Impact Factor ISSN: 1837-9664
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 17; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Special Issues
Introduction
Immunosurveillance in the...
Immune Responses in Cancer
Mechanisms of Immunosuppression...
Immunosuppression mediated by...
Immunosuppression Mediated by...
Immunosuppressive effectors in...
Role of NFκB in tumor...
NFκB inhibition in mice and...
NK cells lyse cancer stem cells,...
Blocking NFκB reverts the...
Induction of split anergy in NK...
Split anergy in NK cells is...
Tumor microenvironment may shape...
Potential role of anergized NK...
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactJ Cancer 2011; 2:443-457. doi:10.7150/jca.2.443 This volume Cite
Review
Tumor Induced Inactivation of Natural Killer Cell Cytotoxic Function; Implication in Growth, Expansion and Differentiation of Cancer Stem Cells
Anahid Jewett ![]() , Han-Ching Tseng
, Han-Ching Tseng
The Jane and Jerry Weintraub Center for Reconstructive Biotechnology, and Division of Oral Biology and Medicine, UCLA School of Dentistry, University of California, Los Angeles, CA 90095, USA
Received 2011-7-2; Accepted 2011-8-5; Published 2011-8-10
Abstract
Accumulated evidence indicates that cytotoxic function of immune effectors is largely suppressed in the tumor microenvironment by a number of distinct effectors and their secreted factors. The aims of this review are to provide a rationale and a potential mechanism for immunosuppression in cancer and to demonstrate the significance of such immunosuppression in cellular differentiation and progression of cancer. To that end, we have recently shown that NK cells mediate significant cytotoxicity against primary oral squamous carcinoma stem cells (OSCSCs) as compared to their more differentiated oral squamous carcinoma cells (OSCCs). In addition, human embryonic stem cells (hESCs), Mesenchymal Stem Cells (hMSCs), dental pulp stem cells (hDPSCs) and induced pluripotent stem cells (hiPSCs) were all significantly more susceptible to NK cell mediated cytotoxicity than their differentiated counterparts or parental cells from which they were derived. We have also reported that inhibition of differentiation or reversion of cells to a less-differentiated phenotype by blocking NFκB or targeted knock down of COX2 in primary monocytes in vivo significantly augmented NK cell function. Total population of monocytes and those depleted of CD16(+) subsets were able to substantially prevent NK cell mediated lysis of OSCSCs, MSCs and DPSCs. Taken together, our results suggest that stem cells are significant targets of the NK cell cytotoxicity. The concept of split anergy in NK cells and its contribution to tissue repair and regeneration and in tumor resistance and progression will be discussed in this review.
Keywords: apoptosis, NFκB, NK, cancer stem cells, differentiation
Introduction
Effective immunosurveillance is important for the prevention of initiation and progression of cancer. Rapid progression of oral leukoplakia to carcinoma was previously observed after immunosuppression [1]. Furthermore, neoplasias of tongue and lip have been widely described in renal transplant patients [2-5], and finally induction of oral cavity cancers was second to liver cancer in patients after bone marrow transplantation [6]. In addition, there is substantial evidence which indicates that immune responses are inhibited by oral tumors, and this may largely be responsible for their induction and progression. This review will focus on the emerging new roles of NK cells in regulation of numbers, resistance and differentiation of cancer stem cells as well as healthy untransformed stem cells. In addition, the significance and the role of anergic NK cells will be discussed in induction of tumor resistance, as well as in shaping the size and differentiation of healthy stem cells.
Immunosuppression and tumor escape from immune recognition are thought to be major factors responsible for the establishment and progression of cancer, however, neither underlying physiological significance nor the exact mechanisms by which immunosuppression occurs are well understood. A number of factors responsible for the suppression of NK cell cytotoxicity in humans has been identified previously [7-12]. It is shown that freshly isolated tumor infiltrating NK cells are not cytotoxic to autologous tumors. Moreover, NK cells obtained from the peripheral blood of patients with cancer have significantly reduced cytotoxic activity [13-16]. In addition, NK cell cytotoxicity is suppressed after their interaction with stem cells [17-19]. In contrast, interaction of NK cells with the resistant tumors leads to a much lower suppression of NK cell cytotoxicity when compared to those dissociated from the NK sensitive target cells [20, 21].
Many mechanisms have been proposed for the functional inactivation of tumor associated NK cells including the over-expression of Fas ligand, the loss of mRNA for granzyme B [8] and decreased CD16 and its associated zeta chain [22] (Table I).
Mechanisms of immune evasion by tumor cells
| Defect | Mechanism | References |
|---|---|---|
| Loss of T cell Recognition | Decreased MHC class I expression/shedding of class I; decreased TCR zeta chain expression, expression of co-stimulatory molecules (B7H1) on tumor cells | [8, 29, 103, 104] |
| Altered NK cell recognition | Decreased expression of NK receptor (NKG2D) ligands, increased MHC class I expression, decreased expression of adhesion molecules, CD16 and zeta chain | [22, 29, 105, 106] |
| Loss of NK cell function | Induction of anergy in NK cells. Activation of upstream transcription factors (NFκB) in differentiated tumors and tumor cell production of inhibitory factors (IL-10, IL-6, IL-1β, PGE2, GM-CSF, IL-8). Decreased IFN-γ secretion by the NK cells when co-cultured with increased NFκB function in tumors | [20, 21, 26, 29, 57, 58, 60, 61, 92, 96-99, 107-110] |
| Enhanced tumor cell survival/resistance to killing | Expression of anti-apoptotic molecules by tumor cells via activation of upstream transcription factors (c-Myc, AP-1, NFκB, STAT3) | [111-116] |
| Increased NK and T cell apoptosis | TNF-α induced apoptosis, Fas ligand expression by tumor cells and membranous vesicles; Fas-mediated apoptosis of responding T cells, expression of DF3 and Muc1 in tumor cells | [10, 56, 117, 118] |
| Inhibition of macrophage/DC maturation and function | Tumor cell production of inhibitory cytokines (VEGF, IL-6, GM-CSF) and activation of STAT3-mediated transcription in DCs. | [58, 62-64, 72, 107, 119-126] |
| Inhibition of T cell chemotaxis to tumor microenvironment | Constitutive STAT3 activation in tumor cells, decreased expression of T cell chemotactic factors (RANTES, IP-10) | [72] |
| Increased recruitment or function of CD14+HLADR-monocytes,Tumor-associated Macrophages, MDSCs, Cancer Associated Fibroblasts, MSCs, Tregs and DCregs | Suppression of cytotoxic immune effectors | [66, 67]; [68, 69]; [70] |
Many metastatic tumor cells exhibit constitutively elevated NFκB activity [23]. We have previously shown that NK resistant primary oral epithelial tumors demonstrate higher nuclear NFκB activity and secrete significant levels of Granulocyte Monocyte-Colony Stimulating Factor (GM-CSF), Interleukin(IL)-1β, IL-6 and IL-8 [24, 25]. Moreover, the addition of Non-steroidal anti-inflammatory drugs (NSAIDs) which inhibit NFκB have the ability to reverse immunosuppression induced by a tobacco-specific carcinogen [26] in addition to their well-established ability to decrease oral dysplasia as well as induction of overt oral cancer in transgenic animals [27]. The significance and exact mechanisms by which NFκB nuclear function in oral tumors modulate and shape the function of key interacting immune effectors is starting to unravel. We have previously shown that inhibition of NFκB in primary OSCCs, or in non-tumorigenic oral keratinocytes (HOK-16B) leads to a significant increase in cytotoxicity and secretion of IFN-γ by the human NK cells [28, 29]. However, it is only now that the underlying significance and the physiological relevance of NFκB modulation in tumors or in primary cells responsible for the alteration of NK cell cytotoxic function is being clarified. Indeed, targeted inhibition of NFκB function in both the intestinal epithelial cells and the myeloid cells was previously shown to result in a significant decrease in the size and the numbers of the tumor cells [30].
