Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2011; 2:386-400. doi:10.7150/jca.2.386 This volume Cite
Research Paper
Complex Alternative Splicing of the Smarca2 Gene Suggests the Importance of Smarca2-B Variants
Min Yang1*, Yuan Sun1, Ling Ma2, Chenguang Wang3, Jian-min Wu2, Anding Bi1, D. Joshua Liao1 ![]()
1. Hormel Institute, The University of Minnesota, Austin, MN 55912, USA
2. Guangxi Veterinary Research Institute, Nanning, Guangxi 530001, P.R. China
3. Department of Stem Cell and Regenerative Medicine, Kimmel Cancer Center, Thomas Jefferson University, 233 S. 10th Street, Philadelphia, PA 19107, USA
*Current address: School of Laboratory Medicine, Chengdu Medical College, Chengdu, Sichuan 610083, P.R. China
Received 2011-4-11; Accepted 2011-6-22; Published 2011-7-6
Abstract
BRM is an ATPase component of the SWI/SNF complex that regulates chromatin remodeling and cell proliferation and is considered a tumor suppressor. In this study we characterized transcripts from the Smarca2 gene that encodes the BRM protein. We found that the human Smarca2 gene (hSmarca2), like its mouse counterpart (mSmarca2), also initiated a short transcript from intron 27 of the long transcript. We name the long and short transcripts as Smarca2-a and Smarca2-b, respectively. Like its human counterpart, mSmarca2-a also underwent alternative splicing at the 54-bp exon 29. The hSmarca2-b had two alternative initiation sites and underwent alternative splicing at three different 3' sites of exon 1 and at exons 2, 3 and/or 5. We identified nine hSmarca2-b mRNA variants that might produce five different proteins. mSmarca2-b also underwent alternative splicing at exon 3 and/or exon 5, besides alternatively retaining part of intron 1 in exon 1. Smarca2-b was expressed more abundantly than Smarca2-a in many cell lines and was more sensitive to serum starvation. Moreover, cyclin D1 also regulated the expression of both Smarca2-a and Smarca2-b in a complex manner. These data suggest that the functions of the Smarca2 gene may be very complex, not just simply inhibiting cell proliferation, and in certain situations may be elicited mainly by expressing the much less known Smarca2-b, not the better studied Smarca2-a and its products BRM proteins.
Keywords: Alternative splicing, Tumor suppressor gene, BRM, Smarca2, SWI/SNF complex, Cancer, Cyclin D1
Introduction
Most genes in a eukaryotic genome consist of exons and introns. During a splicing process, about 91% of a transcript (pre-mRNA) sequence is removed as introns; the remaining 9% as exons are joined to form a mature mRNA for protein translation (1). Transcripts from over 70% of the genes in the human genome undergo alternative splicing to produce different mature mRNAs (2), which greatly diversifies the protein products from individual genes. In a disease situation such as cancer that has gene mutations, even more alternative splicing events may occur because over 14% of disease-causing mutations affect the splicing process (3,4), in part because the mutations occur at splice sites, as in the case of the BRCA2 gene (5).
There are quite a few patterns of alternative splicing, including cassette alternative exons, alternative 5' splice sites, alternative 3' splice sites, mutually exclusive splice sites and retained introns (1). Moreover, one gene can also be transcribed to different pre-mRNAs by using different promoters and initiation sites, and each pre-mRNA may undergo several patterns of splicing, which together further increases the number of mature mRNAs from each individual gene. A well-studied example is the Drosophila Down syndrome cell adhesion molecule (Dscam) gene, which can generate 38,016 mRNA variants by alternatively splicing its 95 exons (6). Another example is the titin gene that is expressed to, on average, about 0.5 kg Titin protein in the muscle of an adult human (7). How the 363 exons of the human titin gene are alternatively spliced is still largely unknown but over one million splice variants are estimated (7).
BRM is an ATPase component of the SWI/SNF complex that regulates chromatin remodeling. It functions as a gatekeeper of proliferation in cancers (8) in part by its collaboration with the retinoblastoma (Rb) protein to repress E2F (9). Most of the known functions of BRM protein suggest that it may be a tumor suppressor (10). However, BRM has also been shown to transactivate the telomerase reverse transcriptase (Tert) gene and further modulate Tert splicing (11), which does not seem to support its tumor-suppressive role because Tert activation is supposed to promote cancer formation and progression. Possibly, the functions of BRM proteins may be complex and may vary among different situations.
Lost or decreased expression of Smarca2, the gene encoding the BRM proteins, has been reported in many cell lines of different types of cancer, which is in line with its putative role as a tumor suppressor (10,12-15). The gene silencing is generally considered to be due to epigenetic mechanisms (14,16), in part because there has not been any critical genetic change identified in those cell lines that have lost BRM expression, although loss of heterozygosity in the Smarca2-containing locus has been reported in some cancers (17).
Like many other genes, Smarca2 may diversify its function by yielding multiple mRNA variants. In the NCBI database, the mouse Smarca2 (mSmarca2) gene has a long transcript designated herein as mSmarca2-a and a short transcript coined herein as mSmarca2-b that is initiated from the intron 27 of the mSmarca2-a, as illustrated in figure 1. The human Smarca2 (hSmarca2) also has two mRNA variants but, unlike the mSmarca2 mRNA variants, they are derived from alternative splicing at the exon 29, designated herein as hSmarca2-a1 with this exon and hSmarca2-a2 without this exon (Fig. 1). Reisman et al mentioned an additional transcription initiation site that is close to the one that initiates the Smarca2-a (10), but we are unable to illustrate it in figure 1 because it is unclear whether this initiation site is identified in the mouse, human or both species.
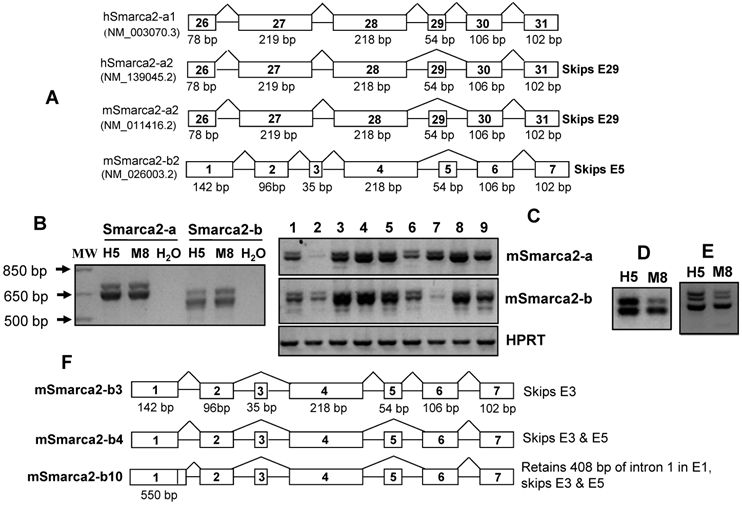
Expression of different Smarca2 mRNA variants. A: Illustration of human (h) and mouse (m) Smarca2 mRNA variants documented in the NCBI database, with access number (such as NM_003070.3) provided. Only several exons at the 3'-part for Smarca2-a or the 5'-part for Smarca2-b are shown. Each box indicates an exon with the exon number inside. The figure below a box indicates the length of that exon in base-pairs (bp). Note that the two human Smarca2 variants differ by the alternative skip of the 54-bp exon 29 and that the two mouse Smarca2 variants are denoted as -a2 or -b2 because they both lack the 54-bp exon, like the hSmarc2-a2. B: RT-PCR detection of mSmarca2 in H5 and M8 cells with F3855+R4524 primers for mSmarca2-a and F'43+R4524 primers for Smarca2-b. As a negative control, the cDNA template was replaced with water. C: Expression of mSmarca2, detected by RT-PCR with the same primer sets as in (B), in a panel of mouse breast cancer or non-malignant cell lines (from lanes 1 to 9): 168FARN, 4T07, 4T1, 66C14, 67RN, NMuMG, HC11, T/M and MT-tgfα. D & E: RT-PCR detection of mSmarca2-b in H5 and M8 cells, with the F'192 and R4524 primers, reveals two (D) or three bands (E) in agarose gel. F: Illustration of the three newly identified mSmarca2-b mRNA variants confirmed by sequence data, designated as mSmarca2-b3, -b4, and -b10, respectively. Note that all three variants differ from the mSmarca2-b2 in the NCBI database (A) by lacking the 35-bp exon 3 and that mSmarca2-b3 retains the 54-bp exon 5 that is absent in the mSmarca2-b2. The exon 1 of mSmarca2-b10 retains 408 bp of intron 1.
