Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2012; 3:310-321. doi:10.7150/jca.4584 This volume Cite
Research Paper
Effect of Tumour Necrosis Factor-Alpha on Estrogen Metabolic Pathways in Breast Cancer Cells
Marwa Kamel1, Samia Shouman1, Mahmoud El-Merzebany1, Gokhan Kilic2, Timothy Veenstra3, Muhammad Saeed4, Mohamed Wagih5, Concepcion Diaz-Arrastia6, Deepa Patel6, Salama Salama6 ![]()
1. Unit of Pharmacology, Department of Cancer Biology, National Cancer Institute, Cairo University, Egypt
2. Department of Obstetrics and Gynecology, University of Texas Medical Branch, Galveston, Texas, United States of America
3. Laboratory of Proteomics and Analytical Technologies, Advanced Technology Program, SAIC-Frederick, Inc., NCI-Frederick, 1050 Boyles Street/P.O. Box B, Frederick, MD 21702, United States of America
4. Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, 986805 Nebraska Medical Center, Omaha, NE 68198, United States of America
5. Department of Pathology, Faculty of Medicine, Beni-Suef University, Egypt
6. Department of Obstetrics and Gynecology, Baylor College of Medicine, One Baylor Plaza MS: BCM 610, Houston, TX 77030, United States of America
Received 2012-5-12; Accepted 2012-6-19; Published 2012-7-6
Abstract
Tumor necrosis factor-alpha (TNF-α) is a proinflammatory cytokine that has been linked to breast cancer development. Estrogen metabolic pathway is also involved in breast carcinogenesis and DNA adducts formation. In this study we investigated the effect of TNF-α on the estrogen metabolic pathway in MCF-7, a breast cancer cell line. Capillary liquid chromatography/mass spectrometry (LC/MS) and High performance liquid chromatography (HPLC) were used for analysis of estrogen metabolites and estrogen-DNA adducts levels respectively. Reporter gene assay, Real time reverse transcription polymerase chain reaction (real time RT-PCR) and Western blot were used to assess the expression of estrogen metabolizing genes and enzymes. TNF-α significantly increased the total EM and decreased the estrone (E1) / 17-β estradiol (E2) ratio. Moreover, it altered the expression of genes and enzymes involved in E2 activation and deactivation pathways e.g. Cytochrome P-450 1A1 (CYP1A1), Cytochrome P-450 1B1 (CYP1B1), Catechol-O-methyl transferase (COMT) and Nicotinamide adenine dinucleotide phosphate-quinone oxidoreductase 1 (NQO1). In addition, there were increased levels of some catechol estrogens e.g. 4-hydroxy-estrone (4-OHE1) and 2-hydroxyestradiol (2-OHE2) with decreased levels of methylated catechols e.g. 2-methoxy estradiol (2-MeOE2). DNA adducts especially 4-OHE1-[2]-1-N3 Adenine was significantly increased. TNF-α directs the estrogen metabolism into more hormonally active and carcinogenic products in MCF-7. This may implicate a new possible explanation for inflammation associated breast cancer.
Keywords: Breast cancer, Tumor necrosis factor-alpha, Estrogen metabolites, Estrogen metabolizing genes and enzymes, DNA adducts.
Introduction
Breast cancer is one of the most common malignancies accounting for nearly 1 in 3 cancers diagnosed among women in the United States, and it is the second leading cause of cancer death among women [1].
It is well established that prolonged exposure to estrogens especially estradiol, is an important risk factor for genesis and promotion of breast cancer [2, 3]. The most acknowledged mechanism of estrogen carcinogenicity is its binding to its specific estrogen receptor alpha (ER-alpha) for exerting a potent mitogenic effect on cell proliferation [4]. However, there is conceivable evidence that estrogen induced breast cancer can also occur by a non ER-alpha mediated mechanism involving the estrogen metabolic pathway [5].
Metabolism of estrogens is characterized by two major pathways: one is hydroxylation at the 16-position and the second is hydroxylation to form the 2- and 4-catechol estrogens. In the catechol pathway (Fig. S1), the metabolism involves further oxidation of 17- β estradiol (E2) to semiquinones and quinones which react with DNA to form depurinating adducts. The apurinic sites obtained by this reaction generate mutations that may lead to the initiation of cancer. Oxidation of catechol estrogens to their quinones normally occurs in homeostasis which minimizes their reaction with DNA. When the homeostasis is disrupted, excessive amounts of catechol estrogen quinones are formed and the resulting increase in depurinating DNA adducts can lead to initiation of cancer [6-9]. Moreover, 4-hydroxylated estrogens may exert proinflammatory roles by inducing reactive oxygen species (ROS) and DNA damage which is possibly decisive in chronic inflammation [10].
Part of the metabolic pathway of estradiol and the role of various enzymes involved: Estradiol is metabolized into 2-hydroxyestradiol (2-OHE2) and 4-hdroxyestradiol (4-OHE2) by CYP1A1 and CYP1B1 respectively. These catechols undergo further oxidation into semiquinones and quinones that react with DNA to form depurinating adducts leading to mutations associated with breast cancer. NQO1 reduces these quinones back to catechols which are detoxified into methoxy derivatives by the action of COMT. This protects the cells against DNA adducts formation and lowers the potential for mutagenic damage.

It has become increasingly clear that inflammation plays a major role in breast cancer pathogenesis. An inflammatory tumor microenvironment consists of infiltrating immune cells and activated fibroblasts, both of which can secrete cytokines, chemokines, and growth factors, as well as DNA-damaging agents [11]. Some studies show evidence that chronic inflammation is linked to breast cancer recurrence and that elevated biomarkers of inflammation are associated with reduced survival among breast cancer patients [12, 13]. In addition, experimental studies clearly indicate that inflammatory mediators promote tumor development in cancer prone animal strains [14]. Moreover, inhibition of TNF-α and NF-kappaB (NF-κB) transcription factor is proved to be protective with respect to chemical induced mammary gland carcinogenesis [15]. Further, the in vitro activation of the TNF-α/NF-κB axis has induced an invasive and malignant behaviour in breast cancer cells [16].
TNF-α is a major inflammatory cytokine shown to be highly expressed in breast carcinomas [17]. Indeed, investigations strongly suggest that the chronic expression of TNF-α in breast tumors actually supports tumor growth. The number of cells expressing TNF-α in inflammatory breast carcinoma was found to be correlated with increasing tumor grade and node involvement, and TNF-α expression was suggested to play a role in the metastatic behavior of breast carcinomas [18]. Furthermore, patients with more progressed tumor phenotypes were shown to have significantly higher TNF-α serum concentration [19]. The tumor-promoting functions of TNF-α may be mediated by its ability to induce proangiogenic functions, to promote the expression of matrix metalloproteinases (MMP) and endothelial adhesion molecules, and to cause DNA damage via reactive oxygen, the overall effect of which is promotion of tumor related processes [20].