In this paper we review the previous studies from our laboratory and those of the others regarding the factors and mechanisms involved in immunosuppression observed in cancer, and furthermore we discuss the emerging view from our laboratory which indicates that the stage of maturation and differentiation of healthy untransformed stem cells as well as transformed tumorigenic cancer stem cells is predictive of their sensitivity to NK cell lysis. In this regard we have previously demonstrated that OSCSCs, which are stem-like oral tumors, are significantly more susceptible to NK cell mediated cytotoxicity; whereas, their differentiated counterpart OSCCs is significantly more resistant [25]. In addition, hESCs and iPSCs as well as a number of other healthy normal stem cells such as hMSCs and hDPSCs were found to be significantly more susceptible to NK cell mediated cytotoxicity than their differentiated counterparts [25]. Based on these results, we propose that NK cells play a significant role in differentiation of the cells by providing critical signals via secreted cytokines as well as direct cell-cell contact. To be conditioned to drive differentiation, NK cells may have to first receive signals either from healthy stem cells or those which have disturbed or defective capabilities to differentiate. In addition, NK cells by targeting other immune inflammatory cells or fibroblasts in the tumor microenvironment may become conditioned to lose cytotoxicity and gain cytokine producing phenotype before they can aid in differentiation of oral squamous cancer stem cells. These alterations in NK cell effector function will ultimately aid in driving differentiation of a population of surviving healthy as well as transformed stem cells. In cancer patients since the majority of NK cells have lost cytotoxic activity, they may eventually contribute rather than halt the progression of cancer by not only driving the differentiation of tumor cells but more importantly, by allowing the growth and expansion of the pool of cancer stem cells.
Immunosurveillance in the Prevention of Cancer
The theory of immunosurveillance was initially set forth by Burnet [31] to indicate that the key thymus dependent effectors were responsible for clearance of tumors [32, 33]. However, since then the concept of immunosurveillance has been expanded to include immunoediting as an important mechanism for the development of cancer [34]; [35]. It was suggested that cancer immunoediting comprises of three phases: elimination, equilibrium and escape [35]. Elimination represents the classical concept of immunosurveillance. However, during equilibrium and escape the interaction and cross signaling between the immune effectors, the tumor cells, and perhaps the effectors of the connective tissue in the tumor microenvironment may result in the generation of tumors which are capable of gradual inactivation and death of the immune effector cells. The final stages of cancer development may result in the induction of less immunogenic tumors in the presence of fewer immune effectors capable of lysing the tumors. Thus, pressures exerted by the tumor cells and immune effectors may eventually shape the microenvironment for the growth, expansion and invasion of tumors. Similarly, some elements of such interactions could also be observed during the interaction of NK cells with healthy non-transformed human stem cells in which case the three phases of interaction may include elimination which marks the decrease in the numbers of stem cells due to the selection of stem cells by the NK cells, induction of tolerance or anergy which denotes the conditioning of NK cells by the stem cells and/or by the other effectors of microenvironment to lose cytotoxicity and support maturation and differentiation of stem cells, and finally the resolution phase which denotes the elimination of anergized NK cells and differentiation of selected stem cells. Similarities and differences between these phases in cancer and during stem cell maturation will be discussed below.
Immune Responses in Cancer
Although limited success in the immunotherapy with cytokines or adaptively transferred effector cells in metastatic melanomas or renal cell carcinomas have previously been observed, Immunotherapy has been found to be ineffective in the treatment of many other cancers [36-38], [39]. The reasons for the failure of known immunotherapeutic modalities in many cancers including head and neck cancers are poorly understood. It has been hypothesized that a widespread paralysis of cytotoxic effectors residing inside the inflammatory infiltrate of advanced cancer patients is the main reason for poor prognosis [40, 41]. Furthermore, freshly isolated tumor infiltrating lymphocytes are not cytotoxic to autologous tumor cells and show a significantly reduced clonogenicity [14-16, 42]. Functional paralysis of cytotoxic cells have also been reported in a variety of cancers, notably breast [9, 10, 43, 44], renal [7], and colon [8]. More importantly, depletion of cytotoxic effectors in the tumor milieu has an unfavorable outcome for survival in cancer patients [43, 45-47]. Indeed, a significantly shorter survival rate is reported for colorectal carcinoma patients with little or moderate NK infiltration as compared to those with extensive infiltration [48]. A five year survival advantage was also seen with higher CD3 positive tumor infiltrating T cells than with lower T cell count in the carcinoma of uterine cervix [47]. Regressing tumor grafts of oral origin contain significantly larger numbers of functional NK and T cells than those associated with the primary tumors [49], while patients with metastasis of head and neck cancers have low NK and T cell activity [50].
Defects in NK, T and DCs have been reported in oral cancer patients. Signaling abnormalities, spontaneous apoptosis and reduced proliferation of circulating T cells, DCs and TILs have been reported in patients with oral cancers [51, 52].
Mechanisms of Immunosuppression by Tumor Cells
Many mechanisms have been proposed for the functional inactivation of tumor associated lymphocytes [7-11] (Table I). Soluble products derived from renal cell carcinoma inhibit proliferative capacity of T cells infiltrating human tumors due to a downregulation of Janus kinase 3 (Jak 3), p56 (lck), p59 (fyn) and zap 70 [7]. Expression of Fas ligand by many human tumor cells including oral tumors has been hypothesized to be a major cause of lymphocyte depletion in the tumor microenvironment [11]. In mice, tumor induced immunosuppression has been associated with a decreased expression of the zeta-chain of the T cell receptor and the loss of mRNA for granzyme B [8]. Indeed, as observed in mice, the frequency of TCR-zeta positive and granzyme positive T cells are decreased in advanced stage head and neck carcinomas in humans, and the restoration of expression during in vitro stimulation suggests the presence of tumor derived suppressive factors [8]. However, caution should be exercised in extending the results from mice cancer model to human disease model in terms of NK cell function due to an array of species specific differences. We have found that freshly isolated NK cells from humans have significant cytotoxic activity against sensitive tumors or healthy stem cells immediately after purification [53], however, freshly isolated NK cells from mice not only lacked any significant cytotoxicity against sensitive tumors such as YAC-1, immediately after purification or after an extended period of incubation, but also they required a much longer period of activation with IL-2 for the acquisition of significant cytotoxicity [54]. In addition, we have found that a greater amount of IL-2 was required for the activation of NK cells from mice to mediate cytotoxicity whereas in humans not only much lower amounts of IL-2 could trigger activation of cytotoxicity but also IL-2 treatment in humans required a very short period of time (hours and not days) for significant increase in cytotoxicity [53, 54]. These differences could be due to the strain of mice used or the source from which purified NK cells were isolated in mice and humans; i.e. spleen vs. peripheral blood or could be due to the inherent differences between human and mice NK cell function. The latter may be likely since species specific differences for the use of key surface receptors for the NK cell function have previously been reported by others [55]. Indeed, it was stated in a previous report that “The extraordinary and unanticipated divergence of human NK cell receptors and MHC class I ligands from their mouse counterparts can in part explain the difficulties experienced in finding informative mouse models of human diseases” Thus, mouse model “has been unimpressive in producing informative and translatable models of human diseases” [55]. It is, however, a very convenient model which can be genetically manipulated, and as such can give a wealth of knowledge if used in parallel with the human disease model confirming the observations obtained in human disease and not vice versa. It is outside the scope of this review to provide a complete description of species specific differences of NK cell receptors. Interestingly, most studies reported for tumor mediated NK cell immunosuppression was conducted using human NK cells. Therefore, decreased CD16 and its associated zeta chains were observed in tumor infiltrating NK cells of patients with cancer [22] (Table I), thus, correlating in phenotype with our in vitro inactivated NK cells by sensitive tumors [20, 56]. Overall both secreted factors and direct cell-cell contact during the interaction of immune effectors with the tumor cells in patients were shown to be responsible for the suppression of immune effector function.
Immunosuppression mediated by secreted factors
Many secreted factors such as PGE2, Interleukin (IL)-6, Vascular Endothelial Growth Factor (VEGF) and Granulocyte Monocyte Colony Stimulating Factor (GM-CSF) are known to induce Immunosuppression in the tumor microenvironment. Immunosuppression linked to enhanced PGE2 synthesis has been documented in many human cancers [57]. Freshly excised human head and neck cancers demonstrated elevated levels of PGE2, transforming growth factor- β1 (TGFβ1) and interleukin-10 (IL-10) secretion which are known to upregulate the expression of Killer Immunoglobulin-like Receptors (KIRs) on the surface of NK, T and DCs and block immune effector function [58]. Furthermore, metastatic head and neck cancers released higher levels of above-mentioned inhibitory factors and lower levels of immune activating factors IFN-γ and IL-2 than did their corresponding primary tumors [58]. PGE2 overproduction in tumor microenvironment was also shown to lead to dendritic cell (DC) abnormalities [59].
IL-6, another immunosuppressive factor is secreted constitutively by oral squamous carcinomas [60], and is found to be elevated in oral cancer patients [61]. IL-6 is known to interfere with IFN-γ signaling by the induction of Th2 differentiation [62]. In support of a role for IL-6 in mediating immune evasion of tumor cells, Menetrier-Caux et al showed that conditioned medium from human renal cell carcinoma cell lines blocked the differentiation of CD34+ bone marrow cells into immature DCs, and this inhibitory effect could be blocked with antibodies against IL-6 [63].
