Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2012; 3:303-309. doi:10.7150/jca.4179 This volume Cite
Mini-Review/Case Report
Malignant Peripheral Nerve Sheath Tumors: Differentiation Patterns and Immunohistochemical Features - A Mini-Review and Our New Findings
Aitao Guo, Aijun Liu ![]() , Lixin Wei, Xin Song
, Lixin Wei, Xin Song
Department of Pathology, the Chinese PLA General Hospital, Beijing 100853, China.
Received 2012-2-1; Accepted 2012-5-28; Published 2012-7-4
Abstract
Malignant peripheral nerve sheath tumors (MPNST) represent a group of highly heterogeneous human malignancies often with multiple histological origins, divergent differentiation patterns, and diverse immunohistochemical presentations. The differential diagnosis of MPNST from other spindle cell neoplasms poses great challenges for pathologists. This report provides a mini-review of these unique features associated with MPNST and also presents the first cases of MPNST with six differentiation patterns.
Keywords: Malignant peripheral nerve sheath tumor, Liposarcomatous, Glandular, Fibrohistiocytoid, Neuroendocrine differentiation, Cartilage, Triton tumor, Gangliocyte.
A. Differentiation Patterns
Malignant peripheral nerve sheath tumor (MPNST), also known as “Malignant schwannoma”, “Neurofibrosarcoma”, or “Neurosarcoma”, is derived from Schwann cells or pluripotent cells of the neural crest [1-5]. Epithelioid or other heterogeneous components can be observed in 15% of MPNSTs [1, 2, 6]: the latter include rhabdomyoblasts [1, 2, 6-12], cartilaginous [6, 13, 14], osseous [6, 12, 13] differentiations and, rarely, smooth muscle [13, 15], glandular [6, 10-12, 16] or liposarcomatous components [6, 12, 17] have been reported. It is rare that there are two or more heterogeneous components in a single MPNST. To our best knowledge, MPNST with four differentiated components has been described only in one case [12]. Herein, we report one case of MPNST with six differentiated components.
MPNSTs can be graded II, III or IV according to WHO classification [1]. They account for only 5% of malignant soft tissue tumors [1, 2, 7]. One half to two thirds arises from neurofibromas, often of plexiform neurofibromas or in the setting of neurofibromatosis type I, which occur frequently on the head or neck. The other MPNSTs arise de novo, which usually involve the peripheral nerves in the buttocks or thighs, mostly the sciatic nerve [1].
MPNST is generally characterized by alternating hypo- and hyper-cell areas or a diffuse growth pattern of spindle-shaped cells which are asymmetrical and fusiform with wavy or comma-shaped hyperchromatic nuclei, arranged in palisades or spiral shapes [1-3, 7, 8, 18, 19]. In about 15% of MPNSTs, epithelioid or heterologous differentiation can be found [1]; the later includes rhabdomyoblasts, smooth muscle, bone, cartilage, and neuroendocrine component [1-24]. The most common heterologous component in MPNST is rhabdomyoblast differentiation [1, 7, 8], being first reported by Masson in 1932 and named "nerve rhabdomyoma", and later renamed by Woodruff as "malignant triton tumor". Additionally, differentiation into cartilage or bone is also not uncommon [6, 12-14], while liposarcomatous differentiation is very rare; to date, only 4 cases have been reported [6, 12, 17]. MPNST also has glandular structures [1, 2, 6, 10-12, 16], and foci of neuroendocrine differentiation are often seen in glandular MPNST [1]. The combination with more than two components is very rare; to our best of known, only one MPNST case simultaneously with epithelioid differentiation, rhabdomyosarcoma, bone, and liposarcoma has been reported [12]. In the present case, six kinds of differentiation, including rhabdomyosarcomatous, chondral, glandular, neuroendocrine, gangliocytic, and liposarcomatous components were observed in the background of a classical MPNST. The histological changes are so complex and diverse that no simple definition can cover such a wide range of differentiation, and a final diagnose of MPNST was made.