In the current study we explored a new mechanism by which inflammation may influence breast carcinogenesis through the estrogen metabolic pathway. We thus investigated the effect of the TNF-α, a hallmark of inflammation, on the estrogen metabolic pathway in MCF-7 estrogen dependent breast cancer cells. Our choice of MCF-7 as our study model was built on several bases: First, studies have shown that regular use of nonsteroidal antiinflammatory drugs (NSAIDS), such as aspirin, significantly reduce the risk of ER-positive but not ER-negative breast cancers [21]. Second, it was recently suggested that inflammation may promote more aggressive ER-positive tumors and that this may be one of the mechanisms by which a portion of ER-positive breast tumors fail to respond to endocrine therapy [22]. In general, we sense that further research is required to fully elucidate the mechanisms of action of TNF-α on breast carcinogenesis because TNF-α activity may vary under different physiological conditions and in a cell-type-dependent manner which contributes to a sense of ambiguity regarding its tumor effects. Most of the reports examining the effects of TNF-α on MCF-7 breast cancer cells demonstrated its ability to induce apoptosis, inhibit proliferation and promote migration, invasion as well as resistance to chemotherapeutic drugs. These effects may however vary with other estrogen dependent (T47D) or independent (MDA-MB-231) cell lines [23].
What encouraged us more to explore this area of research is that not many studies have examined the role of inflammation on the estrogen metabolic pathways in general inspite of this pathway being an important cause of carcinogenesis. In some of these studies, TNF-α has been found to have an important role in regulating estrogen synthesis in peripheral tissues, including normal and malignant breast tissues [24-27]. Other studies have pointed that the effect of TNF-α on estrogen metabolism and homeostasis is mediated by its effect on the coordinated expression of the enzymes that are involved in estrogen biosynthesis and metabolism. For example, it has been suggested that TNF-α increases the local estrogen biosynthesis in human endometrial glandular epithelial cells and directs estrogen metabolic enzymes to produce more hormonally active and carcinogenic metabolites [28].
Collectively, the above mentioned information stimulated our interest to have a closer look on the estrogen metabolic pathway in MCF-7 breast cancer cells as a whole and examine the role played by TNF-α in this pathway aiming to enrich our knowledge about how inflammation may influence breast carcinogenesis. Understanding the mechanisms by which inflammatory mediators promote breast cancer may shed the light on these cytokines being explored as potential therapeutic targets and hopefully will lead to novel therapeutic regimens to treat this devastating disease.
Materials and Methods
Cells and Cell Culture
The human breast cancer cell line MCF-7 was purchased from (Rockville culture collection, Md, USA). The cells were maintained at 37ºC in a 95% humidified, 5% CO2 atmosphere in RPMI-1640 supplemented with 10% heat inactivated fetal bovine serum, 1% L-glutamine, 1% sodium pyruvate, and 1% penicillin and streptomycin (Gibco BRL, Life Technologies, Long Island, NY, USA). To study the effect of TNF-α on the profile of estrogen metabolism and DNA adduct analysis, the cells were grown in serum free, phenol red-free media for 24 hours. The cells were then treated with 10 nM of E2 with or without 5ng/ml TNF-α for 48 hours. The media were collected for estrogen metabolite measurements and DNA adducts analysis.
Determination of estrogen/estrogen metabolites (EM) by capillary LC/MS
The levels of estrogen and estrogen metabolites (EM) were determined by testing the conditioned media from cells treated with E2 alone or with different doses of TNF-α according to the method described by Xu et al [29]. Briefly, 5 µL of the stable isotope labelled estrogen and estrogen metabolite (SI-EM) working internal standard solution (40 pg SI-EM) was added to 400 μl cell medium aliquot followed by 500 μl 0.15 M sodium acetate buffer (pH 4.6) containing 0.25% (w/v) L-ascorbic acid. Dichloromethane (6 ml) was added to the sample, which then underwent inverse extraction at 8 rpm (RKVSD™, ATR, Inc., Laurel, MD, USA) for 30 minutes. After extraction, the aqueous layer was discarded and the organic solvent portion was transferred into a clean glass tube and evaporated to dryness under a stream of nitrogen gas at 60ºC (Reacti-Vap III™, Pierce, Rockford, IL, USA). The dried sample residue was then redissolved in 40 µl of 0.1 M sodium bicarbonate buffer (pH 9.0) and 40 µl of dansyl chloride solution (1 mg/ml in acetone). After sonication, the sample was heated at 60ºC (Reacti-Therm III™ Heating Module, Pierce, Rockford, IL, USA) for 5 minutes to form the EM and SI-EM dansyl derivatives (EM-Dansyl and SI-EM-Dansyl), respectively. After derivatization, all samples were analyzed using capillary LC/MS using an Agilent 1200 series nanoflow LC system (Agilent Technologies, Palo Alto, CA, USA) coupled to a TSQ™ Quantum Ultra triple quadrupole mass spectrometer (Thermo Electron, San Jose, CA, USA). Quantitation of cell medium estrogen metabolites was carried out using Xcalibur™ Quan Browser (Thermo Electron). Absolute concentrations of individual EM were normalized against protein concentrations.
HPLC analysis of estrogen-induced depurinating DNA adduct
Sample preparation
MCF-7 cells were treated as described above. The growth media was used to measure the level of depurinating estrogen−DNA adducts according to the method described by Zahid et al [30]. Briefly, cell culture media was extracted by using Varian C8 Certify II solid phase extraction cartridges (Varian, Harbor City, CA, USA) which were preequilibrated by sequentially passing 1 ml methanol, distilled water, and potassium phosphate buffer (100 mM, pH 8.0) through them. Culture media was adjusted with 1 ml of 1 M potassium phosphate to pH 8 and passed through the cartridge. After washing with 2 ml of 100 mM potassium phosphate, and 5 ml of distilled water, the analytes were eluted with 1 ml of elution buffer consisting of methanol/acetonitrile/water/TFA (8:1:1:0.1). The eluant was evaporated to about 100 ul by using Jouan RC10 Vacuum Concentrator and reconstituted with 100 ul of methanol/water (1:1). The solution was then passed through 5000 MW cut off filters and analyzed on HPLC connected with multiple electrochemical detectors.