Other factors which have been implicated in immunosuppression in cancers are angiogenic factors such as VEGF [64] and cytokines such as G-CSF and GM-CSF. The finding that neutralizing antibodies to VEGF or GM-CSF could partially reverse the inhibitory effects of tumor cell supernatant on DC maturation demonstrated that these factors could interfere with DC differentiation and function [64]. Increased numbers of immature DCs were found in the peripheral blood of cancer patients with elevated levels of circulating VEGF [64].
Immunosuppression Mediated by Direct Cell-Cell Contact
Contact-dependent immune suppression can occur by engagement of MHC class I molecules on CD8+CD28- suppressor cells with immunoglobulin-like transcript (ILT2 and ILT4) inhibitory receptors on DCs. Blocking of both MHC Class I and ILTs by specific antibodies can reverse immunosuppression [65]. Similarly binding of c-type lectin receptors or Killer Immunoglobulin-like Receptors (KIRs) to MHC Class I ligands inhibit NK cell function (Wagtmann et al, 1995; D'Andrea et al, 1995; Lazetic et al, 1996; Carretero et al, 1997;Brooks et al, 1997). In addition, the expression of co-stimulatory molecules, such as B7H1 on tumor cells and inhibitory DCs and T cells can inhibit T cell activation and proliferation (Cuiel, et al., 2003).
Immunosuppressive effectors in tumor microenvironment
The tumor microenvironment consists of a number of heterogeneous cell populations with ability to suppress and limit the function and survival of cytotoxic immune effectors. Patients with cancer often have higher numbers of immature CD14+HLADR- monocytes [66, 67]. Tumor associated Macrophages (TAMs) were previously shown to significantly influence and limit immune activation in the tumor microenvironment [68, 69]. In addition, Myeloid Derived Suppressor Cells (MDSCs) which are comprised of a number of distinct cell populations of myeloid origin and whose roles in immunosuppression have received significant attention in recent years are major cells capable of suppressing the cytotoxic function of T and possibly NK cells [67]. T cell dysfunction is shown to be induced by MDSCs by the secretion of IL-10, TGF-β, reactive oxygen species (ROS), arginase and Nitric Oxide synthase (NOS). T regulatory (Treg) and DC regulatory (DCreg) cells were also recently shown to have significant immunosuppressive roles in the tumor microenvironment [67]. Perhaps one of the most interesting observations regarding the immunosuppressive effectors is the identification of Cancer Associated Fibroblasts (CAFs) and Mesenchymal Stem Cells (MSCs) as two potential tumor promoters. Fibroblasts from tumor tissues demonstrate an activated phenotype and have the ability to secrete many immunosuppressive factors such as TGF-β and VEGF [70]. We have also found that fibroblasts, as well as MSCs and CD14+HLA-DR- monocytes irrespective of their surface expression of CD16 are significantly more susceptible to NK cell mediated cytotoxicity [71], therefore, these cells may condition NK cells to become anergic (please see below). Indeed, in oral epithelial tumors the majority of recruited immune effectors are usually found in the connective tissue area where through cell-cell interaction with the immunosuppressive cells such as fibroblasts, monocytes-macrophages and to a lesser extent T and B cells [71] can condition NK cells to lose cytotoxicity and gain in cytokine secretion capabilities, resulting in differentiation and resistance of oral epithelial tumors (please see below).
Role of NFκB in tumor resistance
Although each one of the secreted factors individually can be responsible in part for the resistance of tumors, previous data obtained from different laboratories [29, 72] indicated that targeting transcription factors may be more effective in decreasing resistance of tumors to immune mediated cytotoxicity. Many tumor cells exhibit constitutively elevated NFκB activity [23]. Human leukemias and lymphomas as well as human solid tumors exhibit constitutively activated NFκB in the nucleus [23].
We have shown previously that NFκB nuclear function in a primary Oral tumor OSCCs and in a non-tumorigenic oral cells (HOK-16B) as well as in an established tumor line, HEp-2 cells known to be Hela contaminant [29, 73-75], modulates and shapes the function of interacting immune effectors [28, 29]. It is believed that HEp-2 cells are Hela contaminants since cells of these lines are shown to contain Hela marker chromosomes (ATCC). Since knock down of NFκB was shown to increase the function of immune inflammatory cells in diverse cell types (please see below) it is not surprising to find similar patterns of immune activation in both oral and non-oral derived cell lines, even in those which have been derived from contaminants such as Hela cells. In addition, since the majority if not all cells increase NFκB during their activation and differentiation, a knock down of NFκB is likely to revert the cells, irrespective of their cellular origin, to their less differentiated phenotypes resulting in the potential activation of the immune effectors in order to aid in their differentiation [25]. Therefore, inhibition of NFκB in oral tumors resulted in a significant activation of human NK cell cytotoxic function and increased IFN-γ secretion [25]. Similarly, inhibition of NFκB by Sulindac increased the functional activation of NK and enhanced anti-tumor cytotoxic activity [28, 29]. Inverse modulation of IFN-γ and IL-6 cytokine secretion was seen in co-cultures of NK cells with NFκB knock down OSCCs and HEp2 cells indicating that blocking NFκB in these cells serves to switch the balance from Th2 type responses to more of a Th1 type response [25, 28].
NFκB inhibition in mice and humans activate immune inflammatory functions
In agreement with our studies, targeted deletion of Iκκ-β in epidermis of mice has previously been shown in one study to lead to inflammatory skin manifestations similar to that seen in patients with Incontinentia Pigmenti (IP) [76]. Elevated levels of cytokines and chemokines have also been demonstrated in the epidermis of patients and animals with Iκκγ and Iκκβ deletions [76, 77]. Blocking of NFκB function by deleting Iκκ-β in intestinal epithelial cells also dramatically decreased the rate of tumor formation without affecting the size of the tumors in colitis-associated cancer model [78, 79]. Moreover, deleting Iκκ-β in myeloid cells in the same model system resulted in the decrease in tumor size. These studies underscore the significance of the cross talk between different subsets of immune effectors with the epithelial cells in induction and progression of the intestinal tumors.
Mice with a keratinocyte-specific deletion of Iκκ-β demonstrated decreased proliferation of epidermal cells and developed TNF-α dependent inflammatory skin disease [76]. In contrast, in other studies in which NFκB function was blocked in dermal keratinocytes by a mutant IκB-α an increased proliferation and hyperplasia [80] and eventual development of cutaneous squamous cell carcinomas of skin were seen if mice were allowed to survive and reach adulthood [81, 82]. It is of interest to note that in these studies with diverse functional outcomes in keratinocytes, blocking TNF-α function resulted in the prevention of both the neoplastic transformation and the inflammatory skin disease [76, 82] . Thus, it appears that TNF-α is the primary factor mediating the pathological processes in both of these studies. Elevated numbers of immune inflammatory cells recruited to the site of epidemis are likely responsible for the increased secretion of TNF-α. Indeed, we have demonstrated that synergistic induction of TNF-α could be observed when NFκB knock down oral tumors were cultured with either PBMCs or NK cells [28].
NK cells lyse cancer stem cells, as well as hESCs, hiPSCs, hMSCs and hDPSCs but not their differentiated counterparts
Increased NK cell cytotoxicity and augmented secretion of IFN-γ were observed when NK cells were co-incubated with OSCSCs which released significantly lower levels of GM-CSF, IL-6 and IL-8 and demonstrated decreased expression of phospho-Stat3, B7H1 and EGFR, and much lower constitutive NFκB activity when compared to differentiated OSCCs [25]. More importantly, OSCSCs expressed CD133 and CD44bright oral stem cell markers [25]. Increase in IFN-γ secretion was correlated with a decrease in secretion of IL-6 in co-cultures of NK cells with OSCSCs as compared to those co-cultured with OSCCs. Therefore, from these results a specific profile for differentiated NK resistant oral tumors emerged which demonstrated increased GM-CSF, IL-6 and IL-8 secretion in the context of decreased IFN-γ secretion during their interaction with the NK cells. In contrast, co-cultures of cancer stem cells with NK cells demonstrated increased IFN-γ in the context of lower GM-CSF, IL-6 and IL-8 secretion [25, 71]. In addition, three brain tumor stem cells which were previously characterized [83-85] were found to be significantly more susceptible to NK cell mediated cytotoxicity when compared to their differentiated counterparts which were significantly more resistant (manuscript submitted). Since OSCSCs and brain stem cells were significantly more susceptible to NK cell mediated cytotoxicity we reasoned that healthy, non-transformed primary stem cells may also be susceptible to NK cell mediated cytotoxicity. We demonstrated previously that NK cells lysed hMSCs, hDPSCs and hESCs significantly. All different types of stem cells became resistant to NK cell mediated cytotoxicity once they were differentiated [25]. In addition, higher sensitivity of hiPSCs to NK cell mediated lysis was also observed when compared to parental line from which they were derived. Increased lysis of cancer stem cells or non-transformed healthy stem cells may be attributed to the use of allogeneic NK cells, however, our previous work using autologous NK cells exhibited similar levels of cytotoxicity against DPSCs when compared to lysis by allogeneic NK cells [71]. Taken together these results indicated that undifferentiated cells were targets of both allogeneic and autologous NK cells. Thus, the stage of differentiation of the cells is predictive of their susceptibility to NK cell mediated cytotoxicity.