In this communication, we report that the long transcript in the mouse also undergoes alternative splicing to generate mSmarca2-a1 that contains exon 29, just like its human counterpart. On the other hand, the hSmarca2 gene also initiates a short transcript, hSmarca2-b, from two different sites in intron 27 of hSmarca2-a. This hSmarca2-b undergoes alternative splicing at three different 3' sites of exon 1 and at exons 2, 3 and/or 5. These complex patterns of transcription initiation and alternative splicing may engender many different hSmarca2-b mRNA variants, and we identified nine of them. mSmarca2-b also had alternative splice variants. Moreover, Smarca2-a and Smarca2-b can be differentially expressed in different cell lines and regulated by cyclin D1 and by serum concentration. These findings suggest that the functions of the Smarca2 gene may be very complex and may be elicited mainly by expressing the Smarca2-b in certain situations.
Materials and Methods
Cell lines and cell culture: 168FARN, 4T07, 4T1, 66C14 and 67RN are cell lines established from the same mouse mammary tumor (18). NMuMG and HC11 are non-malignant mouse mammary epithelial cell lines. ND5 and ND11 are malignant clones of NMuMG cells transformed by cyclin D1 (19). MT-tgfα or T/M is a cell line established by us from a mammary tumor developed in a female MT-tgfα transgenic or MT-tgfα/MMTV-c-myc double transgenic mouse, respectively. H5 and M8 are a pcDNA3.1-hygromycin empty vector and a pcDNA3.1-c-myc expressing clone, respectively, of an Ela-myc transgenic mouse pancreatic cancer cell line (20,21). MCF10A is a non-malignant human breast epithelial cell line, whereas MCF7, MDA-MB231, T47D, and SKBR3 are human breast cancer cell lines. MCF15 is a new human breast cancer cell line in the MCF series (22). GI101A and its parental GILMmix are human breast cancer cell lines provided by Dr. JE Price at M.D. Anderson Cancer Center. AsPC-1, Panc-1, Panc-28, L3.6pL and MiaCapa-2 are human pancreatic cancer cell lines. There were many more mouse and human cell lines that were used in this study but are not listed here because the related data are not presented. All the cells were cultured in routine conditions with DMEM containing 10% serum. The cells were harvested when they reached about 80% confluence, but for the serum starvation study the medium was changed to a new one without serum for additional 48 hours of culture before cell harvest.
Retroviral infection: A human cyclin D1 (D1) cDNA or its K112E mutant (23) tagged with a Flag sequence at the N-terminus was inserted into a pMIG retroviral vector that contains an IRES-driven green fluorescent protein (GFP) sequence after the insert. These constructs and the empty vector were packaged in 293T cells and the resulting viruses were used to infect desired cells, followed by sorting for GFP to enrich the infected cells.
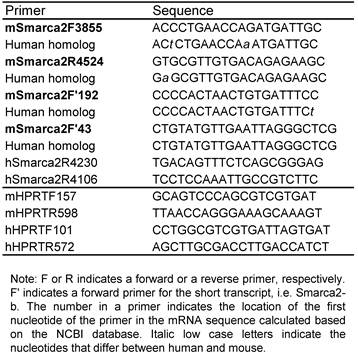
RT-PCR assay: Total RNA samples were extracted from cells by using TRIzol (Invitrogen; catalog number: 15596-026), followed by DNase treatment to remove DNA. Mouse tissues were homogenized with polytron in a buffer containing protease K (24) before RNA extraction with TRIzol. RNA samples from normal human tissues were also purchased from Clontech (www.clontech.com). The RNAs were reverse transcribed (RT) to cDNA using hexamer primers. Amplification by Polymerase chain reaction (PCR) was conducted with the forward and reverse primers listed in table 1.
Primer list
5'RACE and T-A cloning: Total RNA from L3.6pL cells was used for a 5'RACE assay with FirstChoice RLM-RACE Kit (Ambion, Inc. Austin, TX), following the manual. The hSmarca2R4230 (outer) and hSmarca2R4106 (inner) listed in table 1 were used as reverse primers. The RACE products were fractionized in agarose gel, and dominant bands were purified and ligated into a pGEM-T Easy Vector (Promega, Madison, WI). RT-PCR products were also cloned in this way. Bacterial clones transformed by the plasmids were selected and propagated.
DNA sequencing and sequence analysis: The desired band of PCR products in agarose gel was purified with a DNA purification kit from UltraClean Gel DNA Extraction Kit (ISC BioExpress, Kaysville, UT), following the manual. The DNA sample was sent to Genewiz, Inc. (South Plainfield, NJ) for sequencing. Plasmid DNA of desired clones from T-A cloning or 5'RACE was also sequenced. DNA sequence analysis was mainly conducted with DNAStar software.
Western blot assay: Cells were prepared for protein lysates as described before (25). An equal amount of proteins was fractioned in 12% SDS-PAGE and then transferred onto an Immobilon-P Nylon membrane (Millipore, Bedford, MA). After being blocked with 5% milk, the membrane was incubated with a mouse monoclonal (catalog number: sc-166579) or a rabbit polyclonal (catalog number: sc-28710) BRM antibody (Santa Cruz Biotech., Santa Cruz, CA) at 1:1000 dilution and then with a corresponding secondary antibody conjugated with a horseradish peroxidase, with three washes between each blocking. The signal was visualized by luminal-based chemiluminescent substrate (Pierce, Rockford, IL) on X-ray film (ISC BioExpress, Kaysville, UT).
Results
In this study we analyzed in a systematic manner the transcription of mouse and human Smarca2 gene and the splicing of the transcripts. Because the NCBI database documents a long and a short transcripts of Smarca2 in the mouse but only the long transcript in the human, we first determined the alternative splicing of both transcripts in the mouse and then, based on the information from the mouse, explored whether the short transcript was also expressed in the human and, if yes, whether it underwent alternative splicing as well. Once the mRNA variants were confirmed by sequencing, bioinformatic analyses were applied to determine their open reading frames (ORF) and regulation of their expression by cyclin D1 and serum starvation was explored as the mechanistic part of the study. The results obtained were presented below in this logical order.
Alternative splicing of mouse Smarca2-a
Because in the NCBI database hSmarca2-a1 contains the 54-bp exon 29 (Fig. 1A), we wonder whether this exon also appears in mSmarca2-a or mSmarca2-b. Using a forward primer at exon 26 (mSmarca2F3855; table 1) and a reverse primer (mSmarca2R4524) at the 102-bp exon 31 of mSmarca2-a (the exon is numbered based on hSmarca2-a1), we performed PCR to amplify the RT products from many mouse tissues and cell lines. Results from some cell lines are presented in Fig. 1B and 1C, whereas similar data obtained from many other cell lines or tissues are not shown. Purifying and then sequencing the bottom band of mSmarca2-a shown in Fig. 1B revealed that it was the NM_011416.2 sequence in the NCBI database that lacks the 54-bp exon and is thus designated as mSmarca2-a2 (Fig. 1A). Sequencing the top band revealed that it actually contained two cDNA sequences: one contains the 54-bp exon 29, like the hSmarca2-a1 (Fig. 1A), and is thus designated as mSmarca2-a1 (Supplementary Material: Fig. S1), whereas the other one is the mSmarca2-a2 lacking this exon. The mSmarca2-a1 was expressed more abundantly, relative to the mSmarca2-a2, manifested as higher peaks in the sequence graph (not shown).
