and the level of infiltration of macrophages in the primary tumor [78].
In spite of the important recruitment of immune cells in TME, these cells do not represent the major cell population of the tumor stroma. CAFs are the most abundant cells of the tumor microenvironment. CAFs are usually recognized by the expression of α-smooth muscle actin (α-SMA), similar to myofibroblasts present at the site of wound healing and chronic inflammation, which is absent in normal dermal fibroblasts [89, 90]. CAFs might differentiate locally from normal stromal fiibroblasts of surrounding tissue or from bone marrow-derived mesenchymal stem cells recruited to the tumor [91].
Tissue injury triggers fibroblast activation. Activated fibroblasts are responsible for wound contraction, fibrosis, scaring and regulation of inflammatory reaction. Upon activation, fibroblasts trans-differentiate into myofibroblasts. Tumors are frequently regarded as wounds that do not heal. Tumors are commonly associated with desmoplastic stromal myofibroblasts also known as CAFs. CAFs are observed in both primary and metastatic HNSCC. HNSCC stroma is either rich in CAFs dispersed throughout the tumor or has levels of CAFs that are located at the periphery of HNSCC tumors or tumor islands [92-94]. There is evidence to suggest that CAFs use protease and mechanical remodeling of ECM to lay tracks along which HNSCC tumor cell invade [95].
Two dominant pattern, 'spindle' and 'network', have been observed in HNSCC. In the 'network' pattern CAFs are exceptionally abundant and occupy almost the entire tumor stroma whereas 'spindle' pattern is observed at the periphery of a tumor island [96]. Morphological and immunohistochemical studies of oral CAFs demonstrated that there were marked differences between oral CAFs and normal oral fibroblasts. Oral CAFs are long spindle-shaped cells with small cytoplasmic protrusions, whereas, oral fibroblasts are flat-star shaped cells with more pronounced cytoplasmic protrusions. Besides cytokeratins, oral CAFs express vimentin, α-SMA and MMP-2 whereas oral fibroblasts exhibit only positive staining for vimentin. This suggests that some fibroblastic traits are preserved in CAFs. It was also reported that oral CAFs acquire rapid growth, increased proliferation and viability compared with normal oral fibroblasts [97].
It is known that endothelial cells have a significant impact on the progression of HNSCC through secretion of factors involved in tumor proliferation. Endothelial derived factors induce activation of key signaling molecules in HNCCC cells enhancing their motility and inhibiting anoikis [98]. Direct binding of HNSCC cells to endothelial cells is a prerequisite for penetration and metastasis through the vasculature. Furthermore, direct interaction between HNSCC cells and endothelial cells trigger Notch-1 signaling in endothelial cells promoting capillary tubule formation [99]. HNSCC cells and stromal cells secrete cytokines and growth factors including VEGF, PDGF and IL-8 inducing angiogenesis [81, 100-106].
VEGF plays an important role in endothelial survival. Endothelial cell-derived VEGF signals through VEGFR1 and induces the expression of Bcl-2 and the proangiogenic chemokines, CXCL1 and CXCL8, in HNSCC cells [107] On binding to its receptor, VEGF induces expression of Bcl-2 and autocrine signaling through chemokines CXCL1 and CXCL8 facilitating proliferation of endothelial cells and sprouting of new blood vessels [108]. In addition to the formation of new blood vessels, endothelial cells are involved in a crosstalk with squamous cell carcinoma cells resulting in a significant increase in tumor cell survival and migration. Specifically, certain soluble factors secreted by endothelial cells including interleukin (IL)-8, IL-6 and EGF induce phosphorylation of signal transducers and activators of transcription-3, extracellular-regulated kinase and AKT in HNSCC [109].
Gene expression profiling demonstrated that HNSCC induce angiogenesis by either expressing high levels of VEGF/fibroblast growth factor (FGF)-2 and low level of IL-8/CXCL8 or low levels of VEGF/FGF2 and high levels of IL-8/CXCL8. In addition, tumor hypoxia also plays an important role in the release of angiogenic growth factor [110]. Under hypoxic conditions stabilization of the HIF-1α in tumor cells allows transcription of genes involved in angiogenesis [111]. Overexpression of HIF-1α was demonstrated in HNSCC cell lines and tumor tissues [112]. However, HIF-1α expression had no impact on prognosis, while VEGF expression correlated significantly with adverse prognosis of patients with HNSCC [113].
In addition to blood vessels, HNSCC is typically infiltrated by lymphatic vessels a process known as lymphangiogenesis. Lymph vessels are typically distributed throughout the tumor as well as in the peritumoral regions [114-116]. Increased tumor lymphatic vessel density correlates with metastasis to lymph nodes in HNSCC [117]. Metastasis to regional lymph nodes commonly occurs in HNSCC and correlates with poor prognosis. It was found that VEGF-C, a member of VEGF family, plays an important role in tumor lymphangiogenesis [49].
Pericytes are contractile stromal cells closely associated with vascular endothelial cells that stabilize the capillary walls. In the absence of these cells, blood vessels are unstable and undergo regression. Pericytes influence the proliferation, migration and maturation of endothelial cells. However, very few studies have focused on pericytes in HNSCC. The majority of previous studies use markers such as α-smooth muscle actin to stain pericytes associated with endothelial cells via immunohistochemical analysis. An immunohistochemical study demonstrated that newborn vessels in oral cancer are abnormal, showing increased permeability, delayed maturation, and potential for rapid proliferation. Regarding pericyte recruitment, the immature and intermediate vessel types (both negative for α-smooth muscle actin) were the most numerous types of tumor vessels. The mature ones (positive for α-smooth muscle actin) were more numerous at invasive front of tumor, especially in poorly differentiated tumor types [118].
Tumor-stromal crosstalk in HNSCC
The pathogenesis of HNSCC is considered a multistep process with an accumulation of genetic mutations, altered protein expression, leading to the development of a unique microenvironment designed to support tumor growth. While there is limited data regarding the importance of the TME in HNSCC, it is currently a topic of much interest. HNSCC function much like organs with support from multiple cell lineages. Factors and cells that do not support tumor growth are commonly down regulated or mitigated in TME.
Tumor growth, invasion, and metastasis are important aspects of the tumor immune escape. Several mechanisms that mediate immune escape have been identified in cancer. These include down-regulation of major histocompatibility complex (MHC) class I or class II molecules, loss of immune co-stimulatory molecules, defects in processing and presentation of tumor-associated antigens (TAA), down-regulation of TAA and overexpression of immunosuppressive molecules, including TGF-β, IL-6, and IL-10. Collectively, these mechanisms limit the host immune response to cancer cells [119]. Tumor progression is marked by evasion of immunosurveilance, recruitment of bone-marrow-derived cells and their induction to a tumor-promoting phenotype, decreased infiltration and dysfunction of antitumor immune cells, and angiogenesis [120]. Previous studies of HNSCC have linked poor lymphocytic infiltration to local-regional recurrence and decreased overall survival [121, 122]. The worse prognosis of these cancers must certainly link to the fact that HNSCC strongly influences the host immune system. Antitumor responses of HNSCC patients are caused by the presence of functional defects or apoptosis of T-cells, both circulating and tumor infiltrating [123, 124].
Normal cells survive and grow within defined environmental niches and are subjected to microenvironmental control. Normal stroma consists of various connective tissues that act like a supportive framework for tissues and organs. Among all the stromal components, fibroblasts are essential to synthesize and deposit the ECM by producing a variety of collagens and fibronectin. In addition, they are indispensable for the formation of the basement membrane, which separates the epithelium from the stroma by secreting laminin and type IV collagen. They are also an ample source of various soluble paracrine and autocrine growth factors that regulate their growth and those of the surrounding cells [63].
It is generally accepted that invasion is a three-step process involving changes in tumor cell adhesion, proteolytic degradation of the ECM and migration of tumor cells in a proteolytically modified ECM. This is consistent with the idea that the major function of the ECM is to provide a scaffold to support the organization of cells into specific tissues. The ECM structures such as the basement membrane provide a physical barrier against invasion and metastasis. This concept has substantially evolved over the last decade as our understanding of the mechanisms that link cells to the ECM has improved. It is now evident that the ECM is more than a scaffold to which cells are anchored and that the ECM has a significant influence on cell function and behavior [125]. Stromal-tumor interactions in HNSCC involve cell-cell crosstalk via secreted factors and their receptors. The soluble factors can act in both autocrine and paracrine manners. These factors work in coordination with other signaling molecules, such as ECM and integrins, which facilitate not only tumorigenesis, but also tumor progression towards metastasis.
Overexpression of basement membrane components such as type IV collagen was observed in HNSCC [126]. Cell adhesion domain of collagen XVII, Col15, is able to chemotactically attract invasive head and neck cancer cells but not normal keratinocytes. This chemotactic function is mediated by Col15-binding integrins[127]. Different integrins play important roles in the adhesion of head and neck cancer cells to basement membrane components such as laminin and fibronectin [128, 129]. These findings suggest that tumor integrins confer polarity by binding to the ECM. The expression of glycosylated oncofetal fibronectin was increased in the invasive phenotype of oral cancer cell lines. Furthermore, it was revealed that collagens in the connective tissue, appears to stimulate the invasiveness of oral carcinoma cells [130].