Blocking NFκB reverts the cells to more of an undifferentiated phenotype and increases NK cell mediated cytotoxicity against oral tumors
Since the degree of differentiation in the cells is predictive of their sensitivity to NK cell mediated cytotoxicity, we reasoned that blocking NFκB in the cells may de-differentiate and consequently revert the cells to more of undifferentiated phenotype, resulting in their increased susceptibility to NK cell mediated cytotoxicity. Indeed, blocking NFκB in oral tumors was also found to increase CD44 surface receptor expression, which is one of the hallmarks of stem cells (manuscript in prep). In addition, the profiles of cytokines secreted in the co-cultures of NK cells or PBMCs with NFκB knock down tumors resembled those secreted in the co-cultures of NK cells with the stem cells [25, 28, 71]. Since tumorigenic and non-tumorigenic human oral keratinocytes acquire sensitivity to NK cell mediated lysis when NFκB is inhibited, it is likely that this phenomenon is not specific to cancer or oral keratinocytes, and it may occur in other healthy non-transformed cell types. Indeed, when human primary monocytes were differentiated to dendritic cells they too became more resistant to NK cell mediated cytotoxicity [25]. Moreover, knock down of COX2 in primary mouse monocytes [25], or in mouse embryonic fibroblasts (manuscript in prep), resulted in the reversion or de-differentiation of the monocytes and fibroblasts respectively, and the activation of NK cell cytotoxicity. Indeed, it is likely that any disturbance in cellular differentiation may predispose the cells to NK cell mediated cytotoxicity. Since STAT3 is an important factor increased during differentiation, blocking STAT3 is also critical in the activation of immune effectors [72]. In support of a critical role of STAT3 in immune evasion of tumor cells in humans, we and others have recently shown that glioblastoma multiforme (GBM) tumors display constitutive activation of STAT3 (Cacalano and Jewett, unpublished observation) [86], and poorly induce activating cytokines and tumor-specific cytotoxicity in human peripheral blood mononuclear cells (PBMCs) and NK cells. Ectopic expression of dominant-negative STAT3 in the GBM cells increased lysis of the tumor cells by the immune effectors and induced production of IFN-γ by the interacting immune effectors (manuscript in prep).
Since NFκB is shown to regulate IL-6 secretion in OSCCs, HOK-16B and HEp2 cells and secreted IL-6 in tumors is known to activate STAT3 expression and function, increase in NFκB nuclear function could in turn induce STAT3 activation and result in a significant resistance of tumors to NK cell mediated cytotoxicity. Therefore, targeted knock down of STAT3 or signaling pathways upstream of STAT3, such as NFκB, may de-differentiate the cells and predispose the cells to NK cell mediated cytotoxicity.
Induction of split anergy in NK cells after their co-culture with sensitive but not resistant tumors and after the triggering of CD16 on IL-2 treated NK cells
We have previously shown that K562, an NK sensitive tumor, causes loss of NK cell cytotoxicity and induces cell death in a small subset of NK cells [20, 21]. On the other hand NK resistant tumors such as RAJI cells induce much less anergy or loss of NK cell cytotoxicity [20, 21]. Furthermore, following NK cell cultures with sensitive tumor-target cells but not resistant tumors, the target binding NK cells undergo phenotypic and functional changes. Target cell inactivated NK cells express CD16-CD56dim/- CD69+ phenotype [20, 21]. This phenotype has also been observed in several disease manifestations including HIV infection [87]. Significant downmodulation of CD16 receptor expression and decreased NK cell cytotoxic function were also seen in several cancer patients including those of the oral and ovarian cancer patients [88, 89]. In addition, down-regulation of CD16 surface receptors on NK cells was also observed when NK cells were treated with CA125 isolated from ovarian tumor cells [90]. The decrease in CD16 surface receptors was accompanied by a major decrease in NK cell killing activity against K562 tumor cells [90]. These observations suggested that CD16 receptors may play an important role in target cell induced loss of NK cell cytotoxicity. Indeed, CD16:Ig fusion proteins are shown to bind to a variety of tumor-target cells indicating the existence of specific ligands for CD16 receptors on tumor cells [91]. Furthermore, we have previously shown that the triggering of CD16 on untreated or IL-2 treated NK cells was found to result in downmodulation of CD16 receptors and in a great loss of cytotoxicity in NK cells. In addition, a subset of NK cells was programmed to undergo apoptosis [20, 21, 56, 92]. Cell death of NK cells was shown to be regulated, in part, by endogenously secreted TNF-α from the NK cells [56]. Previous studies by other groups have also shown that IL-2 activated NK cells undergo cell death following cross-linking of the CD16 receptor [93, 94]. Thus, we have coined the term “split anergy” for the responses observed by NK cells after their interaction with sensitive target cells or after the triggering of CD16 receptors by the antibody in combination with IL-2 treatment [20, 21, 53, 56, 95]. Indeed, three subpopulations of NK cells; namely Free, Binder and Killer NK cells with varying degrees of loss of cytotoxicity were identified after the formation of conjugates with K562 targets [92, 96-99]. Free cells which did not bind or form conjugates with target cells were inactivated less, or exhibited the most cytotoxicity, whereas both Binder and Killer subsets exhibited significant loss of cytotoxicity. In contrast, Binder and Killer subsets but not Free NK subset secreted significant levels of cytokines and exhibited CD16-CD56dim/-CD69+ phenotype [92, 96-99]. Treatment of NK cells with IL-2 and anti-CD16mAb also induced split anergy by significantly decreasing the NK cell cytotoxicity while increasing the cytokine secretion capabilities of NK cells. Furthermore, NK cells exhibited CD16-CD56dim/-CD69+ phenotype after treatment with the combination of IL-2 and anti-CD16mAb [53, 56, 95, 100]. Loss of cytotoxicity in NK cells was significantly exacerbated when NK cells were either treated with F(ab)'2 fragment of anti-CD16 mAb or treated with a combination of MHC-Class I and anti-CD16 mAb while the same treatments resulted in an increased secretion of cytokines [53, 100]. These results suggested that receptor signaling in NK cells in the presence of IL-2 is likely to result in a decrease in NK cell cytotoxicity while increasing secretion of cytokines by the NK cells. Therefore, three distinct functional outcomes could be observed in NK cells which have either interacted with sensitive tumor-target cells or were treated with anti-CD16 mAb in the presence of IL-2 treatment to induce split anergy, namely; 1-Loss of cytotoxicity, 2-gain in the ability to secrete cytokines and 3- death in a small subset of NK cells.
Split anergy in NK cells is induced by monocytes and those depleted of CD16+ subsets of monocytes
When MSCs or DPSCs were cultured with either viable or irradiated monocytes before they were exposed to IL-2 treated NK cells a significant decrease in NK cell mediated cytotoxicity could be observed against MSCs or DPSCs. Interestingly, significant lysis of MSCs and DPSCs by untreated NK cells was also significantly and reproducibly blocked by the addition of monocytes [71]. To determine whether CD16- subset of monocytes were also able to inhibit the cytotoxic function of NK cells in a 3 way interaction with the stem cells we used both the unsorted, and those sorted to remove CD16+ subsets in a 3 way killing assay with the NK cells. Both the total populations of monocytes and CD16- subsets were capable of inducing inhibition of NK cell cytotoxicity against stem cells [71]. We then determined whether decreased lysis of stem cells by NK cells was due to a competitive lysis of monocytes by the NK cells. We confirmed that monocytes were also lysed by the NK cells significantly. Furthermore, when we co-cultured stem cells with monocytes and sorted to remove the monocytes from the stem cells before assessing the killing function of NK cells, we could still observe significant inhibition of NK cell mediated lysis, arguing against the protection of stem cell lysis by NK cells being solely on the bases of competitive lysis of monocytes [71]. Therefore, even though lysis of monocytes by the NK cells may in part contribute to the prevention of NK cell lysis of stem cells, interaction of monocytes with stem cells can also provide resistance of stem cells against NK cell cytotoxicity. Decrease in NK cell lysis of MSCs and DPSCs by monocytes was paralleled with a significant induction of IFN-γ. Indeed, when MSCs or DPSCs were cultured with IL-2 treated NK cells alone we could observe significant induction of IFN-γ secretion. However, the highest increase was seen when IL-2 treated NK cells were cultured with MSCs or DPSCs in the presence of monocytes. Therefore, although decreased killing of stem cells by the NK cells could be observed in the presence of monocytes, synergistic secretion of IFN-γ by the NK cells in the presence of monocytes and stem cells could be observed, indicating an inverse relationship between cytotoxicity and IFN-γ secretion (split anergy). This was similar to the profiles which we had seen when NK cells were treated with IL-2 and anti-CD16 antibody in which significant decrease in cytotoxicity of NK cells could be observed in parallel with increased secretion of IFN-γ (split anergy) [56].