HPLC analysis of adducts
Analysis of all samples was conducted on an HPLC system equipped with dual ESA Model 580 autosampler, and a 12-channel CoulArray electrochemical detector (ESA, Chelmsford, MA, USA). The two mobile phases used were as follows: A: acetonitrile/methanol/buffer/water (15:5:10:70) and B: acetonitrile/methanol/buffer/water (50:20:10:20). The buffer was a mixture of 0.25 M citric acid and 0.5 M ammonium acetate in triple-distilled water, and the pH was adjusted to 3.6 with acetic acid. The 95ul injections were carried out on a Phenomenex Luna-2 C-18 column (250 x 4.6 mm ID, 5 mm; Phenomenex, Torrance, CA, USA), initially eluted isocartically at 90%A/10%B for 15 minutes, followed by a linear gradient to 90%B/10%A in the next 40 min, and held there for 5 minutes (total 50 minutes gradient) at a flow rate of 1 ml/minute and a temperature of 30 oC. The serial array of 12 coulometric electrodes was set at potentials of -35, 10, 70, 140, 210, 280, 350, 420, 490, 550, 620, and 690 mV. The system was controlled and the data were acquired and processed using the CoulArray software package (ESA). Peaks were identified by both retention time and peak height ratios between the dominant peaks and the peaks in the two adjacent channels. The metabolites, conjugates and depurinating adducts were quantified by comparison of peak response ratios with known amounts of standards. The level of adducts were normalized against cell numbers and the DNA contents.
Reporter assay
Stable transfection of MCF-7 with the reporter plasmids
Luciferase reporter plasmids containing the CYP1A1, CYP1B1, COMTP1 (proximal COMT-promoters) and COMTP2 (distal COMT-promoters) and NQO1 promoters were used in this study. These plasmids were stably transfected into MCF-7 according to the following: MCF-7 cells were cotransfected with individual reporter plasmid (10ug) and Neomycin-expressing vector (1ug) using Fugene 6 (Roche, Indianapolis, IN, USA) following the manufacturer's protocol. Forty-eight hours post-transfection, the media was replaced with Genecitin-containing media (500 μg/ml media). Individual colonies were picked and propagated following 2 weeks of selection and were screened for luciferase activity. Colonies which showed positive luciferase activity indicated that they were successfully stably transfected and thus were selected and maintained in liquid nitrogen for further experiments.
Luciferase assay
Stably transfected cells containing the reporter gene vectors with the promoters of interest were grown in RPMI media, distributed in 6 well plates and treated with different concentrations of TNF-α. After 48 hours, the cells were harvested and luciferase activities were determined using luciferase enzyme assay systems according to the supplier's protocol (Promega, Madison, WI, USA). The luciferase activity was normalized against protein concentration using Bradford protein assay procedure.
Real time RT-PCR
MCF-7 cells were cultured, distributed in 6 well plates and treated with different concentrations of TNF-α. After 48 hours, Total RNA was isolated using RNA STAT-60 kit (Ambsio, lakeforest CA, USA) or RNA aqueous-Micro (Ambion, Foster City, CA, USA) according to the manufacturer's instructions. cDNA synthesis was conducted using High-Capacity cDNA Reverse Transcription kit following manufacturer's protocol. PCR amplification was performed using Tagman Fast Universal PCR Master Mix. Quantitative real-time RT-PCR was carried out using Pre-developed Tagman Assay reagents Control Kits. Kits used were obtained from (Applied Biosystem, Foster city, CA, USA). Each assay was performed in duplicate.
Western blot analysis
MCF-7 cells were cultured, distributed in 6 well plates and treated with different concentrations of TNF-α. After 72 hours, the cells were washed with phosphate buffered saline (PBS) and whole cell lysates were prepared with Radioimmunopreciptation assay (RIPA) lysis buffer. The cell lysates were solubilized in sample buffer [60 mM Tris-HCl pH 6.8, 2% sodium dodecyl sulfate (SDS), 10% glycerol, 0.7 mol/l β-mercaptoethanol; 0.01% bromophenol blue] and subjected to SDS polyacrylamide gel electrophoresis (PAGE). Proteins were electroblotted onto nitrocellulose membranes. Membranes were immunoblotted with the primary antibodies against CYP1B1 and COMT (Chemicon International Inc., Temecula, CA, USA), NQO1 (Novus Biologicals, Inc, Littleton, CO, USA) and β-actin (Sigma-Aldrich Co., St. Louis, MO, USA). After washing, membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Vector Laboratories, Burlingame, CA, USA) and X-ray films where then developed.
Statistical Analysis
Data are presented as Mean±Standard error of the mean (SEM) of at least two or three individual experiments as indicated under the results section. Statistical significance was determined by one-way analysis of variance (ANOVA) followed by Tukey test as a post-ANOVA multiple comparison test. Unpaired t-test was used for comparison between two groups. P<0.05 was considered statistically significant.
Results
TNF-α alters the profile of E2 and/or EM in MCF-7 cells (Table 1)
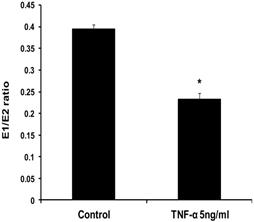
To evaluate the effects of TNF-α on estrogen metabolism, the EM levels were measured in conditioned media from MCF-7 cells treated with E2 (10 nM) alone or in combination with TNF-α (5 ng/ml) using LC/MS. It was found that TNF-α significantly increased the total EM compared with cells treated with E2 alone. The mean rate of formation of total EM for control was 19321.13±1252 pmole /mg protein/48 hours, while in TNF-α treated cells the mean rate was 26600.56±1196.6 pmole/mg protein /48 hours. To further assess the effects of TNF-α on estrogen metabolism, we derived the ratios for E1/E2, which is a commonly used predictor for cancer risk [31]. TNF-α shifted the E1/E2 equilibrium towards the formation of more E2 (Fig. 1). Thus, in MCF-7 cells treated with E2 plus TNF-α, the E1/E2 ratio was 0.232±0.01423 which is significantly lower compared with cells treated with E2 alone where the ratio was 0.395±0.0092. TNF-α also increased the rate of formation of 2-OHE2 accompanied by a decline in the rate of 2-MeOE2 by 75%. Concerning the other methylated catechols, there was non significant increase in the levels of 2-MeoE1 and 4-MeOE2 and non significant decrease in 4-MeOE1. There was however a significant increase in 3-MeOE1 by 2.5 folds. The 4-OHE1 levels were not detected in conditioned media treated with E2 alone, but increased to 63.3±10.623 upon adding TNF-α. On the contrary E3 levels and 16-epiestrol levels were detected upon treatment of the cells with E2 but not in TNF-α treated cells. There was also a non significant increase in the rate of formation of 17-epiestriol compared to control.
