Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2011; 2:324-328. doi:10.7150/jca.2.324 This volume Cite
Short Report
Targeting EGFR in Triple Negative Breast Cancer
Naoto T. Ueno1,2, Dongwei Zhang1,2 ![]()
1. Breast Cancer Translational Research Laboratory, The University of Texas MD Anderson Cancer Center, Houston, TX, USA;
2. Department of Breast Medical Oncology, Morgan Welch Inflammatory Breast Cancer Research Program, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Received 2011-5-16; Accepted 2011-5-28; Published 2011-5-28
Abstract
Our preliminary data show that erlotinib inhibits Triple-negative breast cancer (TNBC) in a xenograft model. However, inhibition of metastasis by erlotinib is accompanied by nonspecific effects because erlotinib can inhibit other kinases; thus, more direct targets that regulate TNBC metastasis need to be identified to improve its therapeutic efficacy.
Keywords: triple negative breast cancer, tyrosine kinase inhibitor, EGFR, erlotinib, metastasis
Targeted Therapy in Triple Negative Breast Cancer (TNBC)
In 2010, 209,060 patients are expected to be diagnosed with breast cancer in the United States.1 At least 20% of breast cancers are characterized by triple-negative receptor status (negative for estrogen receptor [ER], progesterone receptor [PR], and HER2).2 Triple-negative breast cancer (TNBC) is highly proliferative and sensitive to systemic chemotherapies. However, despite its relative sensitivity to chemotherapy, patient outcomes are poor compared with other subtypes of breast cancer (HER2+ or ER+). The cause of death of patients with TNBC is often recurrence (30-40% of TNBC cases), which presents as distant metastasis. This means that chemosensitivity of TNBC does not reflect whether the cancer will metastasize. Therefore, the mechanism of metastasis needs to be studied separately from that of tumorigenicity.
TNBC is the only major type of breast cancer for which no specific FDA-approved targeted therapy is available to improve patient outcomes; it is resistant to targeted therapies such as hormonal therapy and HER2-targeting therapy. A PARP inhibitor is under development, but it is not yet an established standard therapy. Our preliminary data show that erlotinib inhibits TNBC in a xenograft model, a promising result. However, inhibition of metastasis by erlotinib is accompanied by nonspecific effects because erlotinib can inhibit other kinases; thus, more direct targets that regulate TNBC metastasis need to be identified to improve its therapeutic efficacy.
EGFR overexpression in TNBC
EGFR is known to be overexpressed in TNBC.3-5 The question remains whether EGFR is a valid target when many of the Phase II study of EGFR tyrosine-kinase inhibitor in metastatic breast cancer has at most 5% response rate. However, At the European Society for Medical Oncology meeting in October 2010, a major breakthrough in EGFR-targeted therapy was reported.6 In this randomized phase II study, 173 patients with metastatic TNBC were randomized to receive either cetuximab, an anti-EGFR antibody, plus up to six 3-week cycles of cisplatin (N=115) or cisplatin alone (N=58). An overall response rate of 20% was seen in patients who received the cetuximab/cisplatin combination, compared with a response rate of 10.3% in the cisplatin-alone arm. Adding cetuximab to cisplatin doubled the progression-free survival duration from 1.5 to 3.7 months (HR 0.675, P = 0.032). These exciting clinical data suggest that EGFR is an important target in TNBC.6
EGFR inhibitor induced a change from mesenchymal to epithelial phenotype in TNBC cells
Human TNBC may have characteristics of epithelial-mesenchymal transition (EMT).4, 5 EMT is an essential developmental process by which cells of epithelial origin lose epithelial characteristics and acquire a mesenchymal phenotype with fibroblast-like morphology, cytoskeleton reorganization, increased motility, invasiveness, and metastatic capability.7, 8 EMT is characterized by loss of epithelial cell junction proteins (e.g., E-cadherin and cytokeratins) and gain of mesenchymal markers (e.g., vimentin and fibronectin).7, 8 It has been proposed that EMT-like processes allow tumor cells to disassemble and migrate to tissue or organ sites distant from the primary tumor (metastasis).7, 8 It has become increasingly clear that EMT may play a major role in invasion and metastasis of TNBC.3-5
We have previously shown that epidermal growth factor stimulates EGFR-expressing TNBC cells such as SUM149, MDA-MB-468, and BT-20 and induces upregulation of phosphorylation of ERK and/or Akt. Further, when these cells are treated with the EGFR tyrosine kinase inhibitor (TKI) erlotinib, ERK and/or Akt phosphorylation can be downregulated. These data suggest that the EGFR pathway is intact in these cells.9, 10
To further elucidate EGFR pathway activity in TNBC, we examined whether TNBC cells (SUM149) exhibited epithelial or mesenchymal phenotypes. SUM149 cells were plated using a three-dimensional (3D) culture system. The 3D culture was performed with 2% Matrigel in F12 medium containing 5% FBS. Matrigel is a gelatinous protein mixture secreted by mouse tumor cells, which resembles the complex extracellular environment found in many tissues. This 3D culture system can be used to assess cell invasiveness and motility, which reflect part of the epithelial-to-mesenchymal morphological process due to the rapid formation of projections and filopodia. Such projections or filopodia also may strongly correlate with metastatic potential.11-14
In a two-dimensional (2D) culture system, using standard culture plates with F12 medium containing 5% FBS, SUM149 cells showed an epithelial phenotype characterized by the localization of E-cadherin and β-catenin at the sites of cell-cell contact (data not shown). However, in a 3D-culture system, SUM149 cells rapidly formed projections or filopodia and changed to a mesenchymal-like phenotype (Fig. 1A) within 24 hours.