Tumor microenvironment may shape the function and phenotype of the NK cells
The above observations prompted us to speculate regarding the significance of interaction of monocytes with NK cells and stem cells. It is plausible that monocytes may serve as shields against NK cell lysis of stem cells. Similar to anti-CD16 antibody mediated effect on IL-2 treated NK cells, monocytes too can shield stem cells from killing by the NK cells by increasing the total IFN-γ release by the NK cells while decreasing the cytotoxic function of NK cells (split anergy), resulting in an increased protection and differentiation of stem cells. Indeed, monocytes also increased TNF-α, IL-6 and VEGF secretion in the co-cultures of stem cells with NK cells which could augment NFκB and increase differentiation of stem cells. The shielding effect of monocytes could be a more generalized function of other effectors since NK cells can also target fibroblasts and to a much lesser extent the T and B cells [71]. Whether MDSCs or PMNs could also be targeted by the NK cells awaits future investigation. This may have significant implications regarding the role of NK cells in not only limiting inflammation, but also the significance of other immune effectors in shielding and limiting the cytotoxic function of NK cells against cancer or healthy stem cells in order to raise maximally the secretion of key cytokines for speedy and optimal differentiation of stem cells during inflammation. This is precisely what is observed in cancer patients in whom global decrease in NK, cytotoxic T cells and monocytes have all been reported [12].
Potential role of anergized NK cells in differentiation and regeneration of tissues
Even though conditioning of NK cells to support differentiation of cells is discussed in the context of tumors, induction of split anergy in NK cells, we believe, is an important conditioning step responsible for the repair of tissues during pathological processes irrespective of the type of pathology. In tumors since the generation and maintenance of cancer stem cells is higher, the majority if not all of the NK cells may be conditioned to support differentiation and repair of the tissues and as such the phenotype of NK cells in tumor microenvironment as well as in the peripheral blood may resemble that of the anergic NK cells, i.e., decreased NK cell cytotoxicity, acquisition of CD16-/dimCD56-/dimCD69+ phenotype and augmented ability to secrete inflammatory cytokines (Fig. 2). Of course, the degree of the loss of NK cell cytotoxicity may be directly proportional to the load of cancer stem cells. Therefore, our results suggest two very important functions for the NK cells. One function is to limit the number of stem cells by selecting those with a greater potential for differentiation for the repair of the tissues and second to support differentiation of the stem cells and subsequent regeneration of the tissues. To achieve these tasks NK cells have to acquire two different phenotypes and be conditioned to carry out both functions successfully. CD16+CD56+/dimCD69- subsets of NK cells are cytotoxic and will mediate cytotoxicity depending on which sensitive targets they encounter first. In respect to the oral squamous cell carcinomas since the majority of immune effectors can be found at the connective tissue area the chances are that they may first encounter and interact with either the other immune effectors or the effectors of connective tissue such as fibroblasts. However, there is also the possibility that NK cells may first encounter the stem cells at the base of the epithelial layer, in which case by eliminating their bound stem cells, they too can become anergized (Fig. 1). Surprisingly, allogeneic CTLs were also found to target Glioblastoma stem-like cells and not their differentiated counterparts (Veronique Quillien, personal communication). By eliminating a subset of stem cells or after their interaction with other immune inflammatory cells or effectors of connective tissue NK cells could then be in a position to support differentiation of selected population of stem cells since they will be conditioned to lose cytotoxicity, induce cytokine and growth factor secretion and gain the CD16-/dimCD56dimCD69+ phenotype (Fig. 2). It is interesting to note that all of the immune effectors isolated from oral gingival tissues of healthy as well as diseased gingivae have CD69+ phenotype, with the exception that the numbers of immune effectors are much less in the healthy oral gingival tissues when compared to diseased tissues (manuscript in prep). Therefore, our results suggest two very important functions for the NK cells. One function is to kill and the other function is to be conditioned to support differentiation for the repair and regeneration of the tissues.
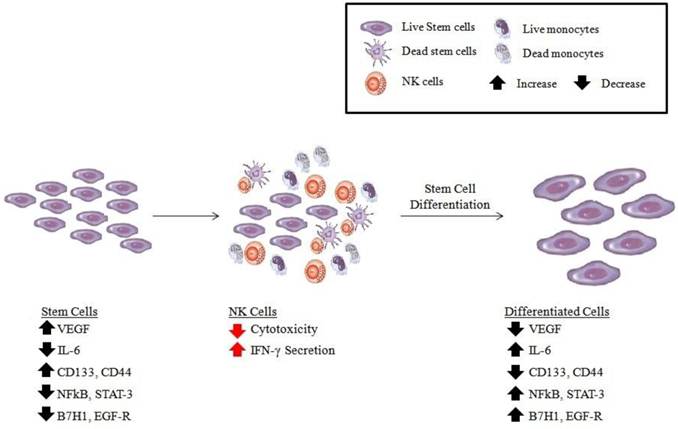
Schematic representation of hypothetical model of oral cancer stem cell differentiation by NK cells. Based on our previous results [25, 71], OSCSCs initially express the phenotype listed in the figure (left panel). Upon conditioning of NK cells to lose cytotoxicity and gain in cytokine secretion and expressing CD16-CD56dim/-CD69+ surface phenotype (middle panel), either by immune inflammatory cells such as monocytes (for more detail see figure 2) or stem cells, the phenotype of stem cells is modified as shown in the right panel, and they become differentiated to support tissue regeneration.
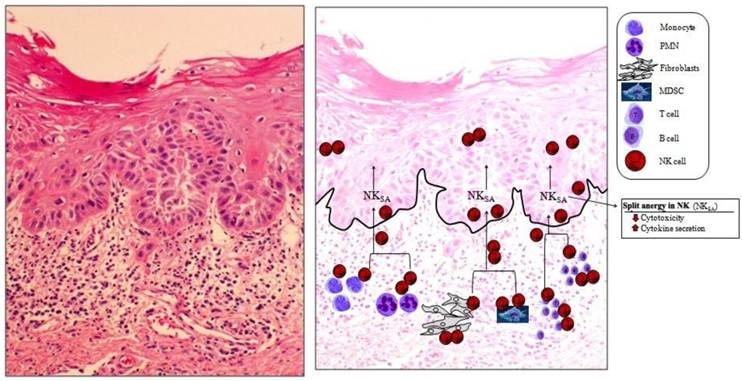
Immune inflammatory cells are mainly concentrated in the connective tissue area right beneath the epithelial layer of OSCC. The slides from OSCC were prepared and stained with H&E (left panel). At the right panel the hypothetical model of NK cell conditioning in the tumor microenvironment is over imposed on the actual slide shown on the left panel. On the left panel significant infiltration of immune effectors right beneath the epithelial layer can be seen in the connective tissue area where the immune inflammatory cells are likely to condition NK cells to lose cytotoxicity and to support differentiation of epithelial cells. At the right panel two hypothetical mechanisms for conditioning of NK cells to support differentiation of stem cells are shown. In one scenario, NK cells, may first encounter and interact with either the other immune effectors as listed in the figure or the effectors of connective tissue such as fibroblasts to undergo split anergy (NKSA). In a second scenario, NK cells may also directly encounter the stem cells at the base of the epithelial layer, in which case by eliminating their bound stem cells, they too can become anergized to support differentiation of other stem cells. Both mechanisms may be operational in the tumor microenvironment.