Effect of TNF-α on the ratio of E1/E2 in MCF-7 cells. Cells were grown in serum-free media and treated with E2 (10 nM) alone or with TNF-α (5 ng/ml) for 48 hours. Media were collected to quantify the mean rate of EM formation in (pmole/mg protein/48 hours) by LC/MS. Data were normalized against protein concentration. E1/E2 ratio was calculated. Values represent the Mean±SEM of three experiments. * indicates significant difference of TNF-α treated cells compared to control at P < 0.05.
The rate and profile of EM in culture media from MCF-7 cells. Cells were grown in serum-free media and treated with E2 (10 nM) alone or with TNF-α (5 ng/ml) for 48 hours. Media were collected to quantify the mean rate of EM formation in (pmole/mg protein/48 hours) by LC/MS. Data were normalized against protein concentration. Values represent the Mean±SEM of three experiments. *indicates significant difference of TNF-α treated cells compared to control at P < 0.05.
| Estrogen and Estrogen metabolites | Measurement (pmole/mg protein/48hours) (% from total EM) | |
|---|---|---|
| Control | TNF-α (5ng/ml) | |
| Parent estrogen | ||
| Estrone (E1) | 4889.02±162.19 (25.3±0.83) | 3939.29±207.68* (14.8±0.78) |
| Estradiol (E2) | 12385.67±511.8 (64.1±2.65) | 17133.73±325.83* (64.4±1.22) |
| 2-Hydroxylation pathway catechols | ||
| 2-Hydroxy estradiol (2-OHE2) | 0 | 1394.1±358.2 (5.24±1.35) |
| 2-Hydroxylation pathway methylated catechols | ||
| 2-Methoxyestrone (2-MeOE1) | 31.25±2.8 (0.16±0.01) | 47.2±5.8 (0.18±0.02) |
| 2-Methoxyestradiol (2-MeOE2) | 25.5±4.28 (0.13±0.02 | 6.34±0.85* (0.02 ±0.0027) |
| 2-Hydroxyestrone-3 methyl ether (3-MeOE1) | 20.122±0.964 (0.11±0.005) | 49.88±4.24* (0.19±0.016) |
| 4-Hydroxylation pathway catechols | ||
| 4-Hydroxy estrone (4-OHE1) | 0 | 63.3±10.623 (0.24±0.04) |
| 4-Hydroxylation pathway methylated catechols | ||
| 4-Methoxy estrone (4-MeOE1) | 13.987±3.96 (0.07±0.02) | 4.49±0.162 (0.02±0.0007) |
| 4-Methoxy estradiol (4-MeOE2) | 49.17±2.73 (0.254±0.014) | 79.11±60.4 (0.3±0.23) |
| 16α-Hydroxylation pathway | ||
| Estriol (E3) | 632.35±545.9 (3.27±2.82) | 0 |
| 17-Epiestriol (17-epi E3) | 1777.481±574.4 (9.2±2.97) | 3669±1130.8 (13.8±4.25) |
| 16-Epiestriol (16-epi E3) | 449.29±418.12 (2.3±2.17) | 0 |
| Total EM | 19321.13±1252 | 26600.56±1196.6* |
TNF-α increases the levels of DNA adducts in MCF-7 cells
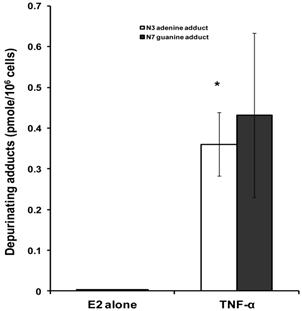
To investigate the implications of the possible effects of TNF-α on E2 metabolism and the formation of depurinating DNA adducts, the levels of DNA adducts (4-OHE1 [2]-1-N3 Adenine and 4-OHE1 [2]-1-N7 Guanine) were explored in culture media from MCF-7 cells treated with either E2 alone or in combination with TNF-α by HPLC. Our results demonstrated that TNF-α significantly increased Adenine adduct levels while there was no significant increase of the Guanine adducts. Thus, in MCF-7 cells treated with E2 alone or E2 plus TNF-α, the levels of N3 Adenine adducts were 0.0025±0.0005, 0.3604±0.078 (pmole/106cells) respectively. Also the levels of N7 Guanine adducts were 0.00293±0.00054 and 0.4319±0.2022 in cells treated with E2 alone or E2 plus TNF-α respectively (Fig 2).
Effect of TNF-α on depurinating DNA adducts formation. Cells were grown in serum-free media and treated with E2 (10 nM) alone or with TNF-α (5 ng/ml) for 48 hours. The media were collected for DNA adducts analysis by HPLC. Values represent the Mean±SEM of three experiments. * indicates significant difference of TNF-α treated cells compared to control at P < 0.05.
Effect of TNF-α on estrogen-metabolizing genes and expression in MCF-7 cells
To elucidate the underlying mechanisms of the differential pattern of estrogen metabolites and the rate of estrogen-induced adducts formation between TNF-α treated and non treated cells, we assessed the effect of TNF-α on the expression of key genes and enzymes involved in estrogen metabolism. Thus, we treated MCF-7 cells with TNF-α and measured its effect on enzyme promoter activity using Reporter gene assays, enzyme mRNA levels using real time RT-PCR and enzyme protein levels using Western blot analysis.
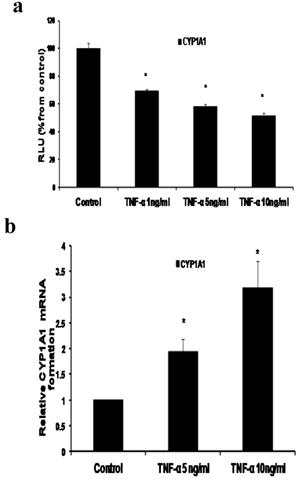
a- TNF-α downregulates CYP1A1 promoter and upregulates CYP1A1 mRNA
In our study we demonstrated that treatment of MCF-7 cells with TNF-α (1, 5 and 10 ng/ml) significantly reduced CYP1A1 to 69.39 ±1.215, 58±1.74 and 51.38±2.091 RLU respectively as indicated by Reporter assay (Fig 3a). However CYP1A1 mRNA was significantly upregulated to 1.938 ±0.23 and 3.1745±0.529 upon treatment with TNF-α 5 ng/ml and 10 ng/ ml respectively as indicated by real time RT-PCR (Fig 3b). This discrepancy between CYP1A1 luciferase activity and mRNA levels will be addressed in the discussion section. It's noteworthy that Western blotting for detecting CYP1A1 enzyme was attempted in this study but unfortunately, commercially available CYP1A1 antibodies didn't give satisfactory results.