A co-culture study demonstrated that type I collagen markedly stimulated the expression of IL-1α, IL-1β, IL-6, TNF-α, and TGF-β in primary and metastatic HNSCC cells. However, type I collagen markedly stimulated certain cytokines in metastatic cancer cells compared with that of the primary cancer cells [131]. An increase in MMP-2 activities was observed in metastatic HNSCC cell lines when they were attached to type I collagen. In contrast, the basement membrane did not obviously enhance MMP-2 activities of these cancer cell lines. Furthermore, an increase in p-ERK and p-p38 but not ERK and p38 was observed when primary and metastatic HNSCC cells were cultured on type I collagen. These findings suggest that phosphorylation of MAPKs plays an important role in ECM-induced MMPs [132].
In addition to cell-ECM interactions, cell-cell interactions are suggested to contribute to tumor growth, especially interactions of stromal fibroblasts with cancer cells. As mentioned above, myofibroblasts are known to induce migration and invasion in a number of contexts both in normal development and tumorigenesis. Such a role for myofibroblasts in HNSCC is supported by observations that myofibroblasts induced by TGF-β1 secrete hepatocyte growth factor (HGF), which promotes the in vitro invasion of HNSCC cells [80, 133]. Moreover, it was demonstrated that mutual interactions between HNSCC cells and myofibroblasts may exist, and that conditioned media from TGF-β1-induced myofibroblasts enhances cell growth of HNSCC cells [134, 135]. The proliferation of HNSCC cells was increased by 15-80% when they were cultured with fibroblasts. Furthermore, this fibroblast enhanced tumor cell growth was suppressed by the fibroblast growth factor receptor (FGF-R) [136, 137].
Extracellular matrix metalloprotease inducer (EMMPRIN) is a cell surface glycoprotein that is overexpressed in many malignant cancers, including HNSCC [138-140]. Elevated EMMPRIN expression levels correlate with tumor proliferation, invasion, angiogenesis and metastasis. It was found that the growth of HNSCC cells in both FGR2-dependent and FGR2 independent fashions depend on EMMPRIN expression on tumor cells. FGFR2 likely plays an important role in the initial stages of oral cancer development in which EMMPRIN expression is also low. Up-regulation of EMMPRIN suppresses FGR2 expression, leading to fibroblast-independent growth. This means that EMMPRIN acquired during tumor progression promote fibroblast-independent tumor growth [137]. Recently, it has been demonstrated that myofibroblasts promote proliferative activity of oral squamous carcinoma cells by up-regulating activin A, a member of the transforming growth factor-β super family of proteins [141].
Taken together, the stroma of HNSCC is significantly different from that of the normal and surrounding tissues in many ways. Experimental data from the last decades do suggest that the stroma is an active participant in malignant processes. Cancer cells may induce profound alterations in the stromal cells and the ECM via various mechanisms (cell-matrix interactions, cell-cell contact, soluble factor, etc.). This underscores the pivotal role of such complex crosstalk in tumor development and metastasis.
HNSCC-associated Inflammation
Inflammatory reactions preceding early stages of neoplastic progression contribute to the creation of an environment, which favors cancer development. Many cancers have been associated with persistent inflammation, including head and neck cancer [142-146]. Aberrant arachidonic (AA) pathway metabolism, especially cyclooxygenase (COX)-2 and 5-lipoxygenase (5-LOX) pathways is activated during HNSCC development [147]. Cycloxygenase (COX) enzymes specifically catalyze the production of prostaglandins (PGs). COX-1 and COX-2 are the two isoforms found in humans. COX-1 is constitutively present in most tissues. In contrast, COX-2 is usually overexpressed in inflammation and in pre-neoplastic lesions and tumors. PGs are increased in HNSCC, and one of the most important members, PGE2, in known to promote growth, and inhibit apoptosis by up-regulating Bcl-2 expression. PGE2 is also known to increase the production of angiogenic factors, resulting in the promotion of invasion and tumor metastasis. Higher expression of COX-2 in tumor cells is seen in parallel with an increased PGE2 expression in HNSCC [148, 149]. Immunohistochemical studies revealed that COX-2 overexpression was positively correlated with the number of tumor-infiltrating Foxp3+ regulatory T cells in the local microenvironment of HNSCC. This suggests that COX-2 facilitated the expansion of the regulatory T cells through PGE2 [150].
Several studies showed the effects of COX inhibitors on cancer cell proliferation, invasion and metastasis. Selective COX-2 inhibitor decreased viability, invasion and adhesion of HNSCC cells by down-regulating MMP-2, MMP-9 and VEGF secretion [151-153].
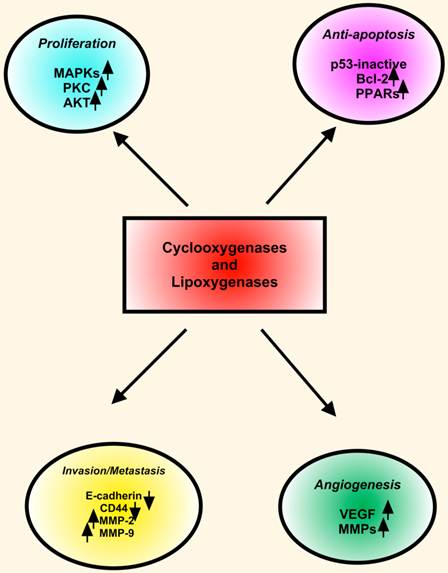
The LOXs convert AA, linoleic, and other polyunsaturated fatty acids into biologically active metabolites that influence cell signaling, structure and metabolism [154]. Many LOX enzymes, including 5-LOX, 12-LOX, 15-LOX-1 and 15-LOX-2 were detected in HNSCC cells derived from primary and metastatic tumors. The expression of 5-LOX was increased in dysplasia and squamous cell carcinoma in animal studies. Topical application of leukotriene (LT) B4, a metabolic product of 5-LOX, enhanced oral carcinogenesis by increasing the incidence and size of the tumors. These findings indicated that in addition to COX-2, the LOX pathway also plays important roles in HNSCC. COX-2 and LOX display similarities in expression and functions in HNSCC. However, COX-2 and 5-LOX may have redundant functions in HNSCC pathobiology. First, COX-2 and 5-LOX enhance tumor cell proliferation. Second, both COX-2 and 5-LOX are proangiogenic with a convergent targeting on VEGF, FGF and MMPs. Third, COX-2 as well as 5-LOX inhibitors arrest cell cycle progression and induce apoptotic cell death in HNSCC cells. Fourth, both COX-2 and 5-LOX are located in the nucleus, and they may function as endogenous ligands for nuclear receptors such as the PPARs (Figure 3) [151].
COX and LOX in HNSCC. COXs and LOXs stimulate proliferation, inhibit apoptosis, induce angiogenesis, and enhance invasion and metastasis in HNSCC. Key proteins including MAPKs, PKC, Bcl-2, PPARs (peroxisome proliferator-activated receptors), VEGF and MMPs involve in downstream effects of COXs and LOXs.
Significant involvement of cytokines and chemokines in formation of tumor microenvironment in HNSCC
Cytokines regulate immunity, inflammation, and hematopoiesis. This family of proteins includes ILs, interferons (IFNs), tumor necrosis factors (TNFs), and other growth factors. Chemokines are super family of small, cytokine-like proteins with chemoattractant and activation properties for different cell types involved in inflammatory reactions. Cytokine and chemokines are key molecules controlling autocrine or paracrine communication within and between individual cells in TME [155, 156]. Altered expression of cytokines and growth factors plays a major role in the malignant transformation of many cancers including HNSCC. A number of such factors are found in HNSCC cell lines as well as in patients' tumor specimens and serum. These include IL-1α, IL-1β, IL-6, IL-8, TNF-α, TGF-β, granulocyte-macrophage colony-stimulating factor (GM-CSF) and VEGF. Decreasing certain cytokine and growth factor levels in serum are associated with response to therapy, whereas increasing levels are related to cancer progression and recurrence [157-160].
Chemokines have been shown to play roles in the regulation or stimulation of cancer progression, neovascularization, immunosurveillance, and metastasis. Chemokine receptors are present on many different cell types. In fact, these receptors were initially identified on leukocytes, where they were found to play an important role in the homing of such cells to sites of inflammation. However, at present, hematopoietic and nonhematopoietic cells have been found to express receptors for various chemokines that are constitutively expressed in distinct tissue microenvironment. The interactions between such receptors and their respective chemokines help coordinate the trafficking and organization of cells to various tissue compartments [161, 162].