The diagnosis of MPNST with multiple mesenchymal differentiations is difficult, and sometimes should be differentiated from rhabdomyosarcoma, osteosarcoma, chondrosarcoma or liposarcoma [6]. In most cases, only regional or focal above-mentioned differentiation can be observed on the background of typical spindle-shaped tumor cells, which is easy to identify. However, rhabdomyosarcomatous differentiation occasionally becomes predominant; differential diagnosis is very difficult, especially in pediatric patients; thus, the localization of the tumor and sufficient sampls of the specimen are very important.
In 5-10% of dedifferentiated liposarcomas, myoblasts, cartilage, and osteosarcomatous differentiation can be observed [6, 9]; however, the predominancy is composed of malignant fibrous histiocytoma and/or leiomyosarcoma, and the ordinary MPNST structure and epithelioid or glandular differentiations should not be visible, and medical history and tumor location are of keypoints for diagnosis. For example, dedifferentiated liposarcoma tends to occur in deep locations such as the retroperitoneal area, and some patients may have surgical resection history of a typical liposarcoma [6, 9].
Additionally, MPNST with glandular differentiation must be differentiated from metastatic carcinoma. In MPNST, the spindle-shaped cells around glandular differentiation are also tumor cells, with thin and red stained cytoplasm, and wavy or comma-shaped hyperchromatic nuclei; however, in metastatic carcinoma, spindle-shaped cells around the glandular structures are often reactive proliferating fibroblasts. Full physical examination is also helpful for differential diagnosis.
MPNST is a tumor associated with an aggressive behavior and its prognosis is poor with death occurring in 63%, usually with 2-year of diagnosis. The 2-year and 5-year overall survival rate are reported to be 57% and 39% [8], with the median survival period is 32 months [3]. Surgical resection is the best available option for treating MPNST [3, 7, 8, 23]. Relatively better prognosis can be seen in the patients with a superficial and smaller tumor, complete excision and/or with family history [1-3, 20, 22, 29]. But the prognosis is poorer in patients with rhabdomyosarcomatous differentiation, the 2-year and 5-year survival rates are 15% and 11% [8], respectively. And the mortality rate is increased to 79% in tumors with glandular differentiation [1]. In this case, MPNST recurred in the original location 10 months after resection, but chest CT and whole body bone scan did not demonstrate metastases in the lungs or bones. The patient is still under follow-up.
B. Immunohistochemical Features
Some neural markers, such as S-100, CD56 and protein gene product 9.5 (PGP 9.5) are considered sensitive markers for peripheral nerve sheath tumors. S-100, which is traditionally regarded as the best marker for MPNST, has limited diagnostic utility and is positive in only about 50-90% of the tumors [8]. In high grade MPNST, only scattered, if any, tumor cells are S-100 positive [1, 23]. On the other hand, although sensitive, CD56 and PGP 9.5 expression is in no way specific for tumors of MPNST [24]. Thereby, MPNSTs per se lack sufficiently specific and sensitive immunohistochemical marker. Recent studies suggest that nestin, which is an intermediate filament protein, is more sensitive for MPNST than other neural markers (S-100, CD56 and PGP 9.5) and immunostains for nestin in combination with other markers could be useful in the diagnosis of MPNST [25]. To cases with divergent differentiation only typical tumor components correspond to special immunophenotyping, as indicated in the present case. The spindle and glandular tumor cells were positive for S-100 and nestin, and most spindle-shaped cells were positive for vimentin. Cytokeratin was expressed in the glandular region, CD56 expressed in most spindle-shaped cells and rosette formations, and Syn in some rosette formations. Actin was positive focally, and other myogenic markers were negative.
MPNST with glandular differentiations should be distinguished from other tumors with dual differentiation such as synovial sarcoma and mesothelioma. Histological separation of MPNST from synovial sarcomas can be difficult and available immunohistochemical markers, such as S-100 and cytokeratin, sometimes give rise to overlapping staining patterns. Immunostaining EMA/CK7 for synovial sarcoma and nestin/S100 for MPNST yielded high specificity and positive predictive values [26]. Recent study considered that expression of HMGA2 is a feature of MPNST but not of synovial sarcoma and immunohistochemical staining of HMGA2 may be a useful marker to separate MPNST from synovial sarcoma [27]. Another study showed the presence of SYT-SSX fusion transcript analyzed by real-time reverse transcription polymerase chain reaction can be useful in the diagnosis of synovial sarcoma [28]. In MPNST goblet cells and neuroendocrine differentiation are often seen in glandular structures, which are seldom found in mesothelioma. Immunohistostaining for Calretinin, in combination with the tumor site, is also helpful to differentiate mesothelioma from MPNST. The expression of Calretinin is uncommon in MPNST.