EGFR inhibitor induced a change from mesenchymal to epithelial phenotype in TNBC cells. A, SUM149 TNBC cells were grown (3D culture) in the presence of 0 or 0.1 μM erlotinib, an EGFR tyrosine kinase inhibitor. Erlotinib (0.1 μM) inhibited the formation of projections in 3D models at 24 h. In 2D culture, no projections formed. B, Immunostaining performed after 24 h of treatment with erlotinib shows upregulation of epithelial marker E-cadherin, cell membrane localization of β-catenin, and inhibition of mesenchymal marker vimentin. The figure is referenced from Clin Cancer Res. 2009;15(21):6639-48.
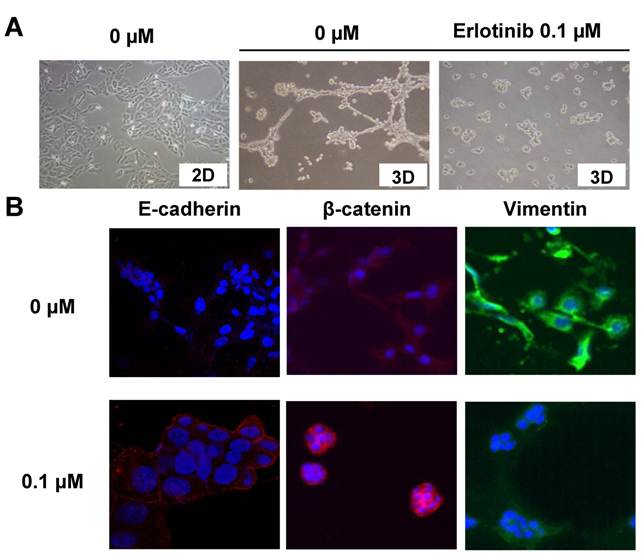
In 3D culture, SUM149 cells with projections exhibited reduced E-cadherin expression and increased vimentin expression compared with SUM149 cells in 2D culture. Also, β-catenin accumulated at the cytoplasm in 3D culture (Fig. 1B).
However, when these cells were treated with the EGFR-TKI erlotinib, the mesenchymal cell phenotype changed back to epithelial-like in the 3D system, E-cadherin was upregulated, vimentin was downregulated, and β-catenin localized to the cell membrane (Fig. 1B). Interestingly, the erlotinib concentration that inhibited the mesenchymal phenotype (0.1 μM) did not have a cytotoxic effect and was one log lower than the concentration that inhibited proliferation (1 μM).15
EGFR tyrosine kinase inhibitor erlotinib inhibited tumor growth and metastasis in a SUM149 xenograft mouse model
After observing that erlotinib reversed the mesenchymal phenotype of SUM149 TNBC cells in vitro, we hypothesized that erlotinib inhibits tumor growth and metastasis in a SUM149 TNBC xenograft model.15 To address this hypothesis, we generated a SUM149 xenograft model by implanting luciferase-expressing SUM149 (SUM149-luc) cells into the mammary fat pads (mfp) of athymic nude mice. SUM149 tumors were readily apparent in the mfp after 14 d and grew rapidly. Beginning 3 w after tumor cell implantation, the mice were treated with erlotinib or a vehicle control administered by oral gavage daily for 4 w. To identify the optimal doses that inhibit tumor growth and/or metastasis, we used three different doses of erlotinib (25, 50, or 100 mg per kilogram of body weight). The doses of 50 and 100 mg/kg/d were chosen based on previous studies of antitumor activity of erlotinib.16, 17 The dose of 25 mg/kg/day was chosen because of its known subtherapeutic effect.