There has been considerable interest regarding the fact that chemokines regulate leukocyte trafficking and recruitment, leading to the hypothesis that tumor cells might use analogous, chemokine-dependent mechanisms for targeting to specific secondary sites. There is increasing evidence that epithelial tumor cells exploit mechanisms that normally regulate leukocyte trafficking and homing. The distinct pattern of chemokine receptor expression by cancer cells has a critical role in determining the site(s) of metastatic spread. Additionally, stromal cells within TME at primary or metastatic sites apparently can regulate tumor progression.
Muller and coworkers demonstrated that up-regulation and activation of CXCR4 and CCR7 in breast cancer cells is capable of inducing actin polymerization, migration and invasion both in vitro and in vivo studies [163]. Stromal cell-derived factor-1 (SDF-1), which is also designated as CXCL12, is a homeostatic chemokine that signals through CXCR4, which in turn plays an important role in tumor pathogenesis [164, 165]. Up-regulation of CXCR4 expression has been observed in cancer cells and neoplastic tissues in HNSCC [132, 166-168]. Furthermore, it was reported that hypoxia enhances CXCR4 expression by activating HIF-1α in oral squamous cell carcinoma [169].
CXCL12-CXCR4 signaling contributes to HNSCC progression by up-regulating the expression of proteases. It was found that MMPs were modulated by CXCL12-CXCR4 interactions. CXCL12 induced rapid phosphorylation of ERK-1/2 in a metastatic HNSCC cell lines. This chemokine could mediate the adhesion of HNSCC cells to fibronectin and collagen and the interaction enhances MMP-9 activation [167]. CXCL12 also promote the invasion of primary and metastatic HNSCC cells. For this reason, it is possible that CXCR4-CXCL12 crosstalk may cause an increase in cellular motility as well as MMP-9 activities of HNSCC cells [132]. However, these effects were not consistent for all HNSCC cell lines, suggesting that other cell-specific factors may modify or modulate the response to CXCL12. In contrast, CXCL12 did not affect MMP-2 and MMP-9 expression in tumor cells of hypopharyngeal squamous cell carcinoma, but was found to induce expression of MMP-13 [170]. In addition, CXCR4 and CXCL12 interactions may enhance invasion of HNSCC cell by up-regulation of IL-6 production [171]. It has been reported that lipopolysaccharides increase the expression of CXCR4, and modulate the morphology and invasive ability of HNSCC in a CXC12-dependent manner [172]. Thus, bacterial infections may create an optimal environment for tumor growth by modulating CXCR4-CXCL12 interactions.
In addition to MMP stimulation, CXCR4 and CXCL12 interaction induces epithelial-mesenchymal transition (EMT) in HNSCC cells. It appears that the crosstalk between CXCL12 and its receptor might be involved in the lymph node metastasis of HNSCC cells [173].In addition to CXCL12, it was reported that HNSCC derived from a lymph node metastasis, but not from a synchronous primary tumor, secreted CXCL5 [174]. Furthermore, CXCL5 also stimulated cell proliferation and the in vitro invasion of metastatic HNSCC cells. It should be noted that not only CXCR4-CXCL12 interactions but also other chemokine crosstalk including CCL5/CCR5 axis might also contribute to the motility of HNSCC cells [175].
Cytokines control the crosstalk between tumor cells and stromal cells. Tumor cells release cytokines that induce ECM remodeling, basement membrane degradation, tumor cell proliferation and angiogenesis. Fibroblasts and endothelial cells in TME play a crucial role in the response to tumor-derived cytokines. Not only do stromal cells respond to signals from tumor cells, but also the stromal cells themselves can induce tumor growth. We found that type I collagen markedly stimulates cytokine expression in metastatic HNSCC cell lines compared with that of the primary cancer cell lines. Furthermore, co-culturing of HNSCC cells with oral fibroblasts caused an increase in cytokine expression, which may enhance the invasive properties of HNSCC cell lines [131].
CAFs co-cultured with oral cancer cells showed an increase in the expression of a number of proinflammatory cytokines and chemokines including CCL7, CXCL1, CXCL2, CXCL3 and CXCL8 when compared with CAF cultured alone. Interestingly, the authors demonstrated that the IL-1α secreted from the tumor cells induces CCL7 secretion in CAFs in a paracrine manner and resulting in cancer progression [176].
Roles of MMPs in the tumor microenvironment of HNSCC
The tissue matrix may be classified into interstitial connective, or stromal matrix, that supports individual cells, and a very specialized structure, which forms a continuous sheet called the basement membrane. This membrane supports cell layers, such as the epithelium and endothelium. The ECM refers to inter-cellular materials of assembled specialized fibrous protein families including fibronectin, laminins, collagens, proteoglycans and tenascin [177].
The remodeling of ECM by MMPs is one of the most crucial steps for cancer progression as well as for the formation of TME. Under normal physiological conditions, the balance between MMPs and their inhibitors, tissue inhibitors of metalloproteinases (TIMPs), keeps the ECM in a well-organized shape. Different MMPs can digest a large set of ECM and non-ECM. They act as processing enzymes that perform highly selective and limited cleavage of specific substrates including growth factors and their receptors, cell adhesion molecules, cytokines, chemokines, apoptotic ligands and angiogenic factors. Data supporting the role of MMPs in cancer progression were obtained from both in vitro and in vivo studies. However, MMP functions are much more complex than initially anticipated. After years of considering MMPs as pro-tumorigenic enzymes, an intriguing observation has prompted re-evaluation of the roles of MMPs in cancer. In fact, some MMPs play a paradoxical protective role in tumor progression whereas others display opposite functions depending on the stage of cancer progression [178, 179]. Recent advances in genomic and proteomic technologies have increased our knowledge on MMP contributions to different processes associated with tumor development such as tumor growth, angiogenesis, invasion and inflammation. Despite their implication in ECM remodeling and growth factor signaling that favor angiogenesis and boost tumor development, some MMPs exert protective effects that retard the tumor development [179].
Many studies revealed that gelatinases (MMP-2, MMP-9), stromelysin (MMP-3, MMP-10, and MMP-11), collagenases (MMP-1 and MMP-13) and membrane-bound MMP (MT-MMP) are all expressed in oral cancer [180]. A degradation of basement membrane is the first step toward invasion and metastasis. Type IV collagen is the main component of basement membrane, and destruction of this structural protein is favored by two MMPs, namely the gelatinase A (MMP-2) and gelatinase B (MMP-9). These MMPs are known to be closely associated with the malignant potential of tumor cells [181].
The ability of MMP-2 and MMP-9 to initiate basement membrane destruction and further degrade the collagenous and non-collagenous components of the ECM suggests that the gelatinases are important in tumor invasion and metastasis. For these reasons, several studies focused on the clinical significance of MMP-2 and MMP-9 in patients with HNSCC. It has been reported a relationship between the expression of these gelatinases and tumor aggressiveness [182, 183]. Latent, active, and total forms and activation ratio of MMP-2 and MMP-9 were significantly elevated in malignant tissues as compared with adjacent normal tissues. Activation ratios (active form/total form) of MMP-2 were significantly higher in malignant tissues of patients with lymph node metastasis. Moreover, plasma MMP-9 levels were significantly lower in responders compared with pretreatment levels [184].
CAFs affect cancer cell invasion through both cell-cell contact and pro-invasive factor secretion. They are also one of the most significant contributors to MMP production, which play a major role in cancer metastasis. The mechanisms by which stromal cell expression of MMPs contribute to HNSCC progression is an area of intense interest. Using co-cultures of live and fixed cells, we demonstrated that direct contact between HNSCC cells and fibroblasts was required to activate MMP-2 and MMP-9 secretion in both tumor cells and fibroblasts [132]. Moreover, it was demonstrated that fibroblasts seem to be responsible for the increased MMP-2 in the co-culture. In addition, fibroblasts or tumor cell conditioned media up-regulated the secretion of MMP-2 and MMP-9 in HNSCC cells. Therefore, our findings suggest that autocrine and paracrine factors augmented MMPs secretion of tumor and/or stromal cells in TME [132].
Recently, the paracrine interaction between HNSCC cells and periodontal ligament (PDL) fibroblasts was found to lead to the up-regulation of different MMPs, including MMP-2, MMP-1 and MMP-13. MMP-2 is secreted in it pro- (inactive-) form mainly by PDL fibroblasts surrounding the tumor cells. Activation of MMP-2 either required MT1-MMP localized on the tumor cells, or αv integrins. Crosstalk between tumor cells and PDL fibroblasts leads to up-regulation of αv integrins in both cell types. Moreover, it was found that TGF-β1 contributed to the up-regulation of MMP-2. In contrast to pro-MMP-2, pro-MMP9 is produced by HNSCC cells, not by PDL fibroblasts. Pro-MMP-9 is activated on the surface of tumor cells. The paracrine interaction between tumor cells and PDL fibroblasts also resulted in the up-regulation of MMP-9 in HNSCC cells, where fibronectin, and its receptor, αvβ6 integrin as well as CD-44 could have been involved [185].