C. Our new findings
Clinical data
A 79-year-old man was hospitalized because of an enlarging mass in his left thigh for 2 years. Intraoperative findings showed that a solid subcutaneous mass, which protruded about one centimeter above the skin surface, closely connected to the lateral femoral nerve and slightly adherent to the vastus lateralis muscle. The tumor mass was completely resected with part of skin covering on it. Previous history of the patient was unremarkable except cholecystectomy due to cholecystitis and cholelithiasis 12 years ago.
Laboratory Tests
The surgical specimen was fixed in 10% neutral formalin, routinely processed, paraffin embedded, sectioned at 3 um thick, and stained conventionally with hematoxylin and eosin. For immunohistochemistry, four-micrometer sections of the paraffin-embedded tissues were deparaffinized, rehydrated in a graded series of alcohol and microwave-treated for 10 min in the citrate buffer (pH 6.0). Endogenous peroxidase activity was blocked using 0.3% hydrogen peroxide. The tissue were processed in an automatic IHC staining machine using the standard protocols (Lab Vision Autostainer, Lab Vision Co., Fremont, CA, USA) with DAKO Real™ EnVision™ Detection System (K5007, DAKO). All antibodies were bought from DAKO Co. The signals were visualized with 3-3′-diaminobenzidine and counterstained with Mayer's hematoxylin. The antigens are summarized in Table 1.
Immunohistochemical reagents and results.
| Antibodies | Clone No. | Pretreatments and dilutions | Expression in different components | |||||
|---|---|---|---|---|---|---|---|---|
| SSC | chondroid | LS | RMS | E/G | NE | |||
| S-100 | multi-Clone | HP 1:2000 | + | + | + | + | ||
| nestin | GN-1401 | HP 1:200 | + | - | + | + | ||
| vimentin | Vim3B4 | HP 1:100 | + | + | + | + | ||
| MBP | 7H11 | NPT 1:100 | - | - | - | |||
| cytokeratin | AE1/AE3 | NPT 1:200 | - | + | + | |||
| CD56 | 1B6 | HP 1:150 | + | + | + | |||
| CD99 | 12E7 | HP 1:100 | + | + | + | |||
| Syn | multi-Clone | HP 1:150 | - | - | + | |||
| CgA | 5H7 | NPT 1:75 | - | - | - | |||
| actin | HHF35 | HP 1:100 | - | + | - | - | ||
| desmin | D33 | NPT 1:100 | - | - | - | |||
| MyoD1 | 5.8A | GE 1:100 | - | - | - | |||
| myoglobin | MY018 | GE 1:100 | - | - | - | |||
| myosin | F5D | HP 1:200 | - | - | - | |||
| SMA | 1A4 | HP 1:100 | - | - | - | |||
| CD34 | QBEnd10 | HP 1:150 | - | - | - | |||
| Bcl-2 | 124 | HP 1:150 | +/- | + | + | |||
MBP myelin basic protein, HP high pressure, GE gastric enzyme, NPT non- pretreatments, SSC spindle shaped cell, LS liposarcomatous, RMS rhabodomyosarcomatous, E/G epithelioid/glandular, NE neuroendocrine, component was not stained with any antibody.