In vivo physiological relevance of above-mentioned observations could be seen in a subpopulation of NK cells in peripheral blood, uterine and liver NK cells which express low or no CD16 receptors, have decreased capacity to mediate cytotoxicity and is capable of secreting significant amounts of cytokines [101, 102]. In addition, 70% of NK cells become CD16 dim or negative immediately after allogeneic or autologous bone marrow transplantation [101]. Since NK cells lose their cytotoxic function and gain in cytokine secretion phenotype and down modulate CD16 receptors after their interaction with tumor cells or the stem cells [20, 56], it is tempting to speculate that in vivo identified CD16- NK cells and in vitro tumor induced CD16- NK cells have similar developmental pathways since they have similar if not identical functional properties.
The ultimate proof of concept in support of this model was recently obtained in our laboratory. We observed that anergized NK cells directly were responsible for the increased differentiation and resistance of a number of different stem cells including cancer stem cells against cytotoxic effectors (manuscript submitted). In addition, we now have evidence which supports the notion that the induction of anergy in NK cells is an active process which is induced by the triggering of CD16 receptor on the NK cells and is not due to degranulation and exhaustion of cytotoxic granules (manuscript in prep).
Our work collectively suggests that anergized NK cells are as important as the non-anergized NK cells in their effector functions. NK cells are not only important for the removal and shaping of the size of the stem cells but also their differentiation, and the ultimate regeneration of the new tissues. The task of NK cells in this regard goes above and beyond their most appreciated function of being the effectors of first line defense against viral infection and malignancies. They too can be effectors of differentiation and tissue regeneration.
Conclusion
Recent advances in our understanding of anti-tumor immune responses and cancer biology have revealed a complex dynamic interaction between the immune effectors and the tumor cells. Effectors of the immune system are known to shape the tumor cells (immunoediting) and to select for cancers with reduced immunogenicity and enhanced capacity to actively induce immunosuppression. However, the same effector mechanisms are likely responsible for the selection of healthy stem cells with enhanced capacity to induce immunosuppression for the ultimate goal of the regeneration of damaged or disturbed tissues and the resolution of inflammation. Much work has been done to identify strategies by which tumor cells evade the function of immune system. Altered expression of MHC molecules which block recognition and activation of T and NK cells are examples of mechanisms by which tumor cells evade the function of immune system. In addition, tumor cells by releasing immunosuppressive factors such as Fas, VEGF, IL-6, IL-10, TNF-α, GM-CSF and IL-1β, induce T and NK cell apoptosis, block lymphocyte homing and activation, and dampen macrophage and dendritic cell function. However, the same effector functions are also important in tissue repair. Furthermore, progress has been made in identification of the upstream mechanisms which control the expression of immunosuppressive factors in tumor cells. Two key control elements, NFκB and STAT3 were identified and shown to coordinately regulate the production of multiple tumor-derived immunosuppressive molecules and play a pivotal role in tumor cell immune suppression. The potential for these two signaling modules to repress immune responses is underscored by the finding that the pathways interact and may even amplify each other. One model for NFκB-STAT3-mediated immunosuppression suggests that NFκB-induced IL-6 expression activates STAT3 in tumor cells and modulates the production of inhibitory cytokines and chemokines resulting in decreased T cell infiltration and activation. However, the same mechanisms are likely to be important for normal tissue regeneration and induction of resistance to NK and T cell mediated cytotoxicity.
Based on the accumulated work presented in this review we suggest that NK cells may have two significant functions; one that relates to the removal of stem cells that are either defective or disturbed or in general more in numbers than are needed for the regeneration of damaged tissue. Therefore, they may select stem cells that are competent and are able to achieve the highest ability to regenerate tissues. Alternatively, or in addition, NK cells may lyse other effectors in the connective tissue area in order to not only decrease inflammation but also to be conditioned to promote tissue regeneration. The second important task for NK cells is therefore, to support differentiation and promote tissue regeneration after altering their phenotype to cytokine secreting cells (Fig. 2). This process will not only remove cells that are damaged and have flaws in the differentiation process, but also it will ensure the regeneration of tissues and the resolution of inflammation. Thus, any disturbance in the NK cell function may result in a chronic inflammatory process, causing continual tissue damage and recruitment of immune effectors to aid in tissue regeneration.
The inability of patient NK cells to contain cancer stem cells due to the flooding of NK cells by proliferating cancer stem cells and conversion of NK cells to cytokine secreting cells may likely be one mechanism by which cancer may progress and metastasize. Therefore, there should be two distinct strategies by the NK cells to eliminate tumors, one which targets stem cells and the other which targets differentiated cells. Since cancer stem cells were found to be more resistant to certain chemotherapeutic drugs but sensitive to NK cell mediated killing while differentiated oral tumors were more resistant to NK cell mediated killing but relatively more sensitive to chemotherapeutic drugs, combination therapy should be considered for the elimination of both undifferentiated and differentiated tumors. In addition, since a great majority of patient NK cells have modified their phenotype to support differentiation of the cells, they may not be effective in eliminating the cancer stem cells. Therefore, these patients may benefit from repeated allogeneic NK cell transplantation at the site of the tumor for elimination of cancer stem cells. In this regard depletion of immunosuppressive effectors in the tumor microenvironment, which condition NK cells to lose cytotoxicity, via radiation or chemotherapeutic drugs should in theory provide a better strategy for successful targeting of tumors by the NK cells.
Acknowledgements
This work was in part supported by RO1-DE12880 from NIDCR-NIH.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Hernandez G. et al. Rapid progression from oral leukoplakia to carcinoma in an immunosuppressed liver transplant recipient. Oral Oncol. 2003;39(1):87-90
2. Bilinska-Pietraszek E. et al. [A case of tongue neoplasm in a 15-year old patient treated with immunosuppressants for renal insufficiency]. Otolaryngol Pol. 2001;55(1):95-7
3. de Visscher JG. et al. Squamous cell carcinoma of the lower lip in renal-transplant recipients. Report of six cases. Int J Oral Maxillofac Surg. 1997;26(2):120-3
4. King GN. et al. Increased prevalence of dysplastic and malignant lip lesions in renal-transplant recipients. N Engl Med J. 1995;332(16):1052-7
5. van Zuuren E.J. et al. Carcinoma of the lip in kidney transplant recipients. J Am Acad Dermatol. 1998;38(3):497-9
6. Bhatia S. et al. Solid cancers after bone marrow transplantation. J Clin Oncol. 2001;19(2):464-71
7. Kolenko V. et al. Tumor-induced suppression of T lymphocyte proliferation coincides with inhibition of Jak3 expression and IL-2 receptor signaling: role of soluble products from human renal cell carcinomas. Immunol J. 1997;159(6):3057-67
8. Mulder W.M. et al. cell receptor-zeta T and granzyme B expression in mononuclear cell infiltrates in normal colon mucosa and colon carcinoma. Gut. 1997;40(1):113-9
9. Camp B.J. et al. In situ cytokine production by breast cancer tumor-infiltrating lymphocytes. Ann Surg Oncol. 1996;3(2):176-84
10. Gimmi C.D. et al. Breast cancer-associated antigen, DF3/MUC1, induces apoptosis of activated human T cells. Nat Med. 1996;2(12):1367-70
11. Bennett M.W. et al. The Fas counterattack in vivo: apoptotic depletion of tumor-infiltrating lymphocytes associated with Fas ligand expression by human esophageal carcinoma. Immunol J. 1998;160(11):5669-75
12. Jewett A, Head C, Cacalano N.A. Emerging mechanisms of immunosuppression in oral cancers. J Dent Res. 2006;85(12):1061-73