The higher doses (50 and 100 mg/kg) of erlotinib showed significant tumor-growth inhibition in the SUM149 xenograft model (Fig. 2A). On day 49, the mean tumor-growth inhibition rates compared with the control group were 84% in the 50-mg/kg group (P < 0.0001) and 103% in the 100-mg/kg group (P < 0.0001). Some tumor growth inhibition (43%) was observed in the 25-mg/kg group, but this inhibition was not significant compared with the control group (P = 0.06) (Fig. 2A). To evaluate the anti-metastatic activity of erlotinib in TNBC, at the endpoint of the animal study, we collected lung tissues from the mice and then performed ex vivo luciferase imaging to detect spontaneous lung metastases (Fig. 2B). The metastatic tumors in bioluminescence-positive lung tissues were further confirmed by hematoxylin and eosin staining (Fig. 2C). Bioluminescence-positive lungs, indicating the presence of metastatic tumors, were detected in 3 of 7 mice (43%) in the control group. However, no bioluminescence-positive lungs were detected in mice in the three erlotinib-treated groups, indicating that erlotinib inhibited lung metastasis of TNBC (P = 0.04) (Fig. 2D).15
Erlotinib inhibited tumor growth and metastasis in a SUM149 xenograft model. A, Tumor volumes in 4 groups of mice (vehicle, 25, 50, and 100 mg/kg erlotinib) were measured weekly. Each data point represents the mean tumor volume of 7 mice per group; bars, SD. B, Findings on ex vivo imaging of lung tissues to detect metastatic tumors in a control (vehicle) mouse and a 25-mg/kg erlotinib-treated mouse. C, Hematoxylin and eosin staining of a lung tissue section from a control mouse at the endpoint of the study showed 2 small deposits of metastatic tumors (arrowheads) in the alveolar septae. D, The incidence of lung metastasis in the 4 groups of mice. E, Immunohistochemical analysis of p-EGFR, p-ERK, E-cadherin, and vimentin in tumor tissues of SUM149 xenografts. The figure is referenced from Clin Cancer Res. 2009;15(21):6639-48.
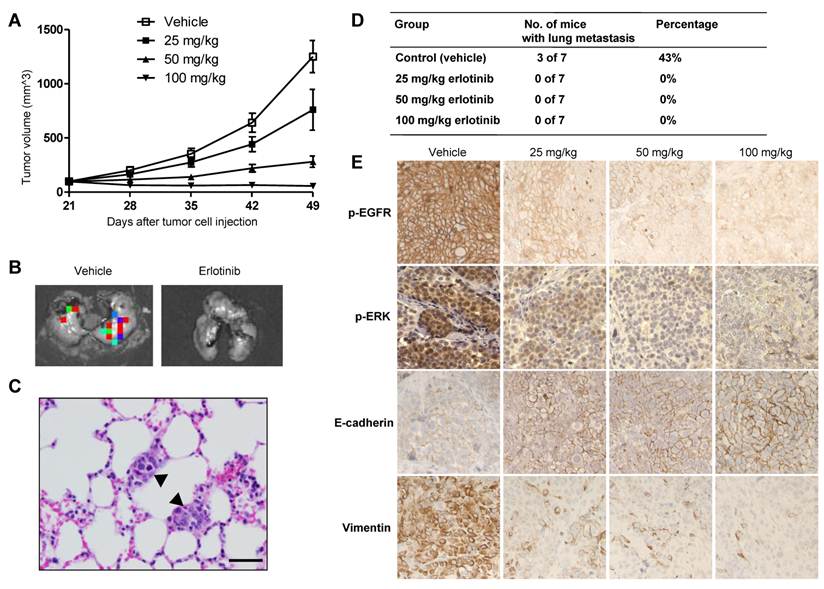
In immunohistochemical analysis, erlotinib increased the expression of the epithelial marker E-cadherin and lowered the expression of the mesenchymal marker vimentin in all treated tumors compared with control tumors (Fig. 2E). In conclusion, inhibition of lung metastasis and reversal from a mesenchymal to an epithelial phenotype were observed even in the low-dose condition (25-mg/kg erlotinib), whereas at that dose primary tumor growth was similar between the treated and control groups.15 These findings suggest that EMT modulation by targeting EGFR may reduce metastasis of TNBC.