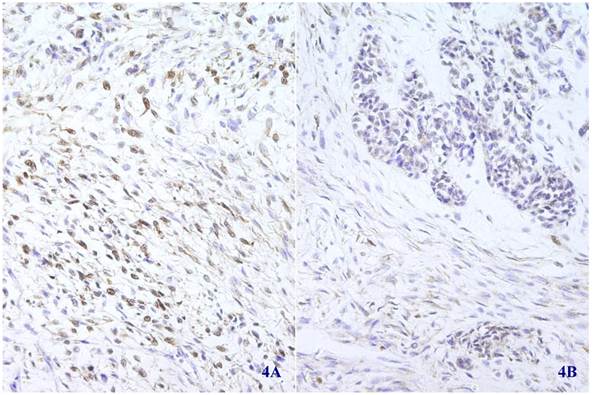
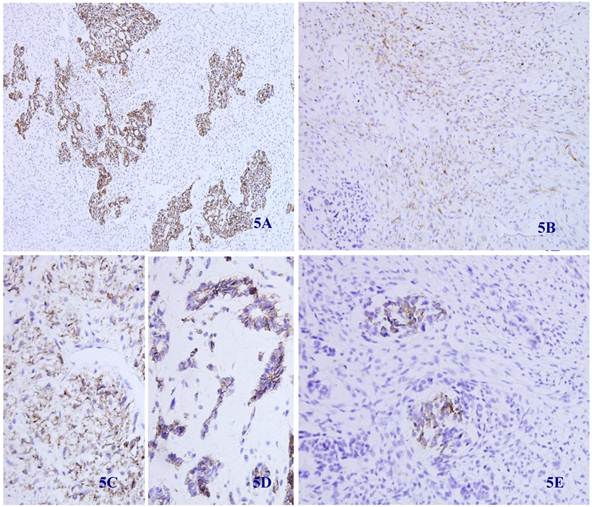
Immunohistochemical staining showed that the spindle and grandular tumor components were positive for S-100 (Figure 4A and 4B). Cytokeratin was expressed in the region of epithelioid differentiation (Figure 5A). Most of the spindle cells were positive for vimentin (Figure 5B) and some positive for CD56 (Figure 5C). Rosette formations areas expressed CD56 (Figure 5D), Syn (Figure 5E) and CD99. Actin was positive focally, and other antibodies were all negative (Table 1).
Pathologic findings
By gross examination, the fusiform mass was 9cm × 6cm × 5cm in size and the covering skin is 8cm x 6cm in size. The cut surface revealed two independent nodules of 5cm × 4.5cm × 3.5cm and 4cm × 2cm × 1.5cm in size, respectively. Both of them were soft in texture, pale-gray and yellowish in colour, and well demarcated without encapsulation.
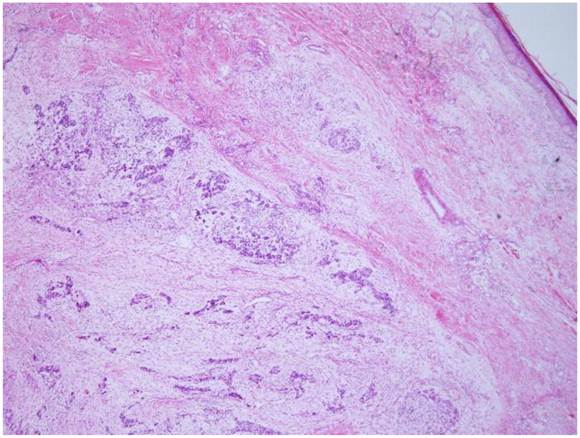
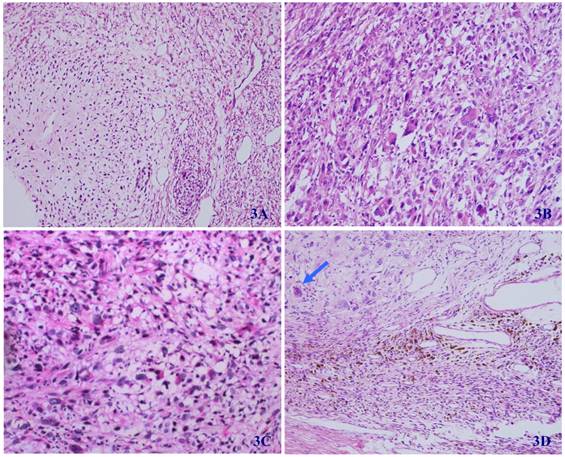
Microscopically, the tumor was well circumscribed without encapsulation and invaded the dermis and subcutis tissues (Figure 1). The tumor was mainly composed of spindle-shaped cells with thin and eosinophilic cytoplasms and atypical nuclei. The tumor cells arranged in bundles and wave pattern in most areas (Figure 2A), and in some areas, arranged radially or in whorls with abundant spoke-like structures (Figure 2B). Myxoid degeneration was visible in some parts of the tumor. Epithelioid differentiation with glandular structures (Figure 2C) appeared in multiple foci, and rosette formation, indicating neuroendocrine differentiation, were also observed (Figure 2D) in these areas. Distinct chondro-differentiation could be seen focally (Figure 3A). In some fields the tumor cells showed eosinophilic cytoplasms with eccentric nuclei, resembling rhabdomyoblasts (Figure 3B), and in the other areas typical lipoblasts were also visible (Figure 3C). Ganglion cells were focally scattered (Figure 3D). Multiple patches of necrosis were observed. Pathological diagnosis was MPNST with heterougenous differentiations, WHO grade IV.