Alternative splicing of mouse Smarca2-b
Wondering whether the short transcript mSmarca2-b also undergoes alternative splicing at the 54-bp exon, we used a forward primer (mSmarca2'F192; table 1) located at the 96-bp exon 2 of mSmarca2-b and the same reverse primer used above (mSmarca2R4524) to perform PCR with cDNA samples from H5 and M8 cells. The PCR products revealed two bands (Fig. 1D) sometimes but three bands (Fig. 1E) at other times in agarose gel. Purifying and then sequencing each of the two bands shown in Fig. 1D revealed that the top band was a variant lacking the 35-bp exon 3 but retaining the 54-bp exon 5, whereas the bottom band was a variant lacking both exons 3 and 5 (Supplementary Material: Fig. S2). We also purified and sequenced each of the three bands appearing in Fig. 1E, and found that the top and the bottom bands were the above two variants, respectively, whereas the middle band contained both of these two variants and thus was a heterodimer that sometimes occurs between two similar sequences in PCR products, according to our experience. We designated the sequence in the NCBI database (NM_026003.2) (Fig. 1A) that contains the 35-bp exon 3 as mSmarca2-b2 and the two new sequences as mSmarca2-b3 and -b4, respectively (Fig. 1F), in case there is a full-length variant mSmarca2-b1 that retains both exons 3 and 5 as seen in the human (see below).
Identification of human Smarca2-b and its splicing pattern
Wondering whether the short transcript, i.e. Smarca2-b, is also expressed in humans, we performed RT-PCR with the forward primer mSmarca2'F43, which is located at exon 1 of mSmarca2-b, and with the reverse primer mSmarca2R4524 that is located at exon 7 of mSmarca2-b (table 1). The PCR products from cDNA samples of multiple human cell lines or tissues revealed multiple bands in agarose gel. Results from some cell lines are presented in the middle panel of Fig. 2A and 2B, whereas similar data from many other cell lines and tissues are not shown.
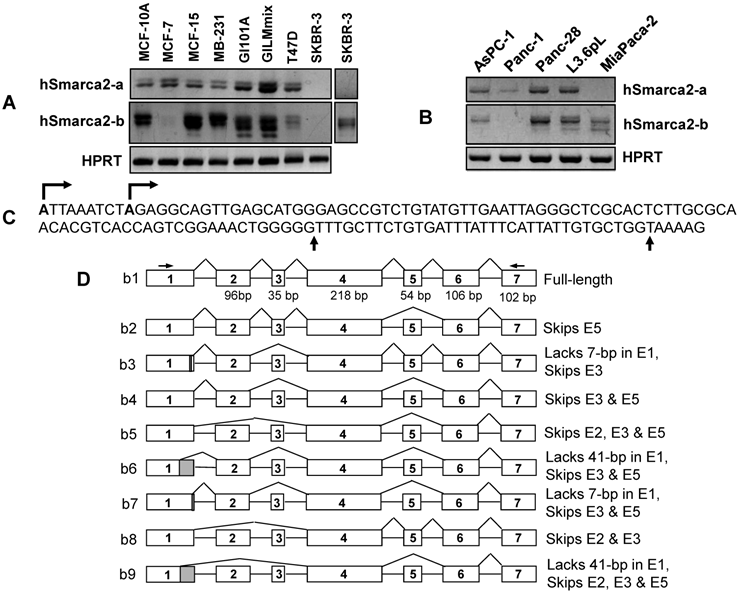
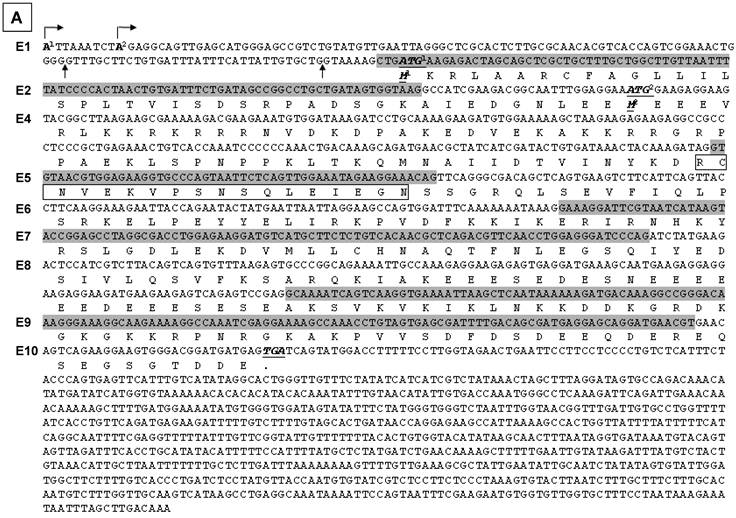
Detection of hSmarca2 mRNA variants. A & B: RT-PCR detection of hSmarca2 in several human breast (A) and pancreatic (B) cancer cell lines or non-malignant (MCF10A) cells with the F3855+R4524 primers for hSmarca2-a and the F'43+R4524 primers for hSmarca2-b. Note that the hSmarca2-b level is very low but still seems to be visible in MCF7 cells. The separate panel in (A) shows that the hSmarca2-b, but not the hSmarca2-a, can still be detected in SKBR3 cells when the amount of cDNA template and the PCR cycles were increased. C: Sequence of the longest (134-bp) exon 1 of the hSmarca2-b determined by 5'RACE. Two transcription initiation sites are indicated by arrows pointing right. Two additional 3' splice sites that result in a deletion of 7-bp or 41-bp, respectively, are indicated by the arrows pointing up. D: Illustration of the nine hSmarca2-b variants confirmed by DNA sequencing. The size of exon 1 is not indicated because it varies, but the skip of 7-bp or 41-bp is indicated by the shaded 3' end.
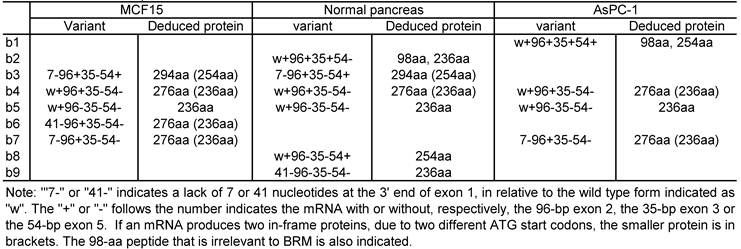
We collectively purified these bands and cloned them into a TA vector. Cloned cDNA products were from RNA samples of MCF15 human breast cancer cells (22), AsPC-1 pancreatic cancer cells, and a commercial RNA sample of normal human pancreas. For each of these three cell lines and tissue, four to six plasmid clones were sequenced and the clones from the same cell line or tissue show different sequences (table 2).
Summary of the hSmarca2-b variants with alternative splice at exons 1, 2, 3 and/or 5 and the deduced protein identified in the normal pancreas as well as MCF15 and AsPC-1 cells.
The sequence results shows that exon 1 has three different 3' splice sites, two of which result in a deletion of 7-bp or 41-bp at the 3'end, relative to the third one (Fig. 2C). Another finding is alternative skip of the 96-bp exon 2, the 35-bp exon 3, and/or the 54-bp exon 5. A total of nine mRNA variants were identified among the 15 plasmid clones sequenced (table 2), designated as hSmarca2-b1, hSmarca2-b2… hSmarca2-b9, respectively, as illustrated in Fig. 2D (sequences in Supplementary Material: Fig. S3).