Endothelins (ETs) are isopeptides, which produced by vascular endothelium. They are encoded by three separate genes and processed to yield 39-residue, 'big ET' molecules which are further processed to the 21 amino acid sequences designated ET-1, ET-2 and ET-3. It has been proposed that ET-1 plays an important role in HNSCC cell and fibroblast interactions in TME. ET-1 is known to modulate the phenotype of human oral fibroblasts. It also contributes to pro-migratory paracrine signaling between stromal fibrobasts and HNSCC cells [186]. ET binds to its receptors on oral fibroblasts, activating ADAM17 and triggering the release from the cell surface of EGFR ligands such as amphiregulin and TGF-α. Soluble EGFR ligands subsequently bind and activate EGFR on HNSCC cells, triggering signaling pathway up regulating COX-2 and promoting migration [187].
Recently, it has been demonstrated that CXCR4 silencing obviously decreased the expression of MMP-9 and MMP-13 expression in HNSCC cell lines. This indicated that CXCR4 specifically modulate the expression of certain MMPs in HNSCC cells. ERK signaling pathway is likely associated with several mechanisms of tumor cell motility mediated by CXCR4, including regulation of the transcriptional levels of MMP-9 and MMP-13 [188].
Interaction of certain cell surface receptors such as integrins with their ECM ligands may also impact on MMP expression. We found that type I collagen enhanced MMP-2 and MMP-9 secretion in both primary and metastatic HNSCC cell lines. Furthermore, it was demonstrated that type I collagen acted through the α2β1 integrin to activate tyrosine kinases, protein kinase C, ERK1/2, and p38, which in turn activated MMP-2 and MMP-9 in the cancer cell lines [132].
Taken together, co-culture studies between HNSCC cells and stromal cells provide a strong support for the concept that stromal cells or more likely fibroblasts play important roles in tumor growth. Furthermore, they underline the complexity of the tumor-host interface that deserves further in-depth investigation.
Concluding remarks
Molecular pathogenesis of HNSCC is a multistep process consisting of genetic mutations and alterations in oncogenes and tumor suppressor genes, manifested as a lack of growth control and increased proliferation, survival, invasion and angiogenesis. However, genetic and cell-biology studies indicate that tumor growth is not just determined by malignant cancer cells themselves, but also by the tumor microenvironment. The progression of HNSCC is the net result of a highly complex, mutual relationship between their parenchymal and stromal components. Tumor and stromal cells may interact with each other through direct cell contact or via paracrine signaling. HNSCC cells may induce profound alterations in stromal cells and ECM via various mechanisms. Cancer cells may alter their stroma by cell- cell contact, soluble factors or by modification of the extracellular matrix. They frequently secrete different molecules including cytokines, growth factors, chemokines, MMPs and inflammatory mediators that may promote their invasiveness. On the other hand, these soluble factors released by the stromal cells also influence the tumor progression via paracrine signaling. However, the mechanisms by which changes in stromal cells facilitate HNSCC growth are not completely understood at present. Increased understanding of the mechanisms involved in the complex crosstalk between cells and the tumor microenvironment hold great promise for designing strategies to target HNSCC effectively (Figure 4).
Tumor-stromal interactions in HNSCC. Tumor-stromal crosstalk leads to the overexpression of growth factors sustaining tumor growth, angiogenic factors promoting angiogenesis, and proteolytic enzymes enhancing the degradation of extracellular matrixes. These autocrine and paracrine factors facilitate tumor cell invasion and finally metastasis.

Competing Interests
The authors have declared that no competing interest exists.
References
1. Duvvuri U, Myers JN. Cancer of the head and neck is the sixth most common cancer worldwide. Curr Probl Surg. 2009;46:114-117
2. Ragin CC, Modugno F, Gollin SM. The epidemiology and risk factors of head and neck cancer: a focus on human papillomavirus. J Dent Res. 2007;86:104-114
3. Napier SS, Speight PM. Natural history of potentially malignant oral lesions and conditions: an overview of the literature. J Oral Pathol Med. 2008;37:1-10
4. van der Waal I. Potentially malignant disorders of the oral and oropharyngeal mucosa; present concepts of management. Oral Oncol. 2010;46:423-425
5. Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963-968
6. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70
7. Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res. 2010;316:1324-1331
8. Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11:9-22
9. Choi S, Myers JN. Molecular pathogenesis of oral squamous cell carcinoma: implications for therapy. J Dent Res. 2008;87:14-32
10. Bartkova J, Lukas J, Muller H, Strauss M, Gusterson B, Bartek J. Abnormal patterns of D-type cyclin expression and G1 regulation in human head and neck cancer. Cancer Res. 1995;55:949-956
11. Koontongkaew S, Chareonkitkajorn L, Chanvitan A, Leelakriangsak M, Amornphimoltham P. Alterations of p53, pRb, cyclin D(1) and cdk4 in human oral and pharyngeal squamous cell carcinomas. Oral Oncol. 2000;36:334-339
12. Miyamoto R, Uzawa N, Nagaoka S, Hirata Y, Amagasa T. Prognostic significance of cyclin D1 amplification and overexpression in oral squamous cell carcinomas. Oral Oncol. 2003;39:610-618
13. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341-354
14. Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177-184
15. Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. 2006;24:2666-2672
16. Zimmermann M, Zouhair A, Azria D, Ozsahin M. The epidermal growth factor receptor (EGFR) in head and neck cancer: its role and treatment implications. Radiat Oncol. 2006;1:11
17. Rogers SJ, Harrington KJ, Rhys-Evans P, P OC, Eccles SA. Biological significance of c-erbB family oncogenes in head and neck cancer. Cancer Metastasis Rev. 2005;24:47-69
18. Numico G, Russi EG, Colantonio I, Lantermo RA, Silvestris N, Vitiello R, Comino A, Abrate M, Zavattero C, Melano A, Merlano M. EGFR status and prognosis of patients with locally advanced head and neck cancer treated with chemoradiotherapy. Anticancer Res. 2010;30:671-676
19. Rabinowits G, Haddad RI. Overcoming resistance to EGFR inhibitor in head and neck cancer: A review of the literature. Oral Oncol. 2012;48:1085-1089
20. Song J, Chen C, Raben D. Emerging role of EGFR-targeted therapies and radiation in head and neck cancer. Oncology (Williston Park). 2004;18:1757-1767
21. Nikitakis NG, Siavash H, Sauk JJ. Targeting the STAT pathway in head and neck cancer: recent advances and future prospects. Curr Cancer Drug Targets. 2004;4:637-651
22. Song JI, Grandis JR. STAT signaling in head and neck cancer. Oncogene. 2000;19:2489-2495
23. Grandis JR, Drenning SD, Zeng Q, Watkins SC, Melhem MF, Endo S, Johnson DE, Huang L, He Y, Kim JD. Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci U S A. 2000;97:4227-4232
24. Masuda M, Suzui M, Yasumatu R, Nakashima T, Kuratomi Y, Azuma K, Tomita K, Komiyama S, Weinstein IB. Constitutive activation of signal transducers and activators of transcription 3 correlates with cyclin D1 overexpression and may provide a novel prognostic marker in head and neck squamous cell carcinoma. Cancer Res. 2002;62:3351-3355
25. van Houten VM, Tabor MP, van den Brekel MW, Kummer JA, Denkers F, Dijkstra J, Leemans R, van der Waal I, Snow GB, Brakenhoff RH. Mutated p53 as a molecular marker for the diagnosis of head and neck cancer. J Pathol. 2002;198:476-486