Summary
The above results support a concept that metastasis and tumorigenicity should be considered separately and justify the need to study the role of EGFR in TNBC metastasis.
Acknowledgements
This work was supported by NIH grant R01 CA123318-01A1, Morgan Welch Inflammatory Breast Cancer Research Program, and State of Texas Rare and Aggressive Breast Cancer Research Program Grant (NT. Ueno).
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277-300
2. Society AC. Cancer Facts and Figures. American Cancer Society. 2010:1-68
3. Gluz O, Liedtke C, Gottschalk N, Pusztai L, Nitz U, Harbeck N. Triple-negative breast cancer--current status and future directions. Ann Oncol. 2009;20(12):1913-27
4. Rodriguez-Pinilla SM, Sarrio D, Honrado E, Moreno-Bueno G, Hardisson D, Calero F. et al. Vimentin and laminin expression is associated with basal-like phenotype in both sporadic and BRCA1-associated breast carcinomas. J Clin Pathol. 2007;60(9):1006-12
5. Sarrio D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68(4):989-97
6. Baselga J. The addition of cetuximab to cisplatin increases overall response rate (ORR) and progression-free survival (PFS) in metastatic triple-negative breast cancer (TNBC): results of a randomized phase II study (BALI-1). Milan: 35th Congress of the European Society for Medical Oncology (ESMO). 2010
7. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127(4):679-95
8. Savagner P. Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays. 2001;23(10):912-23
9. Yamasaki F, Zhang D, Bartholomeusz C, Sudo T, Hortobagyi GN, Kurisu K. et al. Sensitivity of breast cancer cells to erlotinib depends on cyclin-dependent kinase 2 activity. Mol Cancer Ther. 2007;6(8):2168-77
10. Yamasaki F, Johansen MJ, Zhang D, Krishnamurthy S, Felix E, Bartholomeusz C. et al. Acquired resistance to erlotinib in A-431 epidermoid cancer cells requires down-regulation of MMAC1/PTEN and up-regulation of phosphorylated Akt. Cancer Res. 2007;67(12):5779-88
11. Qian F, Vaux DL, Weissman IL. Expression of the integrin alpha 4 beta 1 on melanoma cells can inhibit the invasive stage of metastasis formation. Cell. 1994;77(3):335-47
12. Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26(5):711-24
13. Van Hoof D, Braam SR, Dormeyers W, Ward-van Oostwaard D, Heck AJ, Krijgsveld J. et al. Feeder-free Monolayer Cultures of Human Embryonic Stem Cells Express an Epithelial Plasma Membrane Protein Profile. Stem Cells. 2008;26:2777-2781
14. Ward KR, Zhang KX, Somasiri AM, Roskelley CD, Schrader JW. Expression of activated M-Ras in a murine mammary epithelial cell line induces epithelial-mesenchymal transition and tumorigenesis. Oncogene. 2004;23(6):1187-96
15. Zhang D, LaFortune TA, Krishnamurthy S, Esteva FJ, Cristofanilli M, Liu P. et al. Epidermal growth factor receptor tyrosine kinase inhibitor reverses mesenchymal to epithelial phenotype and inhibits metastasis in inflammatory breast cancer. Clin Cancer Res. 2009;15(21):6639-48
16. Friess T, Scheuer W, Hasmann M. Combination treatment with erlotinib and pertuzumab against human tumor xenografts is superior to monotherapy. Clin Cancer Res. 2005;11(14):5300-9
17. Ouchi KF, Yanagisawa M, Sekiguchi F, Tanaka Y. Antitumor activity of erlotinib in combination with capecitabine in human tumor xenograft models. Cancer Chemother Pharmacol. 2006;57(5):693-702
Author contact
![]() Corresponding author: Prof. Naoto T. Ueno, Phone: 713-792-8754; Fax: 713-794-4385; E-mail: nuenoorg
Corresponding author: Prof. Naoto T. Ueno, Phone: 713-792-8754; Fax: 713-794-4385; E-mail: nuenoorg