The new finding of the alternative skip of the 35-bp exon 3 in mouse Smarca2-b was also observed in the human. Actually, seven out of nine variants identified (b3, b4, b5, b6, b7, b8 and b9 vs b1 and b2 in Fig. 2D) or 12 out of 15 plasmid clones sequenced (table 2) skip this exon, suggesting that the variants without exon 3 are the majority. Similarly, the variants that skip the 54-bp exon 5 are also dominant, since six out of nine variants identified (b2, b4, b5, b6, b7 and b9 vs b1, b3 and b8) or 11 out of 15 plasmid clones sequenced (table 2) skip this exon. These features are even more obvious when plasmid clones sequenced for other purposes are counted (see below).
Identification of different transcription initiation sites of human Smarca2-b
After confirming the existence of hSmarca2-b, we determined its initiation site by performing 5'RACE with RNA samples from L3.6pL human pancreatic cancer cells and with hSmarca2R4230 and hSmarca2R4106 (the number was calculated based on hSmarca2-a since the initiation site of hSmarca2-b was unknown) as the reverse primers that are localized at exon 4, which does not undergo alternative splicing (Fig. 2D). Sequencing five plasmid clones of the 5'RACE products identified two initiation sites that were nine nucleotides apart in intron 27 of hSmarca2-a (Fig. 3C). The sequence results also revealed the 7-bp deletion from the 3'-end of exon 1 and the skip of exon 3 identified by the above described TA cloning (sequences in Supplementary Material: Fig. S3). Thus, a random combination of the two alternative initiation sites and the three 3' alternative splice sites of exon 1 might, theoretically, result in a longest exon 1 of 134-bp and a shortest exon 1 of 84-bp (Fig. 2C). We have not yet identified the 84-bp exon 1 but it may exist in our 5'RACE products. Mathematically, two different transcription initiation sites, three different 3' alternative splice sites of exon 1, and alternative skips of exons 2, 3 and 5 may result in 48 (2x3x2x2x2) different hSmarca2-b variants, although we have only identified nine of them.
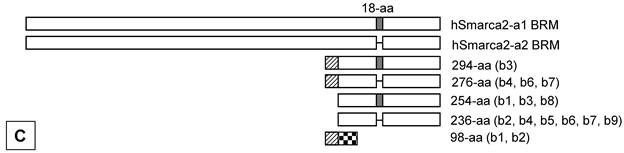
Illustration of deduced open reading frame (ORF) of hSmarca2-b mRNA variants. Every other exon is shaded to show the length of each exon and the boundary of neighbor exons. The number of exon (E) is indicated at the left margin. There are two putative ATG translation start codons, located in exons 2 and 4, respectively. No matter whether exon 1 is initiated from the A1 or A2 site (arrows pointing right) or has or not a 7- or 41-bp deletion at its 3'-end, it does not affect ORF. Those variants that lack exon 2 have only one ORF starting from ATG2 at exon 4 and ending at the only TGA stop codon in exon 10. A: In those variants that lack the 35-bp exon 3, the ATG1 and ATG2 are in the same ORF, which ends at the TGA in exon 10 as well. Thus, the proteins starting from ATG1 have additional 40 amino acids (aa) at the N-terminus compared with those starting from ATG2, whereas those variants retaining the exon 5 have an 18-aa insert (in box). B: In those variants retaining the 35-bp exon 3, ATG1 starts a protein that ends at a TGA in exon 4, thus having only 98 aa. The sequence of its first 31 aa encoded by exon 2 is identical to the corresponding part of those proteins that are also started from ATG1, whereas the remaining 67 aa constitute a sequence that is different from any part of BRM-related proteins, due to the frame-shift caused by the insert of exon 3. Only ATG2 starts the BRM-related proteins. C: Illustration of the BRM and related proteins, with the variants that may produce the protein indicated in brackets. The boxes that are in the same format and vertically aligned at the same position represent identical sequence, with open box as BRM or BRM related sequence and small grey box representing the 18 aa encoded by the 54-bp exon that may be alternatively skipped. The two BRM proteins differ by alternative skip of 18 aa. Note that the 294-aa and 276-aa proteins share the first 31 aa (slashed box) with the 98-aa peptide but differ from the 254-aa and 236-aa proteins by this 31-aa sequence. The remaining 67 aa (checkered box) of the 98-aa peptide does not exist in any other protein.

Expression of Smarca2-a and Smarca2-b in various cell lines
We have determined the expression levels of Smarca2-a in many cell lines but here we only describe those data that are discrepant from or absent in the literature. SKBR3 human breast cancer cells were reported to express relatively high level of BRM proteins (12), but we were not able to detect hSmarca2-a in this cell line (Fig. 2A). On the other hand, we found that the Smarca2-a level in 4T07 mouse breast cancer cells (Fig. 1C) and MiaPaca2 human pancreatic cancer cells (Fig. 2B) was very low or undetectable. Moreover, we detected hSmarca2-a in Panc-1 human pancreatic cancer cells (Fig. 2B).
The short transcript Smarca2-b was expressed abundantly in most mouse and human cell lines and tissues. Smarca2-b was still readily detectable in some cell lines wherein the level of Smarca2-a was very low or undetectable, such as in 4T07 and MiaCapa2 cells (Fig. 1C and 2B). In SKBR3 cells, hSmarca2-b could be detected when the number of PCR cycles or the amount of cDNA template was increased, although the hSmarca2-a was still undetectable (Fig. 2A). However, there still were some cell lines such as HC11, MCF7 and T47D wherein the Smarca2-a level was detected abundantly but the level of the Smarca2-b was very low (Fig. 1C and 2A).
Bioinformatic analyses of ORF of hSmarca2-a and hSmarca2-b
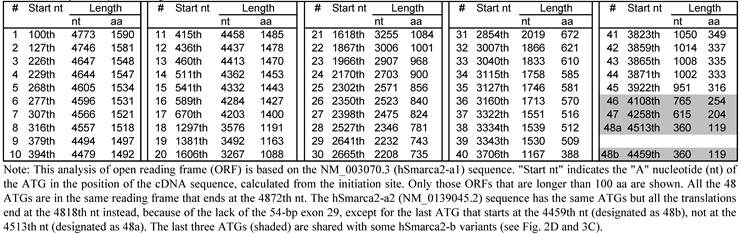
In the NCBI database, hSmarca2-a1 and hSmarca2-a2 encode only a protein of 1590 amino acids (aa) and 1572 aa, respectively. However, ORF analyses with DNAStar software identifies a total of 48 ATG (AUG in mRNA) translation start codons in hSmarca2-a, each of which encodes an ORF longer than 100 aa but all the ORFs are in the same frame and end at the TGA (UGA in mRNA) translation stop codon at the 4872-4875th nucleotides (nt) (table 3). hSmarca2-a2 has the same 48 ATGs but the stop codon occurs earlier (at the 4818-4820th nt TGA), because of the lack of the 54-bp exon 29, which makes the last (the shortest) ORF different from their counterparts in hSmarca2-a1 (table 3). The 48 ORFs in hSmarca2-a1 encode proteins that range from 1590 to 119 aa, which, if expressed, are estimated at about 181 kD to 13 kD on a western blot.
Analysis of open reading frame of human hSmarca2-a1
ORF analysis also shows that those hSmarca2-b variants (b3, b4, b6 and b7 shown in Fig. 2D) that lack exon 3 may be translated from two ATG translation start codons located at exons 2 and 4, respectively (Fig. 3A). These two ATGs are within the same frame that ends at a TGA stop codon in exon 10; the proteins starting from ATG1 have additional 40 aa at the N-terminus compared with those starting from ATG2. Alternative skip of the 54-bp exon 5 deletes 18 aa from these proteins (Fig. 3A). However, in those variants that retain exon 3 (b1 and b2 shown in Fig. 2D), the two different ATGs start different ORFs because exon 3 causes a frame-shift (Fig. 3B). The protein starting from ATG1 at exon 2 will end at a TGA in exon 4, resulting in a 98-aa short peptide (Fig. 3B). The proteins starting from ATG2 still end at the second TGA in exon 10 and may have an alternative deletion of 18 aa because of the skip of exon 5, just like those proteins from the variants without exon 3. Skip of exon 2 in certain variants (b5, b8 and b9 in Fig. 2D) also deletes the ORF starting from ATG1 without creating any new ORF, and thus has only one ORF starting from ATG2 (Fig. 3A and 3B).