13. Tanaka H. et al. Nonsteroidal anti-inflammatory drugs differentially regulate cytokine production in human lymphocytes: up-regulation of TNF, IFN-gamma and IL-2, in contrast to down-regulation of IL-6 production. Cytokine. 1995;7(4):372-9
14. Miescher S. et al. Preferential clonogenic deficit of CD8-positive T-lymphocytes infiltrating human solid tumors. Cancer Res. 1988;48(24 Pt 1):6992-8
15. Qin J, Han B, Pang J. [The relationship between TIL from human primary hepatic carcinoma and prognosis]. Zhonghua Yi Xue Za Zhi. 1997;77(3):167-70
16. Han X. et al. Tumor lymphocytes in patients with advanced ovarian cancer: changes during in vitro culture and implications for immunotherapy. Gynecol Oncol. 1997;65(3):391-8
17. Aggarwal S, Pittenger M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105(4):1815-22
18. Selmani Z. et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress lymphocyte T and natural killer function and to induce CD4+CD25highFOXP3+ regulatory T cells. Stem Cells. 2008;26(1):212-22
19. Spaggiari G.M. et al. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood. 2008;111(3):1327-33
20. Jewett A, Bonavida B. Target-induced inactivation and cell death by apoptosis in a subset of human NK cells. Immunol J. 1996;156(3):907-15
21. Jewett A, Bonavida B. Target-induced anergy of natural killer cytotoxic function is restricted to the NK-target conjugate subset. Cell Immunol. 1995;160(1):91-7
22. Nakagomi H. et al. Decreased expression of the signal-transducing zeta chains in tumor-infiltrating T-cells and NK cells of patients with colorectal carcinoma. Cancer Res. 1993;53(23):5610-2
23. Rayet B, Gelinas C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene. 1999;18(49):6938-47
24. Teruel A. et al. Potential contribution of naive immune effectors to oral tumor resistance: role in synergistic induction of VEGF, IL-6, and IL-8 secretion. Cancer Immunol Immunother. 2008Mar;57(3):359-66
25. Tseng H.C. et al. Increased lysis of stem cells but not their differentiated cells by natural killer cells; de-differentiation or reprogramming activates NK cells. PLoS One. 2010;5(7):e11590
26. Rioux S. et al. Isolation and characterization of LPS mutants of Actinobacillus pleuropneumoniae serotype 1. Curr Microbiol. 1997;35(3):139-44
27. Opitz O.G. et al. A mouse model of human oral-esophageal cancer. J Clin Invest. 2002;110(6):761-9
28. Jewett A. et al. Inhibition of nuclear factor kappa B (NFkappaB) activity in oral tumor cells prevents depletion of cells NK and increases their functional activation. Cancer Immunol Immunother. 2006;55(9):1052-63
29. Jewett A. et al. Cytokine dependent inverse regulation of CD54 (ICAM1) and major histocompatibility complex class I antigens by nuclear factor kappaB in HEp2 tumor cell line: effect on the function of natural killer cells. Hum Immunol. 2003;64(5):505-20
30. Karin M. et al. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301-10
31. Burnet F.M. The concept of immunological surveillance. Prog Exp Tumor Res. 1970;13:1-27
32. Burnet F.M. Immunological surveillance in neoplasia. Transplant Rev. 1971;7:3-25
33. Burnet F.M. Implications of immunological surveillance for cancer therapy. Isr J Med Sci. 1971;7(1):9-16
34. Dunn G.P. et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991-8
35. Dunn G.P, Old L.J, Schreiber R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21(2):137-48
36. Whiteside T.L. et al. Evidence for local and systemic activation of immune cells by peritumoral injections of interleukin 2 in patients with advanced squamous cell carcinoma of the head and neck. Cancer Res. 1993;53(23):5654-62
37. Rivoltini L. et al. In vivo interleukin 2-induced activation of lymphokine-activated killer cells and tumor cytotoxic T-cells in cervical lymph nodes of patients with head and neck tumors. Cancer Res. 1990;50(17):5551-7
38. Cortesina G. et al. Treatment of recurrent squamous cell carcinoma of the head and neck with low doses of interleukin-2 injected perilymphatically. Cancer. 1988;62(12):2482-5
39. Whiteside T.L. et al. Antitumor effects of cytolytic T lymphocytes (CTL) and natural killer (NK) cells in head and neck cancer. Anticancer Res. 1996;16(4C):2357-64
40. Zoller M, Goodman M.L, Cummings C.W. Guidelines for prognosis in head and neck cancer with nodal metastasis. Laryngoscope. 1978;88(1 Pt 1):135-40
41. Hayry P, Totterman T.H. Cytological and functional analysis of inflammatory infiltrates in human malignant tumors. I. Composition of the inflammatory infiltrates. Eur Immunol J. 1978;8(12):866-71
42. Itoh K, Tilden A.B, Balch C.M. Interleukin 2 activation of cytotoxic T-lymphocytes infiltrating into human metastatic melanomas. Cancer Res. 1986;46(6):3011-7
43. Marrogi A.J. et al. Study of tumor infiltrating lymphocytes and transforming growth factor-beta as prognostic factors in breast carcinoma. Int Cancer J. 1997;74(5):492-501
44. Hartveit F, Breast cancer. poor short-term prognosis in cases with moderate lymphocyte infiltration at the tumour edge: a preliminary report. Oncol Rep. 1998;5(2):423-6
45. Wada Y. et al. Clinicopathological study on hepatocellular carcinoma with lymphocytic infiltration. Hepatology. 1998;27(2):407-14
46. Clemente C.G. et al. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 1996;77(7):1303-10
47. Bethwaite P.B. et al. Infiltration by immunocompetent cells in early stage invasive carcinoma of the uterine cervix: a prognostic study. Pathology. 1996;28(4):321-7
48. Coca S. et al. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer. 1997;79(12):2320-8
49. Thomas D.W. et al. Inflammatory cell infiltrate associated with primary and transplanted tumours in an inbred model of oral carcinogenesis. J Oral Pathol Med. 1995;24(1):23-31
50. Schantz S.P. et al. Natural killer cell activity and head and neck cancer: a clinical assessment. J Natl Cancer Inst. 1986;77(4):869-75
51. Hoffmann T.K. et al. Frequencies of tetramer+ T cells specific for the wild-type sequence p53(264-272) peptide in the circulation of patients with head and neck cancer. Cancer Res. 2002;62(12):3521-9
52. Reichert T.E. et al. Signaling abnormalities, apoptosis, and reduced proliferation of circulating and tumor-infiltrating lymphocytes in patients with oral carcinoma. Clin Cancer Res. 2002;8(10):3137-45
53. Jewett A. et al. Rapid and potent induction of cell death and loss of NK cell cytotoxicity against oral tumors by F(ab')2 fragment of anti-CD16 antibody. Cancer Immunol Immunother. 2008;57(7):1053-66
54. Romero-Reyes M. et al. Potent induction of TNF-alpha during interaction of immune effectors with oral tumors as a potential mechanism for the loss of cell viability NK and function. Apoptosis. 2007Nov;12(11):2063-75
55. Parham P. et al. Primate-specific regulation of natural killer cells. J Med Primatol. 2010;39(4):194-212
56. Jewett A, Cavalcanti M, Bonavida B. Pivotal role of endogenous TNF-alpha in the induction of functional inactivation and apoptosis in NK cells. Immunol J. 1997;159(10):4815-22
57. Kojima M. et al. Follicular lymphoma of the salivary gland: a clinicopathological and molecular study of six cases. Int J Surg Pathol. 2001;9(4):287-93
58. Yang F.C. et al. CD27/CD70 interaction directly induces natural killer cell killing activity. Immunology. 1996;88(2):289-93
59. Yang L. et al. Cancer-associated immunodeficiency and dendritic cell abnormalities mediated by the prostaglandin EP2 receptor. J Clin Invest. 2003;111(5):727-35
60. Thomas G.R. et al. Cytokines IL-1 alpha, IL-6, and GM-CSF constitutively secreted by oral squamous carcinoma induce down-regulation of CD80 costimulatory molecule expression: restoration by interferon gamma. Cancer Immunol Immunother. 2004;53(1):33-40
61. Rhodus N.L. et al. NF-kappaB dependent cytokine levels in saliva of patients with oral preneoplastic lesions and oral squamous cell carcinoma. Cancer Detect Prev. 2005;29(1):42-5
62. Diehl S, Rincon M. The two faces of IL-6 on Th1/Th2 differentiation. Mol Immunol. 2002;39(9):531-6
63. Menetrier-Caux C. et al. Inhibition of the differentiation of dendritic cells from CD34(+) progenitors by tumor cells: role of interleukin-6 and macrophage colony-stimulating factor. Blood. 1998;92(12):4778-91
64. Gabrilovich D. et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92(11):4150-66
65. Chang C.C. et al. Tolerization of dendritic cells by T(S) cells: the crucial role of inhibitory receptors ILT3 and ILT4. Nat Immunol. 2002;3(3):237-43
66. Vuk-Pavlovic S. Rebuilding immunity in cancer patients. Blood Cells Mol Dis. 2008;40(1):94-100
67. Greten T.F, Manns M.P, Korangy F. Myeloid derived suppressor cells in human diseases. Int Immunopharmacol. 2011;11(7):802-6
68. Coffelt S.B, Hughes R, Lewis C.E. Tumor-associated macrophages: effectors of angiogenesis and tumor progression. Biochim Biophys Acta. 2009;1796(1):11-8
69. Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22(2):231-7
