Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2011; 2:123-131. doi:10.7150/jca.2.123 This volume Cite
Research Paper
In Vivo Imaging of Human Malignant Mesothelioma Grown Orthotopically in the Peritoneal Cavity of Nude Mice
Mingqian Feng1, Jingli Zhang1, Miriam Anver2, Raffit Hassan1, Mitchell Ho1 ![]()
1. Laboratory of Molecular Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, 37 Convent Drive, Bethesda, MD 20892, USA;
2. Pathology/Histotechnology Laboratory, SAIC-Frederick, National Cancer Institute, P.O. Box B, Frederick, MD 21702, USA
Received 2011-1-22; Accepted 2011-3-1; Published 2011-3-1
Abstract
Malignant mesothelioma (MM) causes significant morbidity and mortality in patients. With increasing efforts devoted to developing therapeutics targeting mesothelioma, a xenograft mouse model with in vivo tumor imaging is especially desired for evaluating anti-tumor therapies. In the present study, we fluorescently labeled the NCI-H226 human mesothelioma cell line by a lentiviral vector harboring a luciferase-GFP (Luc/GFP) fusion gene driven by the RNA polymerase II promoter. After single-cell cloning by flow cytometry, a clone (named LMB-H226-GL) that stably expresses high levels of Luc/GFP was obtained. The in vivo tumorigenicity of Luc/GFP-labeled LMB-H226-GL was determined by using intraperitoneal injections of the cells in nude mice. LMB-H226-GL was found to be able to consistently form solid tumors in the peritoneum of mice. Tumor growth and aggressive progression could be quantitated via in vivo bioluminescence imaging. The model exhibited the pathological hallmarks consistent with the clinical progression of MM in terms of tumor growth and spread inside the peritoneal cavity. To evaluate the in vivo efficacy of drugs targeting mesothelioma, we treated mice with SS1P, a recombinant immunotoxin currently evaluated in Phase II clinical trials for treatment of mesothelioma. All the tumor-bearing mice had a significant response to SS1P treatment. Our results showed that this is a well-suited model for mesothelioma, and may be useful for evaluating other novel agents for mesothelioma treatment in vivo.
Keywords: H226/NCI-H226, mouse xenograft model, in vivo bioluminescence imaging, malignant mesothelioma, mesothelin, monoclonal antibody, immunotoxin, preclinical model
Introduction
Malignant mesothelioma (MM) is a fatal asbestos-associated malignancy. It commonly originates from the mesothelial cells lining the pleural and peritoneal cavities, as well as the pericardium and tunica vaginalis testis [1]. Although combination chemotherapy with pemetrexed plus cisplatin has been shown to be effective, the median overall survival of these patients is only 12 months [1]. One of the promising approaches that may improve patient outcome involves the use of monoclonal antibodies (mAb) [2].
Mesothelin is a glycosyl-phosphatidylinositol (GPI)-anchored cell surface glycoprotein. It was first identified in 1992 by a mAb generated by the immunization of mice with human ovarian carcinoma cells [3]. The mesothelin gene encodes a 71-kDa precursor protein that is processed to a 40-kDa membrane-bound protein and a 31-kDa fragment (a megakaryocyte-potentiating factor) that is released from the cell after furin cleavage [4, 5]. Mesothelin is a differentiation antigen whose expression is limited to mesothelial cells lining the body cavity [6, 7]. It is overexpressed in a variety of cancers including mesothelioma, ovarian cancer, and pancreatic cancer. In addition, mesothelin is expressed on the surface of lung adenocarcinomas [8, 9]. Recently, our preliminary studies have shown that mesothelin is a potential therapeutic target in cholangiocarcinoma (CCA) [10]. We have demonstrated strong immunochemical mesothelin staining in about 30% of the surgically resected CCA specimens. No mesothelin staining is present in hepatocelluar carcinoma or normal liver tissue. Mesothelin is primarily localized to the cellular plasma membrane and the mature form (~40 kDa) is expressed at a high level in CCA tissues. We and others have shown that mesothelin is shed from tumor cells [11, 12]. Given its link to tumor cells, serum mesothelin has been approved by the U.S. Food and Drug Administration for use as a diagnostic biomarker in MM.