The above situations of hSmarca2-b variants, i.e. two different ATGs in the absence of exon 3, the first ATG in the presence of exon 3 that causes an early translation termination, and the alternative skip of exon 5, together can result in five different proteins (Fig. 3C). Four of the five belong to the BRM family and contain 294, 276, 254, and 236 aa, with a calculated molecular weight of 33731.76, 31733.55, 29490.84 and 27492.64 Daltons, respectively, much smaller than the BRM proteins derived from the hSmarca2-a1 (181278.65 Daltons) or the hSmarca2-a2 (179280.45 Daltons). Moreover, the second ATG is in the same ORF of hSmarca2-a. Therefore, the 294-aa and 276-aa proteins that are translated from the first ATG have the same C-terminal sequence as the hSmarca2-a1 and hSmarca2-a2 proteins, whereas the 254-aa and 236-aa proteins translated from the second ATG are identical to a C-terminal part of the hSmarca2-a1 or hSmarca2-a2 derived BRM proteins (Fig. 3C). The 98-aa short peptide, which is translated from the first ATG of the variants containing exon 3 (b1 and b2), is calculated as 11597.91 Daltons. Its first 31-aa sequence encoded by exon 2 is identical to the corresponding part of the 294-aa and 276-aa proteins (Fig. 3C), whereas the remaining sequence of 67 aa differs completely from any part of BRM or BRM-related proteins, due to the frame-shift caused by exon 3.
Small BRM-immunoreactive proteins on western blot
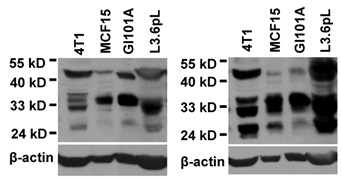
We performed western blot assay using a mouse monoclonal antibody (sc-166579) and a rabbit polyclonal antibody (sc-28710) that were raised against the 1531-1586th aa of hSmarca2-a1, a region shared with Smarca2-b. Both antibodies identified several bands between 24-35 kD and a band slightly higher than 40 kD in cell lines of human and mouse origins. Data from some of the cell lines were shown in Fig. 4. These antibodies also recognized several other bands larger than 50 kD or smaller than 24 kD (data not shown). The identities of these small BRM-immunoreactive proteins are unclear. The BRM proteins of about 180 kD fractioned in a much lower percentage (6%) PAGE were detected only weakly (data not shown), in strong contrast to the high abundance of the much smaller proteins shown in Fig. 4.
Western blot assays show that a rabbit (left panel) and a mouse (right panel) BRM C-terminal antibodies recognize several proteins in the lysates from a mouse (4T1) and several human cancer cell lines. Note that several proteins between 24-35 kD are recognized by both antibodies, in addition to a protein slightly higher than 40-kD. The same membrane was stripped and re-blotted with β-actin to show the protein loading, although no attempt is made to compare the expression levels among the four unrelated cell lines.
Effects of cyclin D1 and serum starvation on Smarca2 expression
We had previously transformed NMuMG mouse mammary epithelial cells with a pcDNA3.1neo-cyclin D1 (D1) construct and established the D1 expressing, malignant ND5 and ND11 clones (19). ND5 and ND11 cells showed a dramatic decrease in the levels of both mSmarca2-a and mSmarca2-b, compared with the parental NMuMG non-malignant cells (Fig. 5A). Smarca2-b was still detectable in ND11 clone, but its splicing was changed as evidenced by different patterns of bands on agarose gel between ND11 and NMuMG cells (Fig. 5A). Interestingly, ND11 cells showed an additional band at a higher molecular weight (arrow in Fig. 5A). Cloning this band to a TA vector followed by sequencing some plasmids revealed that it was an alternative splice variant that retained 408 bp of intron 1 at the 3' end of exon 1 (Supplementary Material: Fig. S2). This variant, which lacks exons 3 and 5 as well, was also expressed at a low abundance in some other mouse cancer cell lines (data not shown). We name this variant “mSmarca2-b10” in case similar retention of intron 1 may later be identified in human Smarca2-b, although not yet by now.
Effect of cyclin D1 (D1) and serum starvation on Smarca2 expression. A: Two malignant clones (ND5 and ND11) of NMuMG cells transformed by stable expression of a pcDNA3.1-D1 construct show dramatic decreases in the levels of mSmarca2-a and mSmarca2-b detected by RT-PCR, compared with the parental NMuMG cells. ND11 cells also manifest a changed pattern of different mSmarca2-b variants and express a variant at a high molecular weight (arrow). B: Comparison of the MCF7 cells infected with a retrovirus containing a Flag-D1 construct (D1), a Flag-K112E mutant (KE) or the empty vector (V) shows that D1 dramatically decreases the level of hSmarca2-a but the KE mutant attenuates this effect of D1. C, D and E: Effect of serum starvation and retroviral infection with D1 or its K112E mutant on the levels of hSmarca2-a and hSmarca2-b, detected by RT-PCR, in MDA-MB231 (C), L3.6pL (D) and AsPC-1 (E) cells. F: Illustration of how serum and D1 regulate the expression and splicing of Smarca2-a and Smarca2-b. Serum derived growth stimuli utilize some transcription factors (TFx) to activate or repress the promoter region (grey box) and the intron 27 (that is also the promoter of Smarca2-b) of the Smarca2 gene (black box). D1, which also acts as a downstream effector of serum, binds to some other transcription factors (TFy) to activate or repress these promoter regions, but whether CDK4 or CDK6 is involved is unclear. The pre-mRNA of Smarca2-a is spliced alternatively to the -a1 or -a2 variants while the Smarca2-b pre-mRNA to -b1, -b2… or -b9. These splicing processes may be regulated by serum via D1 or via other splice factors. The effect of D1 on splicing is more evident in a chronic situation as seen in D1 transformed ND11 clone of NMuMG cells, but whether CDK4 or CDK6 is involved remains elusive.
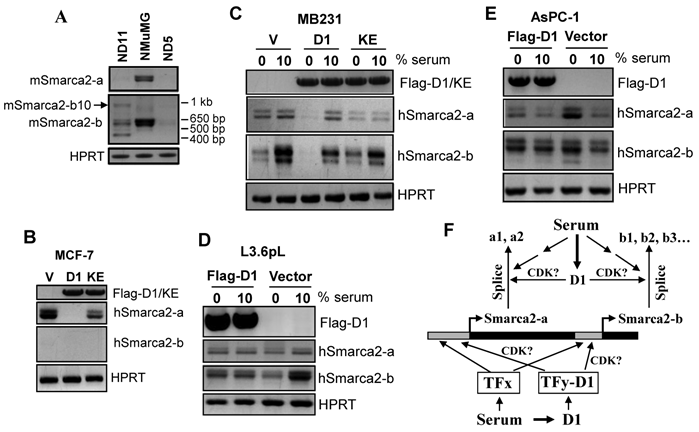
As shown in Fig. 5B, ectopic expression of D1 decreased the Smarca2-a level in MCF7 cells while the Smarca2-b level remained undetectably low under the same condition as seen in the vector infected cells. One major function of D1 is to activate Cyclin Dependent Kinase-4 (CDK4) or -6 (CDK6), but a single nucleotide mutation of D1 that changes the 112th amino acid (K112E) abolishes its activity to activate these CDKs (23). Interestingly, infection of MCF7 cells with a retroviral K112E mutant had only a mild effect on hSmarca2-a (Fig. 5B), indicating that down regulation of hSmarca2-a by D1 largely depends on its activation of CDK4 or CDK6. In MDA-MB231 human breast cancer cells, however, D1 decreased Smarca2-b while affecting Smarca2-a only mildly, if at all, when the cells were cultured with 10% serum (Fig. 5C). K112E mutation also decreased the effect on Smarca2-b, again suggesting its dependence of CDK4 or CDK6 (Fig. 5C).