The tumor is located in the dermis without encapsulated (HE X 40).
In most regions, tumor cells are arranged in bundles with red-stained and scant cytoplasm, and spindle-shaped nuclei (2A); tumor cells grew in storiform as that in fibrous histiocytoma (2B); glandular differentiation (2C) and neuroendcrime differentiation (2D) (HE X 100).

In some regions of the tumor, the differentiation of cartilage (3A), rhabdomyosarcoma (3B), liposarcoma (3C), and ganglion cells (arrow) (3D) can be seen (HE X 100).
Immunohistochemical staining for S-100 was positive in most spindle shaped (4A) (X 100) and glandular (4B) (X 100) tumor cells.
Immunohistochemical staining for Cytokeratin was positive in most epithelioid tumor cells and negative in the fusiform region (5 A) (X 100). Vimentin was positive in the majority of spindle -shaped cells, and negative in the epithelioid region (5 B) (X 100). CD56 was positive in tumor tissue with spindle-shaped cells (5C) and neuroendocrine differentiation (5D) (X 100). Synaptophysin was positive in some neuroendocrine diffentiated tumor cells (5E) (X 100).
Follow-up
The patient was postoperatively treated with radiotherapy and chemotherapy, and 3 months later he received traditional Chinese medicine treatment for 15 days. The tumor recurred in the original site 10 months after tumor resection. Imaging examination demonstrated muscular invasion; however, chest CT and whole body bone scan did not reveal any metastasis.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Danid NL, Hiroko O, Otmar DW. et al. WHO classification of tumors—pathology and genetics of tumors of the nervous system; 4th Edition. WHO. 2007:160.
2. Rodriguez FJ, Folpe AL, Giannini C, Perry A. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295-319
3. Kar M, Deo SV, Shukla NK. et al. Malignant peripheral nerve sheath tumors (MPNST)--clinicopathological study and treatment outcome of twenty-four cases. World J Surg Oncol. 2006;22:55-63
4. Yamaguchi U, Hasegawa T, Hirose T. et al. Low grade malignant peripheral nerve sheath tumour: varied cytological and histological patterns. J Clin Pathol. 2003;56:826-830
5. Rekhi B, Abhijeet I, Rajiv K. et al. Malignant peripheral nerve sheath tumors: Clinicopathological profile of 63 cases diagnosed at a tertiary cancer referral center in Mumbai, India. Indian J Pathol Microbiol. 2010;53:611-618
6. Roberto T, Malcolm G, Robert B. et al. Liposarcomatous differentiation in malignant peripheral nerve shenath tumor:a case report. Pathol Res Practice. 2010;206:138-142
7. Rekhi B, Jambhekar NA, Puri A. et al. Clinicomorphologic features of a series of 10 cases of malignant triton tumors diagnosed over 10 years at a tertiary cancer hospital in Mumbai, India. Annals of Diagnostic Pathology. 2008;12:90-97
8. Stasik CJ, Tawfik O. Malignant Peripheral Nerve Sheath Tumor With Rhabdomyosarcomatous Differentiation (Malignant Triton Tumor). Arch Pathol Lab Med. 2006;130:1878-1881
9. Peter P, Jerome B, Thomas K. et al. Divergent differentiation in malignant soft tissue neoplasms: the paradigm of liposarcoma and malignant peripheral nerve sheath tumor. Int J Surg Pathol. 2005;13:19-28
10. Tetsuro Nagasaka, Raymond Lai, Michihiko Sone. et al. Glandular Malignant Peripheral Nerve Sheath Tumor An Unusual Case Showing Histologically Malignant Glands. Arch Pathol Lab Med. 2000;124:1364-1368