26. Solomon H, Madar S, Rotter V. Mutant p53 gain of function is interwoven into the hallmarks of cancer. J Pathol. 2011;225:475-478
27. Piffko J, Bankfalvi AA, Ofner D, Totsch M, Berens A, Joos U, Bocker W, Schmid KW. Proliferative (MIB1, mdm2) Versus Anti-Proliferative (p53) Markers in Head and Neck Cancer. An Immunohistochemical Study. Pathol Oncol Res. 1996;2:37-42
28. Condon LT, Ashman JN, Ell SR, Stafford ND, Greenman J, Cawkwell L. Overexpression of Bcl-2 in squamous cell carcinoma of the larynx: a marker of radioresistance. Int J Cancer. 2002;100:472-475
29. Kawakami K, Tsukuda M, Mizuno H, Nishimura G, Ishii A, Hamajima K. Alteration of the Bcl-2/Bax status of head and neck cancer cell lines by chemotherapeutic agents. Anticancer Res. 1999;19:3927-3932
30. Lavieille JP, Gazzeri S, Riva C, Reyt E, Brambilla C, Brambilla E. p53 mutations and p53, Waf-1, Bax and Bcl-2 expression in field cancerization of the head and neck. Anticancer Res. 1998;18:4741-4749
31. Trask DK, Wolf GT, Bradford CR, Fisher SG, Devaney K, Johnson M, Singleton T, Wicha M. Expression of Bcl-2 family proteins in advanced laryngeal squamous cell carcinoma: correlation with response to chemotherapy and organ preservation. Laryngoscope. 2002;112:638-644
32. Psyrri A, Cohen E. Oropharyngeal cancer: clinical implications of the HPV connection. Ann Oncol. 2011;22:997-999
33. Marklund L, Hammarstedt L. Impact of HPV in oropharyngeal cancer. J Oncol. 2011;2011:509036
34. Lundberg AS, Weinberg RA. Control of the cell cycle and apoptosis. Eur J Cancer. 1999;35:1886-1894
35. Pande P, Mathur M, Shukla NK, Ralhan R. pRb and p16 protein alterations in human oral tumorigenesis. Oral Oncol. 1998;34:396-403
36. Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15-18
37. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669-676
38. Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264:569-571
39. Compagni A, Wilgenbus P, Impagnatiello MA, Cotten M, Christofori G. Fibroblast growth factors are required for efficient tumor angiogenesis. Cancer Res. 2000;60:7163-7169
40. Giavazzi R, Sennino B, Coltrini D, Garofalo A, Dossi R, Ronca R, Tosatti MP, Presta M. Distinct role of fibroblast growth factor-2 and vascular endothelial growth factor on tumor growth and angiogenesis. Am J Pathol. 2003;162:1913-1926
41. Varner JA, Brooks PC, Cheresh DA. REVIEW: the integrin alpha V beta 3: angiogenesis and apoptosis. Cell Adhes Commun. 1995;3:367-374
42. Nisato RE, Hosseini G, Sirrenberg C, Butler GS, Crabbe T, Docherty AJ, Wiesner M, Murphy G, Overall CM, Goodman SL, Pepper MS. Dissecting the role of matrix metalloproteinases (MMP) and integrin alpha(v)beta3 in angiogenesis in vitro: absence of hemopexin C domain bioactivity, but membrane-Type 1-MMP and alpha(v)beta3 are critical. Cancer Res. 2005;65:9377-9387
43. Pepper MS. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol. 2001;21:1104-1117
44. O-Charoenrat P, Rhys-Evans P, Modjtahedi H, Eccles SA. Vascular endothelial growth factor family members are differentially regulated by c-erbB signaling in head and neck squamous carcinoma cells. Clin Exp Metastasis. 2000;18:155-161
45. Whitelock JM, Murdoch AD, Iozzo RV, Underwood PA. The degradation of human endothelial cell-derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin, and heparanases. J Biol Chem. 1996;271:10079-10086
46. El-Gazzar R, Macluskey M, Ogden GR. Evidence for a field change effect based on angiogenesis in the oral mucosa? A brief report. Oral Oncol. 2005;41:25-30
47. El-Gazzar R, Macluskey M, Williams H, Ogden GR. Vascularity and expression of vascular endothelial growth factor in oral squamous cell carcinoma, resection margins, and nodal metastases. Br J Oral Maxillofac Surg. 2006;44:193-197
48. Macluskey M, Chandrachud LM, Pazouki S, Green M, Chisholm DM, Ogden GR, Schor SL, Schor AM. Apoptosis, proliferation, and angiogenesis in oral tissues. Possible relevance to tumour progression. J Pathol. 2000;191:368-375
49. Sedivy R, Beck-Mannagetta J, Haverkampf C, Battistutti W, Honigschnabl S. Expression of vascular endothelial growth factor-C correlates with the lymphatic microvessel density and the nodal status in oral squamous cell cancer. J Oral Pathol Med. 2003;32:455-460
50. Williams MD. Integration of biomarkers including molecular targeted therapies in head and neck cancer. Head Neck Pathol. 2010;4:62-69
51. Sleeman JP. The metastatic niche and stromal progression. Cancer Metastasis Rev. 2012;31:429-440
52. Geiger TR, Peeper DS. Metastasis mechanisms. Biochim Biophys Acta. 2009;1796:293-308
53. McCawley LJ, Matrisian LM. Matrix metalloproteinases: multifunctional contributors to tumor progression. Mol Med Today. 2000;6:149-156
54. McCawley LJ, Matrisian LM. Matrix metalloproteinases: they're not just for matrix anymore!. Curr Opin Cell Biol. 2001;13:534-540
55. Yu L, Lu S, Tian J, Ma J, Li J, Wang H, Xu W. TWIST expression in hypopharyngeal cancer and the mechanism of TWIST-induced promotion of metastasis. Oncol Rep. 2012;27:416-422
56. Zuo JH, Zhu W, Li MY, Li XH, Yi H, Zeng GQ, Wan XX, He QY, Li JH, Qu JQ, Chen Y, Xiao ZQ. Activation of EGFR promotes squamous carcinoma SCC10A cell migration and invasion via inducing EMT-like phenotype change and MMP-9-mediated degradation of E-cadherin. J Cell Biochem. 2011;112:2508-2517
57. Huang CH, Yang WH, Chang SY, Tai SK, Tzeng CH, Kao JY, Wu KJ, Yang MH. Regulation of membrane-type 4 matrix metalloproteinase by SLUG contributes to hypoxia-mediated metastasis. Neoplasia. 2009;11:1371-1382
58. Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375-379
59. Albini A, Sporn MB. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer. 2007;7:139-147
60. Shaykhiev R, Bals R. Interactions between epithelial cells and leukocytes in immunity and tissue homeostasis. J Leukoc Biol. 2007;82:1-15
61. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539-545
62. Kopfstein L, Christofori G. Metastasis: cell-autonomous mechanisms versus contributions by the tumor microenvironment. Cell Mol Life Sci. 2006;63:449-468
63. Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839-849
64. Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009;9:285-293
65. Jin F, Prestage GP, Mao L, Kippax SC, Pell CM, Donovan B, Cunningham PH, Templeton DJ, Kaldor JM, Grulich AE. Incidence and risk factors for urethral and anal gonorrhoea and chlamydia in a cohort of HIV-negative homosexual men: the Health in Men Study. Sex Transm Infect. 2007;83:113-119
66. Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet. 2007;370:59-67
67. King GN, Healy CM, Glover MT, Kwan JT, Williams DM, Leigh IM, Worthington HV, Thornhill MH. Increased prevalence of dysplastic and malignant lip lesions in renal-transplant recipients. N Engl J Med. 1995;332:1052-1057
68. Van Leeuwen MT, Webster AC, McCredie MR, Stewart JH, McDonald SP, Amin J, Kaldor JM, Chapman JR, Vajdic CM, Grulich AE. Effect of reduced immunosuppression after kidney transplant failure on risk of cancer: population based retrospective cohort study. BMJ. 2010;340:c570
69. Vu HL, Sikora AG, Fu S, Kao J. HPV-induced oropharyngeal cancer, immune response and response to therapy. Cancer Lett. 2010;288:149-155
70. Hoffmann TK, Dworacki G, Tsukihiro T, Meidenbauer N, Gooding W, Johnson JT, Whiteside TL. Spontaneous apoptosis of circulating T lymphocytes in patients with head and neck cancer and its clinical importance. Clin Cancer Res. 2002;8:2553-2562
71. Hoffmann TK, Donnenberg AD, Finkelstein SD, Donnenberg VS, Friebe-Hoffmann U, Myers EN, Appella E, DeLeo AB, Whiteside TL. Frequencies of tetramer+ T cells specific for the wild-type sequence p53(264-272) peptide in the circulation of patients with head and neck cancer. Cancer Res. 2002;62:3521-3529