The limited distribution of mesothelin on normal tissues makes it a promising target for antibody-based immunotherapy. Pastan and colleagues have developed a recombinant immunotoxin (named SS1P) that targets mesothelin-expressing tumors. It contains a murine SS1 Fv fused to a 38-kDa fragment of Pseudomonas exotoxin A (PE38) [13]. Two Phase I clinical trials of single agent SS1P have been completed at the National Cancer Institute (NCI) , National Institutes of Health (NIH) in Bethesda, MD and is currently being evaluated in combination with chemotherapy for treatment of patients with mesothelioma [14, 15]. A chimeric antibody (MORAb-009) containing the same murine SS1 Fv specific for mesothelin was also developed and is currently being examined in a Phase II clinical trial for mesothelioma and pancreatic cancer [7, 16]. We have recently generated a high affinity fully human IgG (named HN1) specific for mesothelin [17]. HN1 kills cancer cells with very strong antibody-dependent cell-mediated cytotoxicity. The HN1 epitope is different from that of SS1. HN1 binds to cell surface-associated mesothelin on human mesothelioma, ovarian cancer, lung adenocarcinoma and pancreatic cancer cells.
Despite such advances in mesothelin-targeted cancer therapy, the biological functions of mesothelin remain largely unknown given that there are no apparent abnormalities in mesothelin knockout mice [18]. Nevertheless, mesothelin interacts with mucin MUC16 (also known as CA125) [19, 20], indicating that mesothelin may play an important role in the metastatic spread of mesothelin-bearing cancer cells in the peritoneal cavity. We have shown that it is primarily the N terminus of the extracellular domain of mesothelin (residues 296-359) that is sufficient and necessary for binding to MUC16 [20]. We have found that the MUC16-binding domain can effectively inhibit heterotypic cancer cell adhesion, indicating that the domain is a good candidate for use as a potential antagonist to prevent or treat peritoneal malignant tumors.
Xenograft models are important tools for preclinical evaluation of SS1P and other anti-mesothelioma targeted therapies. Recently, Yanagihara et al. established an orthotopic implantation mouse model of pleural mesothelioma [21]. In the present study, we engineered the human mesothelioma cell line NCI-H226 (named LMB-H226-GL) to express very high levels of GFP-luciferase fusion proteins (Luc/GFP). Using the resultant LMB-H226-GL cell line, we established an MM model in nude mice. In this model, tumor growth can be precisely measured by non-invasive and very sensitive in vivo bioluminescence imaging. Importantly, the pathological hallmarks of the model were consistent with the clinical progression of MM in terms of tumor growth and spread inside the peritoneal cavity. We further examined the effectiveness of the anti-mesothelin immunotoxin SS1P in our model. We found that all of the mice with tumor burdens had significant response upon SS1P treatment, as evidenced by in vivo imaging and necropsy observance, which strongly supports the role that SS1P may have as a potent anti-mesothelioma drug candidate.
Materials and Methods
Cell lines
The human mesothelioma cell line NCI-H226 was obtained from the American Type Culture Collection (ATCC, Rockville, MD). The cell line was maintained as adherent monolayer cultures in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, UT), 1% L-glutamine, and 1% penicillin-streptomycin (Invitrogen) and incubated in 5% CO2 with a balance of air at 37°C. Cells were harvested and the media were changed twice a week. Cells were confirmed to be negative for mycoplasma.
Generation of the LMB-H226-GL cell line
The NIH Institutional Biosafety Committee approved the Recombinant DNA Registration Document (RD-08-IX-07) for the following experiment and animal studies using the LMB-H226-GL cell line. The lentiviral vectors containing a plasmid (plenti4-B1-pPol2-B3-fluc-eGFP-B2) encoding the Luc/GFP were prepared by Viral Technology Laboratory, Advanced Technology Program, SAIC-Frederick, Inc. as previously described [22].
Lentiviral supernatant was added into a culture plate containing 20-40% confluent parental NCI-H226 cells. The plate was incubated at 37°C and the medium changed the following day. Two days after infection, GFP expression was examined by flow cytometry. At day 10, only cells showing the top 5% of high GFP expression were collected by flow cytometry. After single-cell cloning, the highest GFP expression clone (LMB-H226-GL) was selected and maintained in RPMI 1640 media supplemented with 10% FBS, 1% glutamine, and 1% streptomycin/penicillin.