Like in MDA-MB231 cells, ectopic D1 in L3.6pL cells significantly down regulated Smarca2-b without affecting Smarca2-a (Fig. 5D). However, in AsPC-1 human pancreatic cancer cells, ectopic D1 slightly induced Smarca2-b without significantly affecting Smarca2-a when the cells were cultured with 10% serum (Fig. 5E).
Deprival of MDA-MB231 cells from serum in culture for 48 hours dramatically decreased the level of Smarca2-b without affecting Smarca2-a (Fig. 5C). However, serum starvation together with ectopic D1, but not its K112E mutant, could significantly decrease the Smarca2-a level when compared with the empty vector expressing counterparts, while the Smarca2-b level remained inhibited by serum starvation in D1 or K112E expressing MDA-MB231 cells (Fig. 5C). L3.6pL cells showed a similar decrease in Smarca2-b, but not in Smarca2-a, in response to serum starvation (Fig. 5D). However, unlike in MDA-MB231 cells, serum starvation with a concomitant D1 expression was still unable to significantly change the level of Smarca2-a in L3.6pL cells (Fig. 5D). In opposite to MDA-MB231 and L3.6pL, AsPC-1 cells showed a significant induction of both Smarca2-a and Smarca2-b in response to serum starvation (Fig. 5E). Interestingly, D1 expression attenuated the induction of Smarca2-a by serum starvation, although D1 alone was not able to significantly inhibit Smarca2-a in the presence of 10% serum (Fig. 5E). D1 did not seem to markedly affect Smarca2-b in AsPC-1 cells wherein Smarca2-b seems to be more abundantly expressed than Smarca2-a.
Discussion
Splicing of mSmarca2-a and expression of hSmarcar2-b
The first new finding of our study is that the long transcript in the mouse (mSmarca2-a) also undergoes alternative splicing at the 54-bp exon 29 to produce the -a1 and -a2 variants as seen in the human whereas the human Smarca2 gene also expresses the short transcript, i.e. hSmarca2-b, just like its mouse counterpart. Actually, we identified two transcription initiation sites for hSmarca2-b in the intron 27 of the gene. Another novel finding is that hSmarca2-b can be alternatively spliced at three different 3' sites of exon 1 and at exons 2, 3 and 5. These complex patterns of alternative splicing, together with alternative transcription initiations, may produce 48 different hSmarca2-b mRNA variants, although we have only confirmed nine of them. Thus, in both human and mouse, the Smarca2 gene is expressed to a long (Smarca2-a) and a short (Smarca2-b) transcripts; both transcripts undergo alternative splicing, especially the short one.
Ratios within or among Smarca2-a and Smarca2-b variants
Of the two Smarca2-a variants in both mouse and human, Smarca2-a2 that lacks the exon 29 is expressed at a higher amount in most human and mouse cell lines we studied, relative to Smarca2-a1, since the bottom band of the Smarca2-a duplex in agarose gel is dominant in most cases (Fig. 1B, 1C, 2A and 2B). Skip of this 54-bp exon seems to be prevailing among Smarca2-b variants as well. Therefore, we intend to consider that this exon appears only as a variation whereas the wild type forms of the long and short mRNAs do not contain this exon. Under what condition this exon appears is intriguing but remains to be determined.
One of the surprising findings is that Smarca2-b is detected more abundantly than Smarca2-a in many cell lines, suggesting that the short transcript may be expressed at a higher level in many cases. Therefore, it may be Smarca2-b, not the much better studied BRM proteins translated from Smarca2-a, that better represents the function of the Smarca2 gene. Of the many cell lines studied, MCF7 and T47D breast cancer cells and HC11 mammary epithelial cells are exceptions, as they show the opposite, i.e. a high level of Smarca2-a but a low level of Smarca2-b. Why these exceptions only occur to cell lines derived from mammary epithelium is unclear, probably just fortuitous.
Decristofaro et al report that Panc-1 cells are negative for BRM protein (12) whereas Rosson et al detect relatively high levels of Smarca2-a mRNA and BRM protein in this cell line (15). Like Rosson et al, we detect Smarca2-a mRNA in Panc-1 cells as well. On the other hand, we cannot detect Smarca2-a in SKBR3 cells although DeCristorfard et al detect BRM protein in this cell line (12). Silencing of the Smarca2 gene is generally considered to be due to epigenetic mechanisms (26-28). The same cell lines in different investigators' laboratory may differ in the degree of epigenetic silencing, since epigenetic control such as histone deacetylation can be changed readily. This may be a reason for the discrepant data on the expression status of Smarca2 in Panc-1 and SKBR3 cells observed by different investigators. If so, we may be able to control the behaviors of cancer cells by regulating epigenetic control of the Smarca2 gene, which may have a potential in clinical use eventually.
Complex ORFs encoded by various Smarca2-b variants
The complexity of human Smarca2-b occurs not only at the mRNA level but probably also at the protein level, because our bioinformatic analysis identifies two first ATG translation start codons that may or may not be in the same ORF, depending on whether the variants contain the 35-bp exon 3. Moreover, alternative skip of the 54-bp exon 5 also results in an in-frame deletion of 18 aa. These ORFs encode four different BRM-related proteins with 294, 276, 254 or 236 aa, respectively, in addition to a 98-aa short peptide. In the mouse, mSmarca2-b3 and mSmarca2-b4 encode a 294-aa and a 276-aa protein, respectively, whereas the mSmarca2-b10 may utilize another ATG at the end of exon 1, which adds two more amino acids to the N-terminus of mSmarca2-b4, thus producing a 278-aa protein.
The 98-aa peptide encoded by hSmarca2-b1 and -b2 that contain the 35-bp exon 3 is of interest, if it is really expressed. Because the sequence of its first 31 aa is identical to the corresponding part of the 294-aa and 276-aa proteins (Fig. 3C), it may be considered a member of the BRM family. However, this 31-aa sequence does not exist in BRM or in the 254-aa and 236-aa proteins, and the remaining sequence of 67 aa also differs completely from any part of BRM or BRM related proteins. Therefore, the nature of this 98-aa peptide is uncertain. It needs to be pointed out that the only Smarca2-b mRNA (NM_026002.2) documented in the NCBI database is a mouse sequence that contains the 35-bp exon 3, and its ORF starts from ATG1 in exon 2 and stops earlier, encoding only 72 aa, not 98 aa. The NCBI database shows only the 236-aa protein (NP-080279.1) without documenting this 72-aa peptide, probably because it is too short. It also needs to be mentioned that the variants which contain the 35-bp exon 3 occur only as relatively rare events in both human and mouse in our study. Therefore, the NM_026002.2 sequence in the NCBI database is somewhat misleading as it does not represent the majority. In other words, this 35-bp exon should be considered as an extra one that appears only in minor variants, not in the wild type forms, of hSmarca2-b.
The 254-aa and 236-aa proteins are encoded by the variants that lack exon 2 (b5, b8 and b9) or the variants containing exon 3 (b1 and b2). The possible existence of these proteins somewhat puzzles us because the 254-aa or 236-aa sequence is part of the BRM protein translated from Smarca2-a1 or Smarca2-a2, respectively, as illustrated in Fig. 3C. One of the possibilities is that cells may sometimes prefer a simplified or specified function by the C-terminal part of BRM proteins over the more complex functions by the full-length BRM. The 294-aa and 276-aa proteins encoded by the variants lacking exon 3 but retaining exon 2 (b3, b4, b6 and b7) contain the 31-aa sequence at the N-terminus that does not exist in BRM, thus sharing some functions with BRM on one hand and with the 98-aa peptide on the other. Therefore, these proteins may be a special group of the BRM family and their existence may be meaningful.