70. Yaguchi T. et al. The mechanisms of cancer immunoescape and development of overcoming strategies. Int J Hematol. 2011;93(3):294-300
71. Jewett A. et al. Strategies to rescue mesenchymal stem cells (MSCs) and dental pulp stem cells (DPSCs) from NK cell mediated cytotoxicity. PLoS One. 2010;5(3):e9874
72. Wang T. et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10(1):48-54
73. Murakami J. et al. Effects of histone deacetylase inhibitor FR901228 on the expression level of telomerase reverse transcriptase in oral cancer. Cancer Chemother Pharmacol. 2005;56(1):22-8
74. Murakami J. et al. Influence of CpG island methylation status in O6-methylguanine-DNA methyltransferase expression of oral cancer cell lines. Oncol Rep. 2004;12(2):339-45
75. Abdulkarim B. et al. Antiviral agent Cidofovir restores p53 function and enhances the radiosensitivity in HPV-associated cancers. Oncogene. 2002;21(15):2334-46
76. Pasparakis M. et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature. 2002;417(6891):861-6
77. Berlin A.L, Paller A.S, Chan L.S. Incontinentia pigmenti: a review and update on the molecular basis of pathophysiology. J Am Acad Dermatol. 2002;47(2):169-87
78. Greten F.R. et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118(3):285-96
79. Greten F.R, Karin M. The IKK/NF-kappaB activation pathway-a target for prevention and treatment of cancer. Cancer Lett. 2004;206(2):193-9
80. Seitz C.S. et al. Alterations in NF-kappaB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-kappaB. Proc Natl Acad Sci U S A. 1998;95(5):2307-12
81. van Hogerlinden M. et al. Squamous cell carcinomas and increased apoptosis in skin with inhibited Rel/nuclear factor-kappaB signaling. Cancer Res. 1999;59(14):3299-303
82. van Hogerlinden M. et al. Characterization of the progressive skin disease and inflammatory cell infiltrate in mice with inhibited NF-kappaB signaling. J Invest Dermatol. 2004;123(1):101-8
83. Oka N. et al. VEGF promotes tumorigenesis and angiogenesis of human glioblastoma stem cells. Biochem Biophys Res Commun. 2007;360(3):553-9
84. Inagaki A. et al. Long-term maintenance of brain tumor stem cell properties under at non-adherent and adherent culture conditions. Biochem Biophys Res Commun. 2007;361(3):586-92
85. Soeda A. et al. Epidermal growth factor plays a crucial role in mitogenic regulation of human brain tumor stem cells. J Biol Chem. 2008;283(16):10958-66
86. Rahaman S.O. et al. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002;21(55):8404-13
87. Hu P.F. et al. Natural killer cell immunodeficiency in HIV disease is manifest by profoundly decreased numbers of CD16+CD56+ cells and expansion of a population of CD16dimCD56- cells with low lytic activity. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;10(3):331-40
88. Lai P. et al. Alterations in expression and function of signal-transducing proteins in tumor-associated T and natural killer cells in patients with ovarian carcinoma. Clin Cancer Res. 1996;2(1):161-73
89. Kuss I. et al. Clinical significance of decreased zeta chain expression in peripheral blood lymphocytes of patients with head and neck cancer. Clin Cancer Res. 1999;5(2):329-34
90. Patankar M.S. et al. Potent suppression of natural killer cell response mediated by the ovarian tumor marker CA125. Gynecol Oncol. 2005Dec;99(3):704-13
91. Mandelboim O. et al. Human CD16 as a lysis receptor mediating direct natural killer cell cytotoxicity. Proc Natl Acad Sci U S A. 1999;96(10):5640-4
92. Jewett A. et al. Differential secretion of TNF-alpha and IFN-gamma by human peripheral blood-derived subsets NK and association with functional maturation. J Clin Immunol. 1996;16(1):46-54
93. Ortaldo J.R, Mason A.T, O'Shea J.J. Receptor-induced death in human natural killer cells: involvement of CD16. J Exp Med. 1995;181(1):339-44
94. Azzoni L. et al. Ligand binding to Fc gamma R induces c-myc-dependent apoptosis in IL-2-stimulated NK cells. Immunol J. 1995;154(2):491-9
95. Jewett A. et al. Coengagement of CD16 and CD94 receptors mediates secretion of chemokines and induces apoptotic death of naive natural killer cells. Clin Cancer Res. 2006;12(7 Pt 1):1994-2003
96. Bonavida B, Lebow L.T, Jewett A. Natural killer cell subsets: maturation, differentiation and regulation. Nat Immun. 1993;12(4-5):194-208
97. Bonavida B. et al. Qualitative and quantitative analysis of subpopulations of cytotoxic effector cells by flow cytometry. J Lipid Mediat Cell Signal. 1994;9(1):19-25
98. Jewett A, Bonavida B. Activation of the human immature natural killer cell subset by IL-12 and its regulation by endogenous TNF-alpha and IFN-gamma secretion. Cell Immunol. 1994;154(1):273-86
99. Jewett A, Bonavida B. Interferon-alpha activates cytotoxic function but inhibits interleukin-2-mediated proliferation and tumor necrosis factor-alpha secretion by immature human natural killer cells. J Clin Immunol. 1995;15(1):35-44
100. Jewett A, Bonavida B. MHC-Class I antigens regulate both the function and the survival of human peripheral blood NK cells: role of endogenously secreted TNF-alpha. Clin Immunol. 2000;96(1):19-28
101. Cooper M.A, Fehniger T.A, Caligiuri M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22(11):633-40
102. Nemeth E, Baird A.W, O'Farrelly C. Microanatomy of the liver immune system. Semin Immunopathol. 2009;31(3):333-43
103. Bubenik J, MHC class I down-regulation. tumour escape from immune surveillance? (review). Int Oncol J. 2004;25(2):487-91
104. Curiel T.J. et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9(5):562-7
105. Jewett A. et al. Inhibition of nuclear factor kappa B (NFkappaB) activity in oral tumor cells prevents depletion of cells NK and increases their functional activation. Cancer Immunol Immunother. 2005:1-12
106. Lutz C.T, Kurago Z.B. Human leukocyte antigen class I expression on squamous cell carcinoma cells regulates natural killer cell activity. Cancer Res. 1999;59(22):5793-9
107. Chen Z. et al. Expression of proinflammatory and proangiogenic cytokines in patients with head and neck cancer. Clin Cancer Res. 1999;5(6):1369-79
108. Okada K. et al. Interactions between autologous CD4+ and CD8+ lymphocytes T and human squamous cell carcinoma of the head and neck. Cell Immunol. 1997;177(1):35-48
109. Romano M. et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6(3):315-25
110. Woods K.V. et al. Interleukin-1 regulates interleukin-6 secretion in human oral squamous cell carcinoma in vitro: possible influence of p53 but not human papillomavirus E6/E7. Cancer Res. 1998;58(14):3142-9
111. Bromberg J.F. et al. Stat3 as an oncogene. Cell. 1999;98(3):295-303
112. Calo V. et al. STAT proteins: from normal control of cellular events to tumorigenesis. J Cell Physiol. 2003;197(2):157-68
113. Grandis J.R. et al. Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci U S A. 2000;97(8):4227-32
114. Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19(21):2548-56
115. Kijima T. et al. STAT3 activation abrogates growth factor dependence and contributes to head and neck squamous cell carcinoma tumor growth in vivo. Cell Growth Differ. 2002;13(8):355-62
116. Sriuranpong V. et al. Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res. 2003;63(11):2948-56
117. Gastman B.R. et al. Fas ligand is expressed on human squamous cell carcinomas of the head and neck, and it promotes apoptosis of T lymphocytes. Cancer Res. 1999;59(20):5356-64
118. Kim J.W. et al. Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin Cancer Res. 2005;11(3):1010-20
119. Biswas P. et al. Interleukin-6 induces monocyte chemotactic protein-1 in peripheral blood mononuclear cells and in the U937 cell line. Blood. 1998;91(1):258-65
120. Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4(12):941-52
121. Gabrilovich D.I. et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2(10):1096-103
122. Laxmanan S. et al. Vascular endothelial growth factor impairs the functional ability of dendritic cells through Id pathways. Biochem Biophys Res Commun. 2005;334(1):193-8
123. Nefedova Y. et al. Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. Immunol J. 2005;175(7):4338-46
124. Nefedova Y. et al. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. Immunol J. 2004;172(1):464-74
125. Oyama T. et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. Immunol J. 1998;160(3):1224-32
126. Park S.J. et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. Immunol J. 2004;173(6):3844-54
Author contact
![]() Corresponding author: Division of Oral Biology and Medicine, UCLA School of Dentistry, 10833 Le Conte Ave, Los Angeles, CA. 90095-1668. Telephone: (310) 206-3970; Fax: (310) 794-7109; E-mail: ajewettedu
Corresponding author: Division of Oral Biology and Medicine, UCLA School of Dentistry, 10833 Le Conte Ave, Los Angeles, CA. 90095-1668. Telephone: (310) 206-3970; Fax: (310) 794-7109; E-mail: ajewettedu