Cell growth inhibition assays
Cell growth inhibition was assessed by WST-8 assay using the Cell Counting Kit-8 (Dojindo, Gaithersburg, MD) according to the manufacturer's instructions. Two hundred microliters of either NCI-H226 or LMB-H226-GL cells were seeded at 1x104 cells per well into a 96-well plate, with the addition of SS1P at the indicated concentrations. Cells were incubated at 37°C for 72 hours, followed by a cytotoxicity assay.
Flow cytometry
Cells were harvested in cell dissociation solution (Invitrogen), washed, and resuspended in ice-cold phosphate buffered saline (PBS) containing 5% bovine serum albumin (BSA). Cells were incubated with 5 μg/mL of MN (mesothelin mAb; cat. # 200-301-A88; Rockland, Gilbertsville, PA). Binding was detected with goat anti-mouse IgG conjugated with phycoerythrin (Sigma-Aldrich, St. Louis, MO). The fluorescence associated with the live cells was measured using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA).
Animal experiments
The NCI-Bethesda Animal Study Proposal (LMB-059) was approved by the NIH Animal Care and Use Committee. Six-week old female athymic nude mice (ATHYMIC NCr-nu/nu) obtained from the National Cancer Institute-Frederick Animal Production Area (Frederick, MD) were housed in micro-isolator cages throughout the course of the experiment. The mice were maintained per the institutional guidelines of the NIH. Five (low-dosage group) or 10 (high-dosage group) million LMB-H226-GL cells in 500 μl of growth media were injected intraperitoneally (i.p.) per mouse.
In vivo bioluminescence measurements
Tumor sizes were assessed via in vivo bioluminescence measurement using the IVIS Imaging System (Caliper Life Sciences, Hanover, MD). For the luciferase detection imaging, 200 μl of 15 mg/mL D-luciferin (Caliper Life Sciences) in PBS was injected i.p. before imaging. The photometry of the tumor was calculated by software Living Image 3.1.0, (Caliper Life Sciences) and the results were used to generate the tumor growth curve. To correlate fluorescence intensity and tumor size, tumor dimensions were determined using calipers, and the tumor volume (mm3) was calculated by the formula: length x (width) 2 x 0.4.
Necropsy and histopathology
At the end of the in vivo tumor growth study, a subset of mice from control and treatment groups were euthanized with CO2, the abdominal cavity was opened, and a photo of the tumor in situ was taken. A complete necropsy was performed with collection of all major organs and tissues as well as gross lesions suggestive of neoplasia. Tissues were fixed in 10% buffered neutral formalin, processed to paraffin and sectioned at 5 microns. Slides were stained with hematoxylin and eosin (H&E) or used for immunohistochemistry.
Two antibodies were used: anti-mesothelin to confirm the identity of the cell line and anti-GFP to correlate with in vivo images. Mesothelin (Sigma-Aldrich, cat. #HPA017172) made in rabbit was diluted 1:300 and incubated on sections for 30 minutes at room temperature. Staining was achieved on the Leica Biosystems Bond Autostainer using normal goat serum and biotin-conjugated secondary antibody goat/anti-rabbit from Vector Laboratories (Burlingame, CA). Pretreatment consisted of citrate heat-induced epitope retrieval. The Intense R Detection Kit was used with the Autostainer.
The anti-GFP antibody made in rabbit was obtained from Abcam (cat. #ab6556, Cambridge, MA). Manual staining was performed and the antibody was diluted 1:5000 and incubated on sections for 30 minutes at 37oC. Pretreatment included normal goat serum, and biotin-conjugated goat anti-rabbit secondary antibody from Vector Laboratories was used, with diamino benzidine as chromagen and hematoxylin as a counter-stain.
Pathology evaluation was performed by a board-certified veterinary pathologist and correlated with in vivo images.