Effect of TNF-α on CYP1A1 in MCF-7 cells. (a) Luciferase reporter gene assay. The cells were stably transfected with reporter plasmids containing CYP1A1 promoters. After treatment with different doses of TNF-α, luciferase production was measured 48 hours later. Results represent the Mean±SEM of two experiments. (b) Real time RT-PCR analysis.The cells were treated with TNF-α 5 ng/ml and 10 ng/ml for 24 hours followed by total RNA isolation from the cells and the CYP1A1 mRNA expression was determined by real time RT-PCR. The threshold cycle value of CYP1A1 was normalized based on that of GAPDH. Results represent the Mean±SEM of two experiments. * indicates significant difference of TNF-α treated cells compared to control at P < 0.05.
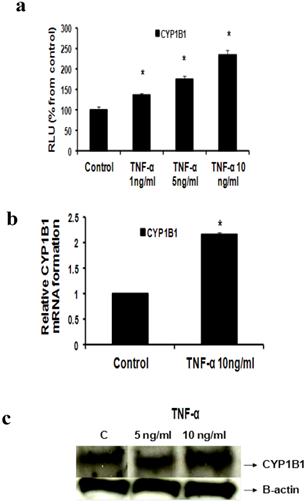
b- TNF-α upregulates CYP1B1
Our study proved that treatment of MCF-7 cells with TNF-α (1, 5 and 10 ng/ml) significantly increased CYP1B1 levels in a dose dependent pattern as indicated by Reporter assay by 35.7 %, 75%, 134 % respectively (Fig 4a) while TNF-α (10 ng/ml) significantly increased CYP1B1 mRNA by 116% as determined by real time RT-PCR (Fig 4b). Western blot data also revealed that CYP1B1 protein expression was upregulated (Fig 4c).
Effect of TNF-α on CYP1B1 in MCF-7 cells: (a) Luciferase reporter gene assay. The cells were stably transfected with reporter plasmids containing CYP1B1 promoters. After treatment with different doses of TNF-α, luciferase production was analyzed 48 hours later. Results represent the Mean±SEM of two experiments. (b) Real time RT-PCR analysis. The cells were treated with TNF-α 10 ng/ml for 24 hours followed by total RNA isolation from the cells, and the CYP1B1 mRNA expression was determined by real time RT-PCR. The threshold cycle value of CYP1B1 was normalized based on that of GAPDH. Results represent the Mean±SEM of two experiments. (c) Western blot analysis. CYP1B1 protein levels were determined 72 hours after treatment with different doses of TNF-α. * indicates significant difference of TNF-α treated cells compared to control at P < 0.05.
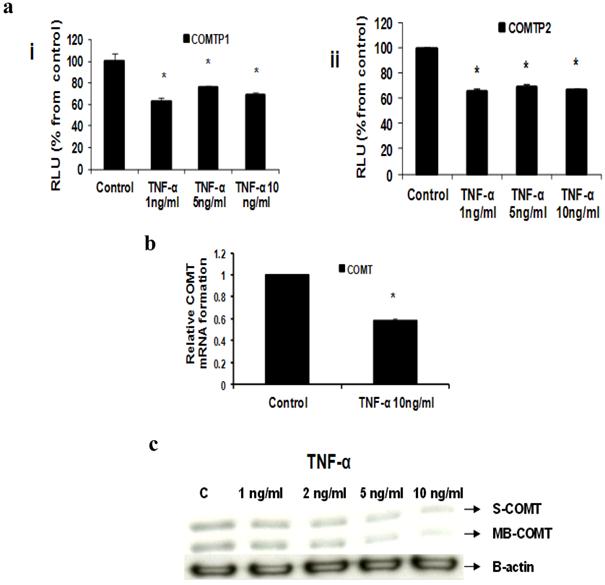
c- TNF-α downregulates COMT
Our Reporter gene assay showed that TNF-α (1, 5 and 10 ng/ml) significantly reduced COMTP1 expression by 36.75%, 23.72% and 31.85% respectively (Fig 5ai). Similarly COMTP2 was significantly reduced by 34%, 30.4% or 33.6% respectively (Fig 5aii). Real time RT-PCR data revealed that COMT mRNA was reduced by 41.7% in cells treated with TNF-α 10 ng/ml compared with untreated control (Fig 5b). Consistent with the above two assays Western blotting confirmed that TNF-α downregulated COMT expression in its two forms: soluble (S-COMT) and membrane bound (MB-COMT) compared with control (Fig 5c).
Effect of TNF-α on COMT in MCF-7 cells: (a) Luciferase reporter gene assay. The cells were stably transfected with reporter plasmids containing COMTP1 (i) and COMTP2 promoters (ii). After treatment with different doses of TNF-α, luciferase production was analyzed 48 hours later. Results represent the Mean±SEM of two experiments. (b) Real time RT-PCR analysis. The cells were treated with TNF-α 10 ng/ml for 24 hours followed by total RNA isolation from the cells and COMT mRNA expression was determined by real time RT-PCR. The threshold cycle value of COMT was normalized based on that of GAPDH. Results represent the Mean±SEM of two experiments. (c) Western blot analysis. Both S-COMT and MB-COMT protein levels were determined 72 hours after treatment with different doses of TNF-α. * indicates significant difference of TNF-α treated cells compared to control at P < 0.05.