72. Reichert TE, Strauss L, Wagner EM, Gooding W, Whiteside TL. Signaling abnormalities, apoptosis, and reduced proliferation of circulating and tumor-infiltrating lymphocytes in patients with oral carcinoma. Clin Cancer Res. 2002;8:3137-3145
73. Pretscher D, Distel LV, Grabenbauer GG, Wittlinger M, Buettner M, Niedobitek G. Distribution of immune cells in head and neck cancer: CD8+ T-cells and CD20+ B-cells in metastatic lymph nodes are associated with favourable outcome in patients with oro- and hypopharyngeal carcinoma. BMC Cancer. 2009;9:292
74. Albers A, Abe K, Hunt J, Wang J, Lopez-Albaitero A, Schaefer C, Gooding W, Whiteside TL, Ferrone S, DeLeo A, Ferris RL. Antitumor activity of human papillomavirus type 16 E7-specific T cells against virally infected squamous cell carcinoma of the head and neck. Cancer Res. 2005;65:11146-11155
75. Hoffmann TK, Arsov C, Schirlau K, Bas M, Friebe-Hoffmann U, Klussmann JP, Scheckenbach K, Balz V, Bier H, Whiteside TL. T cells specific for HPV16 E7 epitopes in patients with squamous cell carcinoma of the oropharynx. Int J Cancer. 2006;118:1984-1991
76. Nitsch SM, Pries R, Wollenberg B. Head and neck cancer triggers increased IL-6 production of CD34+ stem cells from human cord blood. In Vivo. 2007;21:493-498
77. Grote K, Salguero G, Ballmaier M, Dangers M, Drexler H, Schieffer B. The angiogenic factor CCN1 promotes adhesion and migration of circulating CD34+ progenitor cells: potential role in angiogenesis and endothelial regeneration. Blood. 2007;110:877-885
78. Marcus B, Arenberg D, Lee J, Kleer C, Chepeha DB, Schmalbach CE, Islam M, Paul S, Pan Q, Hanash S, Kuick R, Merajver SD, Teknos TN. Prognostic factors in oral cavity and oropharyngeal squamous cell carcinoma. Cancer. 2004;101:2779-2787
79. Kesselring R, Thiel A, Pries R, Trenkle T, Wollenberg B. Human Th17 cells can be induced through head and neck cancer and have a functional impact on HNSCC development. Br J Cancer. 2010;103:1245-1254
80. Barth PJ, Schenck zu Schweinsberg T, Ramaswamy A, Moll R. CD34+ fibrocytes, alpha-smooth muscle antigen-positive myofibroblasts, and CD117 expression in the stroma of invasive squamous cell carcinomas of the oral cavity, pharynx, and larynx. Virchows Arch. 2004;444:231-234
81. Sawatsubashi M, Yamada T, Fukushima N, Mizokami H, Tokunaga O, Shin T. Association of vascular endothelial growth factor and mast cells with angiogenesis in laryngeal squamous cell carcinoma. Virchows Arch. 2000;436:243-248
82. Iamaroon A, Pongsiriwet S, Jittidecharaks S, Pattanaporn K, Prapayasatok S, Wanachantararak S. Increase of mast cells and tumor angiogenesis in oral squamous cell carcinoma. J Oral Pathol Med. 2003;32:195-199
83. Li C, Shintani S, Terakado N, Nakashiro K, Hamakawa H. Infiltration of tumor-associated macrophages in human oral squamous cell carcinoma. Oncol Rep. 2002;9:1219-1223
84. El-Rouby DH. Association of macrophages with angiogenesis in oral verrucous and squamous cell carcinomas. J Oral Pathol Med. 2010;39:559-564
85. Chang KP, Kao HK, Yen TC, Chang YL, Liang Y, Liu SC, Lee LY, Kang CJ, Chen IH, Liao CT, Yu JS. Overexpression of macrophage inflammatory protein-3alpha in oral cavity squamous cell carcinoma is associated with nodal metastasis. Oral Oncol. 2011;47:108-113
86. Wang W, Goswami S, Sahai E, Wyckoff JB, Segall JE, Condeelis JS. Tumor cells caught in the act of invading: their strategy for enhanced cell motility. Trends Cell Biol. 2005;15:138-145
87. Horst HA, Horny HP. Tumor-infiltrating lymphoreticular cells. Histologic and immunohistologic investigations performed on metastasizing squamous cell carcinomas of the head and neck. Cancer. 1991;68:2397-2402
88. Liu SY, Chang LC, Pan LF, Hung YJ, Lee CH, Shieh YS. Clinicopathologic significance of tumor cell-lined vessel and microenvironment in oral squamous cell carcinoma. Oral Oncol. 2008;44:277-285
89. Shimoda M, Mellody KT, Orimo A. Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Semin Cell Dev Biol. 2010;21:19-25
90. Rasanen K, Vaheri A. Activation of fibroblasts in cancer stroma. Exp Cell Res. 2010;316:2713-2722
91. Xouri G, Christian S. Origin and function of tumor stroma fibroblasts. Semin Cell Dev Biol. 2010;21:40-46
92. Bello IO, Vered M, Dayan D, Dobriyan A, Yahalom R, Alanen K, Nieminen P, Kantola S, Laara E, Salo T. Cancer-associated fibroblasts, a parameter of the tumor microenvironment, overcomes carcinoma-associated parameters in the prognosis of patients with mobile tongue cancer. Oral Oncol. 2011;47:33-38
93. Vered M, Dayan D, Yahalom R, Dobriyan A, Barshack I, Bello IO, Kantola S, Salo T. Cancer-associated fibroblasts and epithelial-mesenchymal transition in metastatic oral tongue squamous cell carcinoma. Int J Cancer. 2010;127:1356-1362
94. Vered M, Dobriyan A, Dayan D, Yahalom R, Talmi YP, Bedrin L, Barshack I, Taicher S. Tumor-host histopathologic variables, stromal myofibroblasts and risk score, are significantly associated with recurrent disease in tongue cancer. Cancer Sci. 2010;101:274-280
95. Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392-1400
96. Thode C, Jorgensen TG, Dabelsteen E, Mackenzie I, Dabelsteen S. Significance of myofibroblasts in oral squamous cell carcinoma. J Oral Pathol Med. 2011;40:201-207
97. Liu Y, Hu T, Shen J, Li SF, Lin JW, Zheng XH, Gao QH, Zhou HM. Separation, cultivation and biological characteristics of oral carcinoma-associated fibroblasts. Oral Dis. 2006;12:375-380
98. Campos MS, Neiva KG, Meyers KA, Krishnamurthy S, Nor JE. Endothelial derived factors inhibit anoikis of head and neck cancer stem cells. Oral Oncol. 2012;48:26-32
99. Zeng Q, Li S, Chepeha DB, Giordano TJ, Li J, Zhang H, Polverini PJ, Nor J, Kitajewski J, Wang CY. Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell. 2005;8:13-23
100. Bran B, Bran G, Hormann K, Riedel F. The platelet-derived growth factor receptor as a target for vascular endothelial growth factor-mediated anti-angiogenetic therapy in head and neck cancer. Int J Oncol. 2009;34:255-261
101. Ninck S, Reisser C, Dyckhoff G, Helmke B, Bauer H, Herold-Mende C. Expression profiles of angiogenic growth factors in squamous cell carcinomas of the head and neck. Int J Cancer. 2003;106:34-44
102. Schultz JD, Rotunno S, Riedel F, Anders C, Erben P, Hofheinz RD, Faber A, Thorn C, Sommer JU, Hormann K, Sauter A. Synergistic effects of imatinib and carboplatin on VEGF, PDGF and PDGF-Ralpha/ss expression in squamous cell carcinoma of the head and neck in vitro. Int J Oncol. 2011;38:1001-1012
103. Liss C, Fekete MJ, Hasina R, Lam CD, Lingen MW. Paracrine angiogenic loop between head-and-neck squamous-cell carcinomas and macrophages. Int J Cancer. 2001;93:781-785
104. Liss C, Fekete MJ, Hasina R, Lingen MW. Retinoic acid modulates the ability of macrophages to participate in the induction of the angiogenic phenotype in head and neck squamous cell carcinoma. Int J Cancer. 2002;100:283-289
105. Ali MA. Lymphatic microvessel density and the expression of lymphangiogenic factors in oral squamous cell carcinoma. Med Princ Pract. 2008;17:486-492
106. Li C, Shintani S, Terakado N, Klosek SK, Ishikawa T, Nakashiro K, Hamakawa H. Microvessel density and expression of vascular endothelial growth factor, basic fibroblast growth factor, and platelet-derived endothelial growth factor in oral squamous cell carcinomas. Int J Oral Maxillofac Surg. 2005;34:559-565
107. Kaneko T, Zhang Z, Mantellini MG, Karl E, Zeitlin B, Verhaegen M, Soengas MS, Lingen M, Strieter RM, Nunez G, Nor JE. Bcl-2 orchestrates a cross-talk between endothelial and tumor cells that promotes tumor growth. Cancer Res. 2007;67:9685-9693
108. Karl E, Zhang Z, Dong Z, Neiva KG, Soengas MS, Koch AE, Polverini PJ, Nunez G, Nor JE. Unidirectional crosstalk between Bcl-xL and Bcl-2 enhances the angiogenic phenotype of endothelial cells. Cell Death Differ. 2007;14:1657-1666