Immunotoxin treatment
Two treatment groups and one control group were set up with eight mice in each group. Each mouse was initially i.p. injected with 10 million LMB-H226-GL cells in 500 μl of growth media. Each mouse in the treatment groups received 0.4 mg/kg SS1P in PBS buffer and the control group received PBS. One treatment group (treatd1) was started on day one (the day when the mice received the cells was set as day 1) then received four further doses of SS1P every other day thereafter. The second treatment group (treatd9) was started on day nine, and then received three further doses of SS1P as in the treatd1 group. Suppression of tumor growth was measured by in vivo imaging.
Statistical analysis
Statistical analysis was performed with Prism (version 5) for Windows (GraphPad Software). Raw data were analyzed by “analysis of variance” with Dunnett's and Newman-Keuls multiple comparison post tests. p values < 0.05 were considered statistically significant.
Results
Generation and characterization of the LMB-H226-GL cell line
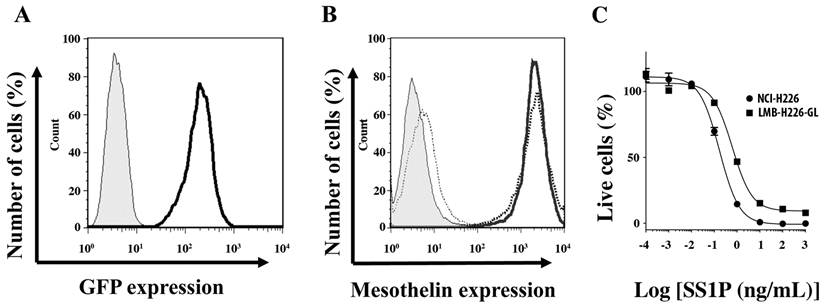
NCI-H226, as one of the NCI-60 cell lines, is a well characterized mesothelioma cell line with high expression levels of mesothelin. Deemed suitable for establishing our model, the NCI-H226 cells were transfected with a lentiviral vector that harbors the Luc/GFP fusion gene under the control of RNA polymerase II promoter (Pol2). After three rounds of sorting by flow cytometry and single-cell cloning, a clone (named LMB-H226-GL) with a very high level of GFP expression was obtained (Fig. 1A). The resultant LMB-H226-GL cell line was then evaluated for cell surface expression of mesothelin and sensitivity to the SS1P immunotoxin. No differences were observed in mesothelin expression levels between NCI-H226 and LMB-H226-GL (Fig. 1B). In addition, there were no significant differences between the IC50 values of NCI-H226 and LMB-H226-GL. The IC50 value for LMB-H226-GL was 0.6 ng/mL and 0.4 ng/mL for NCI-H226 (Fig. 1C). Thus, we determined that the LMB-H226-GL line may be appropriately used for in vivo studies of mesothelin-targeted drugs.
In vitro characterization of LMB-H226-GL cells. A. FACS analysis showing GFP fluorescence in the LMB-H226-GL cell line. Shaded area: parental NCI-H226; heavy solid line: LMB-H226-GL. B. Mesothelin expression on LMB-H226-GL. Shaded area: NCI-H226 without anti-mesothelin staining; thin dotted line: LMB-H226-GL without staining; heavy solid line: NCI-H226 with staining; heavy dotted line: LMB-H226-GL with staining. C. WST-8 assay of NCI-H226 and LMB-H226-GL. Various amounts of SS1P immunotoxin were used to treat NCI-H226 and LMB-H226-GL cells.