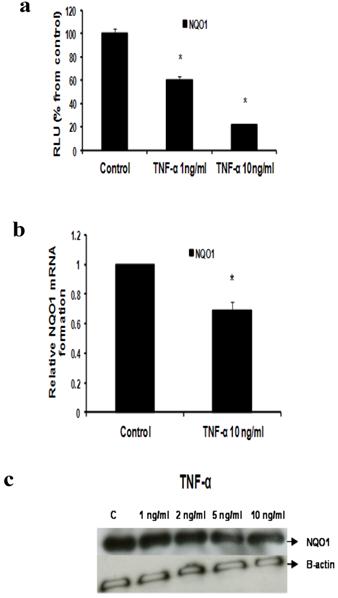
d- TNF-α downregulates NQO1
Our data reflected that the effect of TNF-α on NQO1 occurred on the transcriptional and translational levels as indicated by Reporter, real time RT-PCR and Western blotting. In Reporter assay, treatment with TNF-α 1 ng/ml and 10 ng/ml resulted in significant downregulation of NQO1 promoter activity to 60.3±2.609 and 21.45±0.9192 RLU respectively (Fig 6a). In real time RT-PCR assay NQO1 mRNA levels were reduced by 30.5% using TNF-α 10 ng/ml (Fig 6b). Also, TNF-α decreased NQO1 protein levels as indicated by Western blotting (Fig 6c).
Effect of TNF-α on NQO1 in MCF-7 cells: (a) Luciferase reporter gene assay. The cells were stably transfected with reporter plasmids containing NQO1 promoters. After treatment with different doses of TNF-α, luciferase production was measured 48 hours later. Results represent the Mean±SEM of duplicate experiments. (b) Real time RT-PCR analysis. The cells were treated with TNF-α 10 ng/ml for 24 hours followed by total RNA isolation from the cells, and the NQO1 mRNA expression was determined by real time RT-PCR. The threshold cycle value of NQO1 was normalized based on that of GAPDH. Results represent the Mean±SEM of two experiments. (c) Western blot analysis. NQO1 protein levels were determined 72 hours after treatment with different doses of TNF-α. * indicates significant difference of TNF-α treated cells compared to control at P < 0.05.
Discussion
Chronic inflammation represents a major risk factor for many cancer types, including liver, breast, prostate, pancreas, ovary, skin, gastric, colorectal and pulmonary carcinomas [32].
TNF-α was identified as a keyplayer in the cytokine network and a major mediator of cancer related inflammation. It exerts its action via activation of NF-κB which is the pivotal regulator of cellular inflammatory responses [33-36].
Numerous studies have linked TNF-α to breast cancer progression. As a result, the mechanisms by which TNF-α promotes breast cancer have been recently explored using both in vitro and in vivo models [37]. In this study we are trying to focus on the influence of TNF-α on the estrogen metabolic pathway in MCF-7 cells. This may help us better understand the mechanisms by which inflammation affects breast cancer.
One of the interesting findings in our study is that the total EM was significantly higher in MCF-7 treated cells compared to control. One reason for this may be due to that TNF-α increased the local estrogen biosynthesis in MCF-7 cells through stimulating aromatase expression as suggested by previous reports [38, 39]. However, based on our results we are adding a new mechanism where by TNF-α may be directly interfering with the estrogen metabolic pathway. Besides increasing total EM, we also found that TNF-α significantly decreased the E1/E2 ratio due to a decrease in the level of estrogen as E1 with a concomitant increase in E2 concentration. The interconversion of weakly active estrone (E1) into highly potent estradiol (E2) and their relative abundance dictate the estrogenic environment and may be contributing to the development of breast cancer. This alteration in E1/E2 ratio could be explained in light of the experimental evidence that TNF-α modulates the expression of hydroxysteroid (17β) -dehydrogenase 1 (HSD17β1) in breast cancer, an enzyme that efficiently catalyzes the conversion of E1 into E2 [31]. Also, 4-OHE1, which in our study was detected only in TNF-α treated cells, was previously shown to be tumorigenic in animal models [40]. It's noteworthy that 4-hydroxy catechols have greater carcinogenic potential than 2-hydroxy catechols due to different mechanisms of adduction leading to higher DNA adducts [9].
To understand why TNF-α altered the rate and pattern of estrogen metabolites, we further explored the effect of TNF-α on the expression of key genes involved in estrogen metabolism. Our data revealed that TNF-α increases the expression levels of CYP1A1 and CYP1B1. This was associated with a concomitant effect on the expression of detoxification enzymes COMT and NQO1 where TNF-α reduced the expression levels of these two enzymes. In agreement with our results several studies reported similar effects on these enzymes [28, 41, 42]. Surprisingly, TNF-α was found to upregulate mRNA of CYP1A1 and to downregulate it's promoter activity as indicated by luciferase assay. This effect may be similar to that explained by Morel et al [43] where increasing the CYP1A1 activity in hepatoma cells elicited an oxidative stress and led to the repression of a reporter gene driven by the CYP1A1 gene promoter in a negative feedback loop. Our results also indicated that TNF-α induced the rate of formation of the catechol estrogen 2-hydroxy estradiol (2-OHE2). This catechol estrogen was found to redox cycle and to generate hydrogen peroxide (H2O2) and hydroxyl radicals in MCF-7. Depending on the localized concentrations of catechol estrogens and enzymes that mediate redox cycling, this may be an important mechanism contributing to the development of breast cancer [44]. Given that our results also indicated a significant downregulation of the detoxifying enzyme COMT accompanied by a reduction in the anticarcinogenic, antiangiogenic metabolite 2-MeOE2 [45], this may create a deleterious influence on estrogen carcinogenesis. Interestingly, we also found that E3 was detected in cells treated with E2 alone while it was not detected in cells treated with TNF-α. The effect of this hormone on breast cancer has been controversial. Some studies indicate that there's increased risk of development of breast cancer with E3 therapy [46]. Other studies however, support the hypothesis that E3 exert protective roles by antagonizing the carcinogenic effects of E2 [47]. Recent studies indicate that E3 acts as a G-protein-coupled receptor 30 (GPR30) antagonist in estrogen receptor-negative breast cancer cells. GPR30 has been recently involved in rapid signalling triggered by estrogens [48].
Studies on estrogen metabolism, formation of DNA adducts, carcinogenicity, cell transformation, and mutagenicity have led to the hypothesis that reaction of certain estrogen metabolites, predominantly catechol estrogen-3,4-quinones, with DNA forms depurinating adducts [4-OHE1(E2)-1-N3Adenine and 4-OHE1(E2)-1-N7Guanine]. Formation of these adducts and the concomitant apurinic sites in DNA have been shown to induce mutations that are associated with initiation of breast cancer [49, 50]. Indeed, our study demonstrated that treatment with TNF-α is associated with a significant increase in estrogen-induced depurinating adducts especially the Adenine adducts. It is likely that this resulted from the increased level of some catechol estrogens accompanied by downregulation of the detoxification enzymes COMT and NQO1 as demonstrated in this report.