Mechanisms for the appearance of small BRM immunoreactive proteins
Using two BRM C-terminal antibodies, we show for the first time western blot data of multiple proteins of 24-35 kD, which may be 1) degraded or proteolyzed products of the 180-kD BRM proteins, 2) proteins that are irrelevant to Smarca2 but immunoreactive to the BRM C-terminal antibodies, i.e. nonspecific bands on the blot, 3) Smarca2-b proteins as they are within the estimated molecular weights, or 4) Smarca2-a proteins translated from some of the 48 in-frame ATGs (AUG is referred to as ATG herein to abide to NCBI database that presents mRNA as DNA sequence, including the Smarca2 mRNA variants). The first possibility is less likely because the 180-kD BRM proteins are detected much less abundantly compared with the 24-35 kD proteins and western blots of β-actin and other proteins (data now shown) confirm the high quality of the protein preparation without obvious degradation. The second or third possibility needs to be determined by more studies. If Smarca2-b proteins exist, each Smarca2-b mRNA variant needs to be causally linked to one or several specific bands on the western blot. Cloning the full-length cDNA of various Smarca2-b variants that encode different ORFs and raising the corresponding antibodies are prerequisites for this line of work, which are tedious and thus beyond this communication. The fourth possibility is of interest as this type of situation is rarely discussed. Generally speaking, some of the in-frame ATGs in many genes, like the 48 in-frame ATGs of hSmarca2-a, may be functional via a non-classical cap-dependent or even cap-independent mechanism of protein translation, including leaky scanning, reinitiation, and IRES (internal ribosome entry site) (29-31). In the case of IRES, whether an in-frame ATG is functional depends in a large part on ITAFs (IRES trans-acting factor) expression of which varies among different cells or cellular situations (32,33). For instance, an IRES existing in the exon 1 of the c-myc oncogene is 20-fold more active in Hela cells than in MCF7 cells (34). In silico analyses suggest that up to 10% of cellular genes contain IRES (33), although it remains to be determined whether Smarca2 mRNAs encompass an IRES. Besides IRES, some relative upstream ATGs may be used as “upstream ATGs” to control translation from the ATGs at their downstream via a leaky scanning or reinitiation mechanism (30,31). By using these non-classical cap-dependent or even cap-independent translation mechanisms, cells can regulate mRNA decay or translation efficiency or can produce different protein isoforms that differ in the N-terminus and thus in some functions (29-31,35).
Regulation of Smarca2 expression by D1 and serum
Few factors have been identified that regulate the expression of Smarca2, especially the Smarca2-b. Serum starvation inhibits cell growth while D1 accelerates it, both via the pRb-E2F pathway. Moreover, D1 is known to act as a sensor of cells to deliver extracellular growth signals, such as those derived from serum, to the cell cycle machinery (Fig. 5F) (36,37). For these reasons, we originally anticipated that serum starvation should induce Smarca2 whereas D1 should inhibit it, since BRM is considered to inhibit cell proliferation by mediating the pRb-E2F pathway (8,9). We indeed observe an induction of hSmarca2-a by serum starvation in AsPC-1 cells and an inhibition of hSmarca2-a by D1 in MCF7 cells as anticipated. Unexpectedly, however, serum starvation does not induce hSmarca2-a in MDA-MB231 and L3.6pL cells whereas D1 alone does not inhibit it in AsPC-1, MDA-MB231 and L3.6pL cells. More surprisingly, although serum starvation significantly changes the levels of Smarca2-b in all three cell lines studied (Fig. 5C, 5D and 5E), these cell lines actually respond quite differently and even oppositely. Obviously, the short transcript is more sensitive to serum-derived factors and thus may be a more important regulator than the Smarca2-a for cell cycle progression, which may be why Smarca2-b is more abundantly expressed than Smarca2-a in many cell lines. Nevertheless, these unexpected results suggest a complex regulation of Smarca2 by serum and D1 and the complexity may occur at two levels: 1) There may be many genes other than D1 that also act as downstream effectors of serum-derived factors to control cell growth by regulation of Smarca2 and these genes show different patterns of changes in different cell lines. 2) D1 per se utilizes multiple mechanisms to regulate Smarca2 expression and splicing (Fig. 5F). As reviewed by two of us recently (36,37), D1 functions via three different mechanisms, i.e. a) binding to CDK4/6 to form a holoenzyme to phosphorylate pRb or other substrates, b) forming a complex with CDK4/6 without involving their kinase activity, or c) acting via a CDK4/6-independent mechanism. The third mode of action has so far been observed only in regulation of genes' transcription, which is achieved by binding of D1 to some transcriptional factors (Fig. 5F) such as steroid hormone receptors. Few, if any, of these transcriptional regulations involve CDK4/6, to our knowledge. For this reason, it is surprising that the K112E mutant that cannot activate CDK4/6 is not as effective as the wild type D1 in regulation of Smarac2-a and Smarca2-b mRNA levels, as it implies the involvement of these CDKs. More studies are needed to further determine whether the effects of D1 on the expression of Smarca2-a or Smarca2-b occur at the transcriptional level.
Although the above discussed short-term or temporary effects of D1 on Smarca2 expression are somewhat less evident and different among the retrovirus infected MCF7, MDA-MB231, L3.6pL and AsPC-1 cells, two clones (ND5 and ND11) of D1 transformed NMuMG cells consistently show that the chronic effects of D1 are dramatic decreases in both Smarca2-a and Smarca2-b expression or altered splicing pattern of Smarca2-b. Whether and how D1 plays a role in RNA splicing is an interesting question that has rarely been studied (Fig. 5F), but it has been reported that D1 regulates splicing of fibroblast growth factor 1 receptor (38) and D1b, a splice variant of D1, utilizes a critical splice factor ASF/SF2 as a downstream effector to regulate splicing (39). These chronic effects seen in ND5 and ND11 cells are likely to be indirect events mediated by other genes, although whether CDK4 or CDK6 is involved remains elusive (Fig. 5F). Nevertheless, these effects not only dovetail with the notion that the Smarca2-a products BRM proteins are tumor suppressors but also further suggest that some of the Smarca2-b splice variants may be tumor suppressive as well.
Conclusions
In summary, mSmarca2-a also engenders two alternative splice variants with or without exon 29, just like hSmarca2-a, whereas the hSmarca2 gene produces the short transcript hSmarca2-b as well, just like its mouse counterpart. Nine hSmarca2-b mRNA variants are identified and many more anticipated, due to two alternative initiation sites as well as alternative splicing at three different 3' splice sites of exon 1 and at exons 2, 3 and 5. mSmarca2-b also undergoes alternative splicing at exon 3 and/or exon 5, besides a retention of part of intron 1 by exon 1. Some of the hSmarca2-b mRNA variants may encode two different ORFs, which together may produce five different proteins. The expression of Smarca2-a and Smarca2-b may be regulated by serum concentration and by D1 in a complex manner, and Smarca2-b is expressed more abundantly than Smarca2-a in many cell lines. These complex patterns of transcription initiation, expression regulation, and alternative splicing collectively suggest that in some situations the functions of the Smarca2 gene may be very complex, not just simply inhibiting cell proliferation or tumor formation, and be exerted mainly by expressing the short transcript Smarca2-b.
Supplementary Material
Fig. S1: Sequence of the region between exon 26 and exon 31 of the mSmarca2-a1 cDNA. Fig. S2: Part of the sequences of the three novel mSmarca2-b variants. Fig. S3: Sequences of the nine hSmarca2-b variants and two of the original 5'RACE sequences.