109. Neiva KG, Zhang Z, Miyazawa M, Warner KA, Karl E, Nor JE. Cross talk initiated by endothelial cells enhances migration and inhibits anoikis of squamous cell carcinoma cells through STAT3/AKT/ERK signaling. Neoplasia. 2009;11:583-593
110. Brennan PA, Mackenzie N, Quintero M. Hypoxia-inducible factor 1alpha in oral cancer. J Oral Pathol Med. 2005;34:385-389
111. Sun Q, Zhou H, Binmadi NO, Basile JR. Hypoxia-inducible factor-1-mediated regulation of semaphorin 4D affects tumor growth and vascularity. J Biol Chem. 2009;284:32066-32074
112. Beasley NJ, Leek R, Alam M, Turley H, Cox GJ, Gatter K, Millard P, Fuggle S, Harris AL. Hypoxia-inducible factors HIF-1alpha and HIF-2alpha in head and neck cancer: relationship to tumor biology and treatment outcome in surgically resected patients. Cancer Res. 2002;62:2493-2497
113. Kyzas PA, Stefanou D, Batistatou A, Agnantis NJ. Hypoxia-induced tumor angiogenic pathway in head and neck cancer: an in vivo study. Cancer Lett. 2005;225:297-304
114. Audet N, Beasley NJ, MacMillan C, Jackson DG, Gullane PJ, Kamel-Reid S. Lymphatic vessel density, nodal metastases, and prognosis in patients with head and neck cancer. Arch Otolaryngol Head Neck Surg. 2005;131:1065-1070
115. O'Donnell RK, Feldman M, Mick R, Muschel RJ. Immunohistochemical method identifies lymphovascular invasion in a majority of oral squamous cell carcinomas and discriminates between blood and lymphatic vessel invasion. J Histochem Cytochem. 2008;56:803-810
116. Zhao D, Pan J, Li XQ, Wang XY, Tang C, Xuan M. Intratumoral lymphangiogenesis in oral squamous cell carcinoma and its clinicopathological significance. J Oral Pathol Med. 2008;37:616-625
117. Beasley NJ, Prevo R, Banerji S, Leek RD, Moore J, van Trappen P, Cox G, Harris AL, Jackson DG. Intratumoral lymphangiogenesis and lymph node metastasis in head and neck cancer. Cancer Res. 2002;62:1315-1320
118. Margaritescu C, Simionescu C, Pirici D, Mogoanta L, Ciurea R, Stepan A. Immunohistochemical characterization of tumoral vessels in oral squamous cell carcinoma. Rom J Morphol Embryol. 2008;49:447-458
119. Lorusso G, Ruegg C. The tumor microenvironment and its contribution to tumor evolution toward metastasis. Histochem Cell Biol. 2008;130:1091-1103
120. Tong CC, Kao J, Sikora AG. Recognizing and reversing the immunosuppressive tumor microenvironment of head and neck cancer. Immunol Res. 2012;54:266-274
121. Ogino T, Shigyo H, Ishii H, Katayama A, Miyokawa N, Harabuchi Y, Ferrone S. HLA class I antigen down-regulation in primary laryngeal squamous cell carcinoma lesions as a poor prognostic marker. Cancer Res. 2006;66:9281-9289
122. Reichert TE, Scheuer C, Day R, Wagner W, Whiteside TL. The number of intratumoral dendritic cells and zeta-chain expression in T cells as prognostic and survival biomarkers in patients with oral carcinoma. Cancer. 2001;91:2136-2147
123. Duray A, Demoulin S, Hubert P, Delvenne P, Saussez S. Immune suppression in head and neck cancers: a review. Clin Dev Immunol. 2010;2010:701657
124. Allen CT, Judd NP, Bui JD, Uppaluri R. The clinical implications of antitumor immunity in head and neck cancer. Laryngoscope. 2012;122:144-157
125. DeClerck YA. Interactions between tumour cells and stromal cells and proteolytic modification of the extracellular matrix by metalloproteinases in cancer. Eur J Cancer. 2000;36:1258-1268
126. Cao XL, Xu RJ, Zheng YY, Liu J, Teng YS, Li Y, Zhu J. Expression of type IV collagen, metalloproteinase-2, metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 in laryngeal squamous cell carcinomas. Asian Pac J Cancer Prev. 2011;12:3245-3249
127. Parikka M, Nissinen L, Kainulainen T, Bruckner-Tuderman L, Salo T, Heino J, Tasanen K. Collagen XVII promotes integrin-mediated squamous cell carcinoma transmigration--a novel role for alphaIIb integrin and tirofiban. Exp Cell Res. 2006;312:1431-1438
128. Al-Hazmi N, Thomas GJ, Speight PM, Whawell SA. The 120 kDa cell-binding fragment of fibronectin up-regulates migration of alphavbeta6-expressing cells by increasing matrix metalloproteinase-2 and -9 secretion. Eur J Oral Sci. 2007;115:454-458
129. Pattaramalai S, Skubitz AP. Promotion of human oral squamous cell carcinoma adhesion in vitro by the carboxy-terminal globular domain of laminin. Arch Oral Biol. 1994;39:925-933
130. Nielsen JD, Moeslund M, Wandall HH, Dabelsteen S. Influences of tumor stroma on the malignant phenotype. J Oral Pathol Med. 2008;37:412-416
131. Koontongkaew S, Amornphimoltham P, Yapong B. Tumor-stroma interactions influence cytokine expression and matrix metalloproteinase activities in paired primary and metastatic head and neck cancer cells. Cell Biol Int. 2009;33:165-173
132. Koontongkaew S, Amornphimoltham P, Monthanpisut P, Saensuk T, Leelakriangsak M. Fibroblasts and extracellular matrix differently modulate MMP activation by primary and metastatic head and neck cancer cells. Med Oncol. 2012;29:690-703
133. Lewis MP, Lygoe KA, Nystrom ML, Anderson WP, Speight PM, Marshall JF, Thomas GJ. Tumour-derived TGF-beta1 modulates myofibroblast differentiation and promotes HGF/SF-dependent invasion of squamous carcinoma cells. Br J Cancer. 2004;90:822-832
134. Kellermann MG, Sobral LM, da Silva SD, Zecchin KG, Graner E, Lopes MA, Kowalski LP, Coletta RD. Mutual paracrine effects of oral squamous cell carcinoma cells and normal oral fibroblasts: induction of fibroblast to myofibroblast transdifferentiation and modulation of tumor cell proliferation. Oral Oncol. 2008;44:509-517
135. Kellermann MG, Sobral LM, da Silva SD, Zecchin KG, Graner E, Lopes MA, Nishimoto I, Kowalski LP, Coletta RD. Myofibroblasts in the stroma of oral squamous cell carcinoma are associated with poor prognosis. Histopathology. 2007;51:849-853
136. Sweeny L, Liu Z, Lancaster W, Hart J, Hartman YE, Rosenthal EL. Inhibition of fibroblasts reduced head and neck cancer growth by targeting fibroblast growth factor receptor. Laryngoscope. 2012;122:1539-1544
137. Liu Z, Hartman YE, Warram JM, Knowles JA, Sweeny L, Zhou T, Rosenthal EL. Fibroblast growth factor receptor mediates fibroblast-dependent growth in EMMPRIN-depleted head and neck cancer tumor cells. Mol Cancer Res. 2011;9:1008-1017
138. Dean NR, Knowles JA, Helman EE, Aldridge JC, Carroll WR, Magnuson JS, Clemons L, Ziober B, Rosenthal EL. Anti-EMMPRIN antibody treatment of head and neck squamous cell carcinoma in an ex-vivo model. Anticancer Drugs. 2010;21:861-867
139. Rosenthal EL, Shreenivas S, Peters GE, Grizzle WE, Desmond R, Gladson CL. Expression of extracellular matrix metalloprotease inducer in laryngeal squamous cell carcinoma. Laryngoscope. 2003;113:1406-1410
140. Rosenthal EL, Zhang W, Talbert M, Raisch KP, Peters GE. Extracellular matrix metalloprotease inducer-expressing head and neck squamous cell carcinoma cells promote fibroblast-mediated type I collagen degradation in vitro. Mol Cancer Res. 2005;3:195-202
141. Sobral LM, Bufalino A, Lopes MA, Graner E, Salo T, Coletta RD. Myofibroblasts in the stroma of oral cancer promote tumorigenesis via secretion of activin A. Oral Oncol. 2011;47:840-846
142. Hujoel PP, Drangsholt M, Spiekerman C, Weiss NS. An exploration of the periodontitis-cancer association. Ann Epidemiol. 2003;13:312-316
143. Le Bitoux MA, Stamenkovic I. Tumor-host interactions: the role of inflammation. Histochem Cell Biol. 2008;130:1079-1090
144. Tezal M, Sullivan MA, Reid ME, Marshall JR, Hyland A, Loree T, Lillis C, Hauck L, Wactawski-Wende J, Scannapieco FA. Chronic periodontitis and the risk of tongue cancer. Arch Otolaryngol Head Neck Surg. 2007;133:450-454
145. Wang F, Arun P, Friedman J, Chen Z, Van Waes C. Current and potential inflammation targeted therapies in head and neck cancer. Curr Opin Pharmacol. 2009;9:389-395
146. Yao SG, Fine JB. Periodontitis and cancer..a link? A review of the recent literature. Compend Contin Educ Dent. 2010;31:436-442