11. Li H, Carmen E, Ronald W. et al. Malignant peripheral nerve sheath tumor with divergent differentiation. Arch Pathol Lab Med. 2003;127:e147- e150
12. Suresh TN, Harendra Kumar ML, Prasad CSBR. et al. Malignant peripheral nerve sheath tumor with divergent differentiation. Indian J Patho Microbiology. 2009;52:74-76
13. Janczar K, Tybor K, Józefowicz M. et al. Low grade malignant peripheral nerve sheath tumor with mesenchymal differentiation: a case report. Pol J Pathol. 2011;62:278-281
14. Tetsuji Yamamoto, Rieko Minami, Chiho Ohbayashi. Subcutaneous malignant epithelioid schwannoma with cartilaginous differentiation. J Cutan Pathol. 2001;28:486-491
15. Rodriguez FJ, Scheithauer BW, Abell-Aleff PC. et al. Low grade malignant peripheral nerve sheath tumor with smooth muscle differentiation. Acta Neuropathol. 2007;113:705-709
16. Woodruff JM, Christensen WN. Glandular peripheral nerve sheath tumors. Cancer. 1993;72:3618-3628
17. Samruay S, Sunpetch B, Soottiporn C. Malignant mesenchymoma of median nerve: combined nerve shenath sarcoma and liposarcoma. J surg oncol. 1984;25:119-123
18. A Takeuchi & S Ushigome1. Diverse differentiation in malignant peripheral nerve sheath tumours associated with neurofibromatosis-1: an immunohistochemical and ultrastructural study. Histopathology. 2001;39:298-309
19. Allison KH, Patel RM, Goldblum JR, Rubin BP. Superficial Malignant Peripheral Nerve Sheath Tumor. Am J Clin Pathol. 2005;124:685-692
20. Zhu B, Liu XG, Liu ZJ. et al. Malignant peripheral nerve sheath tumours of the spine: clinical manifestations, classification, treatment, and prognostic factors. Eur Spine J. 2012;21:897-904
21. Ducatman BS, Scheithauer BW. Malignant peripheral nerve sheath tumors with divergent differentiation. Cancer. 1984;54:1049-1057
22. Cashen DV, Parisien RC, Raskin K. et al. Survival data for patients with malignant schwannoma. Clin Orthop Relat Res. 2004;426:69-73
23. Ramanathan RC, Thomas J. M Malignant peripheral nerve sheath tumours associated with von Recklinghausen's neurofibromatosis. Eur J Surg Oncol. 1999;25:190-193
24. Campbell LK, Thomas JR, Lamps LW. et al. Protein gene product 9.5 (PGP 9.5) is not a specific marker of neural and nerve sheath tumors: an immunohistochemical study of 95 mesenchymal neoplasms. Mod Pathol. 2003;16:963-969
25. Shimada S, Tsuzuki T, Kuroda M. et al. Nestin expression as a new marker in malignant peripheral nerve sheath tumors. Pathol Int. 2007;57:60-67
26. Olsen SH, Thomas DG, Lucas DR. Cluster analysis of immunohistochemical profiles in synovial sarcoma, malignant peripheral nerve sheath tumor, and Ewing sarcoma. Mod Pathol. 2006;19:659-668
27. Hui P, Li N, Johnson C. et al. HMGA proteins in malignant peripheral nerve sheath tumor and synovial sarcoma: preferential expression of HMGA2 in malignant peripheral nerve sheath tumor. Mod Pathol. 2005;18:1519-1526
28. Miettinen M. From morphological to molecular diagnosis of soft tissue tumors. Adv Exp Med Biol. 2006;587:99-113
29. Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57:2006-2021
Author contact
![]() Corresponding author: Dr. Aijun Liu, Department of Pathology, the General Hospital of PLA, Beijing 100853, China. Email:liuajcom.cn. Telephone: +86-10-66936253.
Corresponding author: Dr. Aijun Liu, Department of Pathology, the General Hospital of PLA, Beijing 100853, China. Email:liuajcom.cn. Telephone: +86-10-66936253.