Taken altogether, it's clear from this study that TNF-α alters the balanced homeostatic set of activating and deactivating pathways of estrogen metabolism in MCF-7 breast cancer cells. It influences key genes and enzymes involved in estrogen metabolism leading to increased DNA adduct products. This may implicate a new potential mechanism for inflammation associated breast cancer and provide a plausible explanation for the effect of TNF-α on the advancement of breast cancer. Also it can be suggested that compounds which are known to have positive regulatory effects on estrogen metabolic pathways, e.g. indole-3-carbinol, can be useful in inflammation associated breast cancer [51].
For future research we would like to see whether the effect of TNF-α on estrogen metabolic pathways plays a role in ER-positive breast cancer patients who are resistant to endocrine therapy. Also whether it's involved in higher resistance to endocrine therapy among obese breast cancer patients who are also resistant to endocrine therapy. No doubts that further evaluation of human breast cancer samples and experimental models as well as development of novel in vivo models designed to specifically study inflammation in mammary tumorigenesis will help to elucidate many of these questions.
Acknowledgements
We express our gratitude to Dr. Simon D. Spivack (Laboratory of Human Toxicology and Molecular Epidemiology. New York State Department of Health, Albany, NY) and Barbara C. Spink (Wadsworth Center, New York State Department of Health, Albany, NY) for generously providing us with CYP1A1 and CYP1B1 Luciferase Reporter plasmids.
Competing Interests
The authors have declared that no competing interest exists.
References
1. DeSantis C, Siegel R, Bandi P, Jemal A. Breast cancer statistics, 2011. CA Cancer J Clin. 2011;61(6):409-18
2. Henderson IC, Canellos GP. Cancer of the breast: the past decade. N Engl J Med. 1990;302:17-30
3. Henderson HS, Ross RK, Bernstein L. Estrogens as a cause of human cancer: The Richard and Hinda Rosenthal Foundation Award Lecture. Cancer Res. 1988;48:246-53
4. Russo j, Russo IH. Cellular basis of breast cancer susceptibility. Oncol Res. 1999;11(4):169-78
5. Bocchinfuso WP, Korach KS. Mammary gland development and tumorigenesis in estrogen receptor knockout mice. J Mammary Gland Biol Neoplasia. 1997;2:323-334
6. Cavalieri EL, Stack DE, Devanesan PD, Todorovic R, Dwivedy I, Higginbotham S, Johansson SL, Patil KD, Gross ML, Gooden JK, Ramanathan R, Cerny RL, Rogan EG. Molecular origin of cancer: catechol estrogen- 3,4-quinones as endogenous tumor initiators. Proc Natl Acad Sci USA. 1997;94(20):10937-42
7. Bradlow HL, Hershcopf R, Martucci C, Fishman J. 16 alpha-hydroxylation of estradiol: a possible risk marker for breast cancer. Ann NY Acad Sci. 1986;464:138-151
8. Mooberry SL. Mechanism of action of 2-methoxyestradiol: new developments. Drug Resist Update. 2003;6:355-361
9. Cavalieri EL, Rogan EG. Unbalanced metabolism of endogenous estrogens in the etiology and prevention of human cancer. J Steroid Biochem Mol Biol. 2011;125(3-5):169-80
10. Straub RH. The complex role of estrogens in inflammation. Endocr Rev. 2007;28:521-574
11. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860-867
12. Cole SW. Chronic inflammation and breast cancer recurrence. J Clin Oncol. 2009;27(21):3418-9
13. Pierce BL, Ballard-Barbash R, Bernstein L, Baumgartner RN, Neuhouser ML, Wener MH, Baumgartner KB, Gilliland FD, Sorensen BE, McTiernan A, Ulrich CM. Elevated biomarkers of inflammation are associated with reduced survival among breast cancer patients. J Clin Oncol. 2009;27(21):3437-44
14. Rao VP, Poutahidis T, Ge Z, Nambiar PR, Boussahmain C, Wang YY, Horwitz BH, Fox JG, Erdman SE. Innate immune inflammatory response against enteric bacteria Helicobacter hepaticus induces mammary adenocarcinoma in mice. Cancer Res. 2006;66(15):7395-400
15. Connelly L, Barham W, Onishko HM, Sherrill T, Chodosh LA, Blackwell TS, Yull FE. Inhibition of NF-κB activity in mammary epithelium increases tumor latency and decreases tumor burden. Oncogene. 2011;30(12):1402-12
16. Balkwill F. Tumor necrosis factor and cancer. Nat Rev Cancer. 2009;9(5):361-71
17. Leek RD, Landers R, Fox SB, Ng F, Harris AL, Lewis CE. Association of tumor necrosis factor alpha and its receptors with thymidine phosphorylase expression in invasive breast carcinoma. Br J Cancer. 1988;77:2246-2251
18. Miles DW, Happerfield LC, Naylor MS, Bobrow LG, Rubens RD, Balkwill FR. Expression of tumor necrosis factor (TNF-α) and its receptors in benign and malignant breast tissue. Int J Cancer. 1994;56:777-782
19. Sheen-Chen SM, Chen WJ, Eng HL, Chou FF. Serum concentration of tumor necrosis factor in patients with breast cancer. Breast Cancer Res Treat. 1997;43:211-215
20. Balkwill F. Tumor necrosis factor or tumor promoting factor? Cytokine Growth Factor. 2002;13:135-141
21. Terry MB, Gammon MD, Zhang FF, Tawfik H, Teitelbaum SL, Britton JA, Subbaramaiah K, Dannenberg AJ, Neugut AI. Association of frequency and duration of aspirin use and hormone receptor status with breast cancer risk. JAMA. 2004;291:2433-2440
22. Baumgarten SC, Frasor J. Inflammation: an instigator of more aggressive estrogen receptor ER-positive breast cancers. Mol Endocrinol. 2012;26(30):360-71
23. Goldberg JE, Schwertfeger KL. Proinflammatory cytokines in breast cancer: mechanisms of action and potential targets for therapeutics. Curr Drug Targets. 2010;11(9):1133-46
24. Purohit A, Newman SP, Reed M. The role of cytokines in regulating estrogen synthesis: implications for the etiology of breast cancer. Breast Cancer Res. 2002;4(2):65-9