147. Ondrey FG. Arachidonic acid metabolism: a primer for head and neck surgeons. Head Neck. 1998;20:334-349
148. Camacho M, Leon X, Fernandez-Figueras MT, Quer M, Vila L. Prostaglandin E(2) pathway in head and neck squamous cell carcinoma. Head Neck. 2008;30:1175-1181
149. Mauro A, Lipari L, Leone A, Tortorici S, Burruano F, Provenzano S, Gerbino A, Buscemi M. Expression of cyclooxygenase-1 and cyclooxygenase-2 in normal and pathological human oral mucosa. Folia Histochem Cytobiol. 2010;48:555-563
150. Sun DS, Zhao MQ, Xia M, Li L, Jiang YH. The correlation between tumor-infiltrating Foxp3+ regulatory T cells and cyclooxygenase-2 expression and their association with recurrence in resected head and neck cancers. Med Oncol. 2012;29:707-713
151. Koontongkaew S, Leelahavanichkul K. Arachidonic acid metabolism and its implication on head and neck cancer. In: (ed.) Agulnik M. Head and neck cancer. Rijecka: InTech. 2012:167-184
152. Koontongkaew S, Monthanapisut P, Saensuk T. Inhibition of arachidonic acid metabolism decreases tumor cell invasion and matrix metalloproteinase expression. Prostaglandins Other Lipid Mediat. 2010;93:100-108
153. Wang YH, Wu MW, Yang AK, Zhang WD, Sun J, Liu TR, Chen YF. COX-2 Gene increases tongue cancer cell proliferation and invasion through VEGF-C pathway. Med Oncol. 2011;28(Suppl 1):S360-366
154. Shureiqi I, Lippman SM. Lipoxygenase modulation to reverse carcinogenesis. Cancer Res. 2001;61:6307-6312
155. Wilson J, Balkwill F. The role of cytokines in the epithelial cancer microenvironment. Semin Cancer Biol. 2002;12:113-120
156. Frederick MJ, Clayman GL. Chemokines in cancer. Expert Rev Mol Med. 2001;3:1-18
157. Chen Z, Malhotra PS, Thomas GR, Ondrey FG, Duffey DC, Smith CW, Enamorado I, Yeh NT, Kroog GS, Rudy S, McCullagh L, Mousa S, Quezado M, Herscher LL, Van Waes C. Expression of proinflammatory and proangiogenic cytokines in patients with head and neck cancer. Clin Cancer Res. 1999;5:1369-1379
158. Lathers DM, Young MR. Increased aberrance of cytokine expression in plasma of patients with more advanced squamous cell carcinoma of the head and neck. Cytokine. 2004;25:220-228
159. Sparano A, Lathers DM, Achille N, Petruzzelli GJ, Young MR. Modulation of Th1 and Th2 cytokine profiles and their association with advanced head and neck squamous cell carcinoma. Otolaryngol Head Neck Surg. 2004;131:573-576
160. Woods KV, El-Naggar A, Clayman GL, Grimm EA. Variable expression of cytokines in human head and neck squamous cell carcinoma cell lines and consistent expression in surgical specimens. Cancer Res. 1998;58:3132-3141
161. Strieter RM, Belperio JA, Burdick MD, Sharma S, Dubinett SM, Keane MP. CXC chemokines: angiogenesis, immunoangiostasis, and metastases in lung cancer. Ann N Y Acad Sci. 2004;1028:351-360
162. Zlotnik A. Chemokines in neoplastic progression. Semin Cancer Biol. 2004;14:181-185
163. Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50-56
164. Kryczek I, Wei S, Keller E, Liu R, Zou W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am J Physiol Cell Physiol. 2007;292:C987-995
165. Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107:1761-1767
166. Delilbasi CB, Okura M, Iida S, Kogo M. Investigation of CXCR4 in squamous cell carcinoma of the tongue. Oral Oncol. 2004;40:154-157
167. Samara GJ, Lawrence DM, Chiarelli CJ, Valentino MD, Lyubsky S, Zucker S, Vaday GG. CXCR4-mediated adhesion and MMP-9 secretion in head and neck squamous cell carcinoma. Cancer Lett. 2004;214:231-241
168. Lee JI, Jin BH, Kim MA, Yoon HJ, Hong SP, Hong SD. Prognostic significance of CXCR-4 expression in oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;107:678-684
169. Ishikawa T, Nakashiro K, Klosek SK, Goda H, Hara S, Uchida D, Hamakawa H. Hypoxia enhances CXCR4 expression by activating HIF-1 in oral squamous cell carcinoma. Oncol Rep. 2009;21:707-712
170. Tan CT, Chu CY, Lu YC, Chang CC, Lin BR, Wu HH, Liu HL, Cha ST, Prakash E, Ko JY, Kuo ML. CXCL12/CXCR4 promotes laryngeal and hypopharyngeal squamous cell carcinoma metastasis through MMP-13-dependent invasion via the ERK1/2/AP-1 pathway. Carcinogenesis. 2008;29:1519-1527
171. Tang CH, Chuang JY, Fong YC, Maa MC, Way TD, Hung CH. Bone-derived SDF-1 stimulates IL-6 release via CXCR4, ERK and NF-kappaB pathways and promotes osteoclastogenesis in human oral cancer cells. Carcinogenesis. 2008;29:1483-1492
172. Takabayashi T, Takahashi N, Okamoto M, Yagi H, Sato M, Fujieda S. Lipopolysaccharides increase the amount of CXCR4, and modulate the morphology and invasive activity of oral cancer cells in a CXCL12-dependent manner. Oral Oncol. 2009;45:968-973
173. Onoue T, Uchida D, Begum NM, Tomizuka Y, Yoshida H, Sato M. Epithelial-mesenchymal transition induced by the stromal cell-derived factor-1/CXCR4 system in oral squamous cell carcinoma cells. Int J Oncol. 2006;29:1133-1138
174. Miyazaki H, Patel V, Wang H, Edmunds RK, Gutkind JS, Yeudall WA. Down-regulation of CXCL5 inhibits squamous carcinogenesis. Cancer Res. 2006;66:4279-4284
175. Chuang JY, Yang WH, Chen HT, Huang CY, Tan TW, Lin YT, Hsu CJ, Fong YC, Tang CH. CCL5/CCR5 axis promotes the motility of human oral cancer cells. J Cell Physiol. 2009;220:418-426
176. Jung DW, Che ZM, Kim J, Kim K, Kim KY, Williams D. Tumor-stromal crosstalk in invasion of oral squamous cell carcinoma: a pivotal role of CCL7. Int J Cancer. 2010;127:332-344
177. Bourboulia D, Stetler-Stevenson WG. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Semin Cancer Biol. 2010;20:161-168
178. Noel A, Jost M, Maquoi E. Matrix metalloproteinases at cancer tumor-host interface. Semin Cell Dev Biol. 2008;19:52-60
179. Noel A, Gutierrez-Fernandez A, Sounni NE, Behrendt N, Maquoi E, Lund IK, Cal S, Hoyer-Hansen G, Lopez-Otin C. New and Paradoxical Roles of Matrix Metalloproteinases in the Tumor Microenvironment. Front Pharmacol. 2012;3:140
180. Thomas GT, Lewis MP, Speight PM. Matrix metalloproteinases and oral cancer. Oral Oncol. 1999;35:227-233
181. Bjorklund M, Koivunen E. Gelatinase-mediated migration and invasion of cancer cells. Biochim Biophys Acta. 2005;1755:37-69
182. de Vicente JC, Fresno MF, Villalain L, Vega JA, Hernandez Vallejo G. Expression and clinical significance of matrix metalloproteinase-2 and matrix metalloproteinase-9 in oral squamous cell carcinoma. Oral Oncol. 2005;41:283-293
183. Kato K, Hara A, Kuno T, Kitaori N, Huilan Z, Mori H, Toida M, Shibata T. Matrix metalloproteinases 2 and 9 in oral squamous cell carcinomas: manifestation and localization of their activity. J Cancer Res Clin Oncol. 2005;131:340-346
184. Patel BP, Shah SV, Shukla SN, Shah PM, Patel PS. Clinical significance of MMP-2 and MMP-9 in patients with oral cancer. Head Neck. 2007;29:564-572
185. Fullar A, Kovalszky I, Bitsche M, Romani A, Schartinger VH, Sprinzl GM, Riechelmann H, Dudas J. Tumor cell and carcinoma-associated fibroblast interaction regulates matrix metalloproteinases and their inhibitors in oral squamous cell carcinoma. Exp Cell Res. 2012;318:1517-1527
186. Hinsley EE, Kumar S, Hunter KD, Whawell SA, Lambert DW. Endothelin-1 stimulates oral fibroblasts to promote oral cancer invasion. Life Sci. 2012;91:557-561
187. Hinsley EE, Hunt S, Hunter KD, Whawell SA, Lambert DW. Endothelin-1 stimulates motility of head and neck squamous carcinoma cells by promoting stromal-epithelial interactions. Int J Cancer. 2012;130:40-47
188. Yu T, Wu Y, Helman JI, Wen Y, Wang C, Li L. CXCR4 promotes oral squamous cell carcinoma migration and invasion through inducing expression of MMP-9 and MMP-13 via the ERK signaling pathway. Mol Cancer Res. 2011;9:161-172
Author contact
![]() Corresponding author: Sittichai Koontongkaew Email: koontongkaewcom, koontongkaewcom.
Corresponding author: Sittichai Koontongkaew Email: koontongkaewcom, koontongkaewcom.