Acknowledgements
We would like to thank Dr. Fred Bogott at the Medical Center of Austin of Minnesota, for his excellent English editing of this manuscript. This work was supported by a NIH grant (RO1CA100864) to D.J. Liao.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Stamm S, Ben-Ari S, Rafalska I. et al. Function of alternative splicing. Gene. 2005;344:1-20
2. Johnson JM, Castle J, Garrett-Engele P. et al. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302:2141-4
3. Kim E, Goren A, Ast G. Alternative splicing and disease. RNA Biol. 2008;5:17-9
4. Kim E, Goren A, Ast G. Insights into the connection between cancer and alternative splicing. Trends Genet. 2008;24:7-10
5. Pensabene M, Spagnoletti I, Capuano I. et al. Two mutations of BRCA2 gene at exon and splicing site in a woman who underwent oncogenetic counseling. Ann Oncol. 2009;20:874-8
6. Park JW, Graveley BR. Complex alternative splicing. Adv Exp Med Biol. 2007;623:50-63
7. Guo W, Bharmal SJ, Esbona K, Greaser ML. Titin diversity--alternative splicing gone wild. J Biomed Biotechnol. 2010;2010:753675
8. Shen H, Powers N, Saini N. et al. The SWI/SNF ATPase Brm is a gatekeeper of proliferative control in prostate cancer. Cancer Res. 2008;68:10154-62
9. Trouche D, Le CC, Muchardt C, Yaniv M, Kouzarides T. RB and hbrm cooperate to repress the activation functions of E2F1. Proc Natl Acad Sci U S A. 1997;94:11268-73
10. Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene. 2009;28:1653-68
11. Ito T, Watanabe H, Yamamichi N. et al. Brm transactivates the telomerase reverse transcriptase (TERT) gene and modulates the splicing patterns of its transcripts in concert with p54 nrb. Biochem J. 2007;411:201-9
12. Decristofaro MF, Betz BL, Rorie CJ, Reisman DN, Wang W, Weissman BE. Characterization of SWI/SNF protein expression in human breast cancer cell lines and other malignancies. J Cell Physiol. 2001;186:136-45
13. Reisman DN, Sciarrotta J, Wang W, Funkhouser WK, Weissman BE. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res. 2003;63:560-6
14. Yamamichi N, Inada K, Ichinose M. et al. Frequent loss of Brm expression in gastric cancer correlates with histologic features and differentiation state. Cancer Res. 2007;67:10727-35
15. Rosson GB, Bartlett C, Reed W, Weissman BE. BRG1 loss in MiaPaCa2 cells induces an altered cellular morphology and disruption in the organization of the actin cytoskeleton. J Cell Physiol. 2005;205:286-94
16. Glaros S, Cirrincione GM, Muchardt C, Kleer CG, Michael CW, Reisman D. The reversible epigenetic silencing of BRM: implications for clinical targeted therapy. Oncogene. 2007;26:7058-66
17. Gunduz E, Gunduz M, Ali MA. et al. Loss of heterozygosity at the 9p21-24 region and identification of BRM as a candidate tumor suppressor gene in head and neck squamous cell carcinoma. Cancer Invest. 2009;27:661-8
18. Heppner GH, Miller FR, Shekhar PM. Nontransgenic models of breast cancer. Breast Cancer Res. 2000;2:331-4
19. Wang Y, Thakur A, Sun Y. et al. Synergistic effect of cyclin D1 and c-Myc leads to more aggressive and invasive mammary tumors in severe combined immunodeficient mice. Cancer Res. 2007;67:3698-707
20. Biliran HJr, Wang Y, Banerjee S. et al. Overexpression of cyclin D1 promotes tumor cell growth and confers resistance to cisplatin-mediated apoptosis in an elastase-myc transgene-expressing pancreatic tumor cell line. Clin Cancer Res. 2005;11:6075-86
21. Biliran HJr, Banerjee S, Thakur A. et al. c-Myc-induced chemosensitization is mediated by suppression of cyclin D1 expression and nuclear factor-kappa B activity in pancreatic cancer cells. Clin Cancer Res. 2007;13:2811-21
22. Shen KC, Miller F, Tait L. et al. Isolation and characterization of a breast progenitor epithelial cell line with robust DNA damage responses. Breast Cancer Res Treat. 2006;98:357-64
23. Hinds PW, Dowdy SF, Eaton EN, Arnold A, Weinberg RA. Function of a human cyclin gene as an oncogene. Proc Natl Acad Sci U S A. 1994;91:709-13
24. Durnam DM, Palmiter RD. A practical approach for quantitating specific mRNAs by solution hybridization. Anal Biochem. 1983;131:385-93
25. Wang Y, Thakur A, Sun Y. et al. Synergistic effect of cyclin D1 and c-Myc leads to more aggressive and invasive mammary tumors in severe combined immunodeficient mice. Cancer Res. 2007;67:3698-707
26. Yamamichi N, Yamamichi-Nishina M, Mizutani T. et al. The Brm gene suppressed at the post-transcriptional level in various human cell lines is inducible by transient HDAC inhibitor treatment, which exhibits antioncogenic potential. Oncogene. 2005;24:5471-81
27. Yamamichi-Nishina M, Ito T, Mizutani T, Yamamichi N, Watanabe H, Iba H. SW13 cells can transition between two distinct subtypes by switching expression of BRG1 and Brm genes at the post-transcriptional level. J Biol Chem. 2003;278:7422-30
28. Tyagi A, Ryme J, Brodin D, Ostlund Farrants AK, Visa N. SWI/SNF associates with nascent pre-mRNPs and regulates alternative pre-mRNA processing. PLoS Genet. 2009;5:e1000470
29. Van Der KK, Beyaert R, Inze D, De VL. Translational control of eukaryotic gene expression. Crit Rev Biochem Mol Biol. 2009;44:143-68
30. Iacono M, Mignone F, Pesole G. uAUG and uORFs in human and rodent 5'untranslated mRNAs. Gene. 2005;349:97-105
31. Ivanov IP, Atkins JF, Michael AJ. A profusion of upstream open reading frame mechanisms in polyamine-responsive translational regulation. Nucleic Acids Res. 2010;38:353-9
32. King HA, Cobbold LC, Willis AE. The role of IRES trans-acting factors in regulating translation initiation. Biochem Soc Trans. 2010;38:1581-6
33. Spriggs KA, Stoneley M, Bushell M, Willis AE. Re-programming of translation following cell stress allows IRES-mediated translation to predominate. Biol Cell. 2008;100:27-38
34. Stoneley M, Subkhankulova T, Le Quesne JP. et al. Analysis of the c-myc IRES; a potential role for cell-type specific trans-acting factors and the nuclear compartment. Nucleic Acids Res. 2000;28:687-94
35. Nyiko T, Sonkoly B, Merai Z, Benkovics AH, Silhavy D. Plant upstream ORFs can trigger nonsense-mediated mRNA decay in a size-dependent manner. Plant Mol Biol. 2009;71:367-78
36. Wang C, Tai Y, Lisanti MP, Liao DJ. c-Myc induction of programmed cell death may contribute to carcinogenesis: a perspective inspired by several concepts of chemical carcinogenesis. Cancer Biol Ther. 2011;11:615-26
37. Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle. 2011;10:57-67
38. Chen G, Wang J, Liu Z, Kornmann M. Exon III splicing of fibroblast growth factor receptor 1 is modulated by growth factors and cyclin D1. Pancreas. 2008;37:159-64
39. Olshavsky NA, Comstock CE, Schiewer MJ. et al. Identification of ASF/SF2 as a critical, allele-specific effector of the cyclin D1b oncogene. Cancer Res. 2010;70:3975-84
Author contact
![]() Corresponding author: D. Joshua Liao, Hormel Institute, University of Minnesota, Austin, MN 55912, USA. Tel: 507-437-9665; Fax: 507-437-9606; Email: djliaoumn.edu
Corresponding author: D. Joshua Liao, Hormel Institute, University of Minnesota, Austin, MN 55912, USA. Tel: 507-437-9665; Fax: 507-437-9606; Email: djliaoumn.edu