25. Herrmann M, Scholmerich J, Straub RH. Influence of cytokines and growth factors on distinct steroidogenic enzymes in vitro: a short tabular data collection. Ann NY Acad Sci. 2002;966:166-186
26. Hong CY, Park JH, Ahn RS, Im SY, Choi HS, Soh J, Mellon SH, Lee K. Molecular mechanism of suppression of testicular steroidogenesis by proinflammatory cytokine tumor necrosis factor-alpha. Mol Cell Biol. 2004;24:2593-2604
27. Mikhaylova IV, Kuulasmaa T, Jaaskelainen J, Voutilainen R. Tumor necrosis factor-alpha regulates steroidogenesis, apoptosis, and cell viability in the human adrenocortical cell line NCI-H295R. Endocrinol. 2007;148:386-392
28. Salama SA, Kamel MW, Diaz-Arrastia CR, Xu X, Veenstra TD, Salih S, Botting SK, Kumar R. Effect of TNF-α on estrogen metabolism and endometrial cells: potential physiological and pathological relevance. J Clin Endocrinol Metab. 2008;94(1):285-93
29. Xu X, Keefer LK, Ziegler RG, Veenstra TD. A liquid chromatography-mass spectrometry method for the quantitative analysis of urinary endogenous estrogen metabolites. Nat Protoc. 2007;2(6):1350-5
30. Zahid M, Gaikwad NW, Ali MF, Lu F, Saeed M, Yang L, Rogan EG, Cavalieri EL. Prevention of estrogen-DNA adducts formation in MCF-10F cells by resveratrol. Free Radic Biol Med. 2008;45(2):136-45
31. Zhang CY, Chen J, Yin DC, Lin SX. The contribution of 17beta-hydroxysteroid dehydrogenase type 1 to the estradiol-estrone ratio in estrogen-sensitive breast cancer cells. PLoS One. 2012;7(1):e29835
32. Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. Inflammation and cancer: how hot is the link? Biochem Pharmacol. 2006;72(11):1605-21
33. Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, Holdsworth H, Turner L, Rollins B, Pasparakis M, Kollias G, Balkwill F. Tumor necrosis factor-alpha deficient mice are resistant to skin carcinogenesis. Nature Med. 1999;5(7):828-31
34. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-κB functions as a tumor promoter in inflammation associated cancer. Nature. 2004;431(7007):461-6
35. Balkwill F. TNF-α in promotion and progression of cancer. Cancer Metastasis Rev. 2006;25(3):409-416
36. Sethi G, Sung B, Aggarwal BB. TNF: a master switch from inflammation to cancer. Front Biosci. 2008;13:5094-107
37. Goldberg JE, Schwertfeger KL. Proinflammatory cytokines in breast cancer: mechanisms of action and potential targets for therapeutics. Curr Drug Targets. 2010;11(9):1133-46
38. Macdiarmid F, Wang DY, Duncan LJ, Purohit A, Ghilchik MW, Reed MJ. Stimulation of aromatase activity in breast fibroblasts by tumor necrosis factor- alpha. Molec Cell Endocr. 1994;106:17-21
39. Morris PG, Hudis CA, Giri D, Morrow M, Falcone DJ, Zhou XK, Du B, Brogi E, Crawford CB, Kopelovich L, Subbaramaiah K, Dannenberg AJ. Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prev Res (Phila). 2011;4(7):1021-9
40. Liehr JG, Ricci MJ. 4-hydroxylation of estrogens as markers of human mammary tumors. PNAS. 1996;93:3294-3296
41. Gharavi N, El-Kadi AO. Role of nitric oxide in downregulation of Cytochrome P-450 1A1 and NADPH Quinone oxidoreductase 1 by tumor necrosis factor-alpha and lipopolysaccharide. J Pharm Sci. 2007;96(10):2795-807
42. Tchivileva IE, Nackley AG, Qian L, Wentworth S, Conrad M, Diatchenko LB. Characterization of NF-κB-mediated inhibition of Catechol-O-methyltransferase. Mol Pain. 2009;5:13
43. Morel Y, Mermod N, Barouki R. An autoregulatory loop controlling CYP1A1 gene expres-sion: role of H2O2 and NFI. Mol Cell Biol. 1999;19(10):6825-32
44. Fussell KC, Udasin RG, Smith PJ, Gallo MA, Laskin JD. Catechol metabolites of endogenous estrogens induce redox cycling and generate reactive oxygen species in breast epithelial cells. Carcinogenesis. 2011;32(8):1285-93
45. Pribluda VS, Gubish ER Jr, Lavallee TM, Treston A, Swartz GM, Green SJ. 2-Methoxyestradiol: an endogenous antiangiogenic and antiproliferative drug candidate. Cancer Metastasis Rev. 2000;19(1-2):173-9
46. Rosenberg LU, Magnusson C, Lindström E, Wedrén S, Hall P, Dickman PW. Menopausal hormone therapy and other breast cancer risk factors in relation to the risk of different histological subtypes of breast cancer: a case-control study. Breast Cancer Res. 2006;8(1):R11
47. Cole P, MacMahon B. Estrogen fractions during early reproductive life in the etiology of breast cancer. Lancet. 1969;1:604-606
48. Lappano R, Rosano C, De Marco P, De Francesco EM, Pezzi V, Maggiolini M. Estriol acts as a GPR30 antagonist in estrogen receptor-negative breast cancer cells. Mol Cell Endocrinol. 2010;320(1-2):162-70
49. Mailander PC, Meza JL, Higginbotham S, Chakravarti D. Induction of AT to GC mutations by erroneous repair of depurinated DNA following estrogen treatment of the mammary gland of ACI rats. J Steroid Biochem Mol Biol. 2006;101(4-5):204-15
50. Naushad SM, Reddy CA, Rupasree Y, Pavani A, Digumarti RR, Gottumukkala SR, Kuppusamy P, Kutala VK. Cross-talk between one-carbon metabolism and xenobiotic metabolism: implications on oxidative DNA damage and susceptibility to breast cancer. Cell Biochem Biophys. 2011;61(3):715-23
51. Aggarwal BB, Ichikawa H. Molecular targets and anticancer potential of indole-3-carbinol and its derivatives. Cell Cycle. 2005;4(9):1201-15
Author contact
![]() Corresponding author: Department of Obstetrics and Gynecology, Baylor College of Medicine, One Baylor Plaza MS: BCM 610, Houston, TX 77030, United States. Tel.: +1 832 824 4188; fax: +1 832 825 4190. E-mail addresses: salamaedu, salamascom
Corresponding author: Department of Obstetrics and Gynecology, Baylor College of Medicine, One Baylor Plaza MS: BCM 610, Houston, TX 77030, United States. Tel.: +1 832 824 4188; fax: +1 832 825 4190. E-mail addresses: salamaedu, salamascom