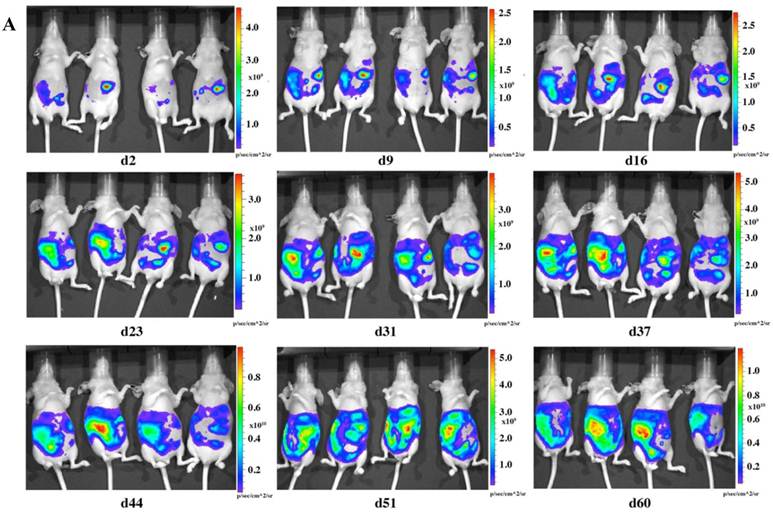
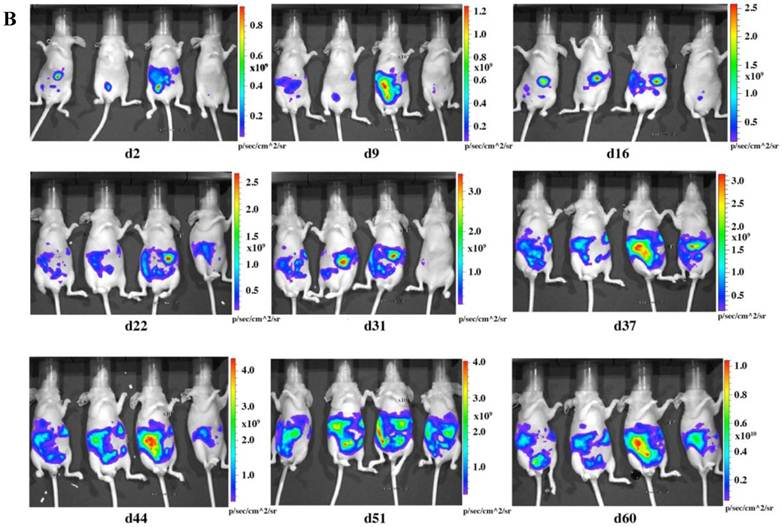
In vivo imaging of tumor growth
To check the tumorigenicity of LMB-H226-GL, we designed two groups of mice based on the numbers of cells injected: the high-dosage group (10 million cells per mouse) (Fig. 2a) and the low-dosage group (5 million cells per mouse) (Fig. 2b). The growth rate of the high-dosage group was similar to that of the low-dosage group. The tumor size in the high-dosage group was relatively larger (Fig. 3a) at the same time point.
In vivo imaging of LMB-H226-GL tumors in nude mice. A. High-dosage group, each mouse received 10 million cells. Animals were imaged the following day (d2) and then once every week thereafter. B. Low-dosage group, each mouse received five million cells and were imaged following the same schedule as the high-dosage group.
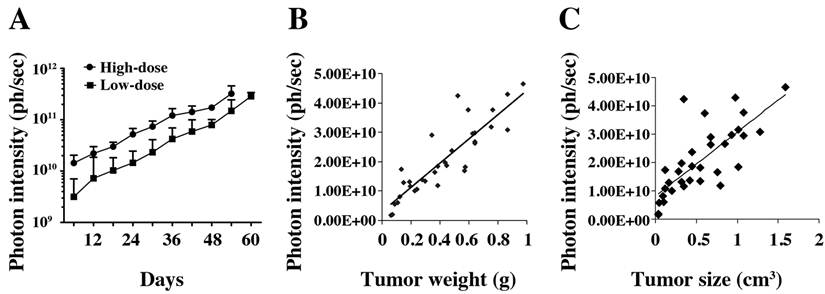
In vivo tumor growth curve. A. Tumor growth was measured by bioluminescence photometry. The day when the mice were injected with the tumor cells was set as d1. The photometry of the in vivo imaging was accumulated by software Living Image 3.1.0 (Caliper Life Sciences) and the resulting photon counts were plotted as the Y-axis. Error bars represent the standard deviation from measurement of four mice in each corresponding group. At the end of the study, mice were sacrificed; tumors were immediately removed and measured by weight, size and fluorescence. Association of photon intensity with tumor weight (B) or size (C) was also performed. A stronger correlation between bioluminescence and tumor weight is shown (correlation coefficients are 0.77 for weight and 0.57 for size).
The distribution of luminescence was irregular in the abdomen at the beginning and the tumors eventually spread to fill the entire abdomen (Fig. 2). At day 60 (high-dosage group) or day 67 (low-dosage group), most of the mice were thin, sick, and less active, indicating the end-point of the experiment. During the whole study, only one mouse was found to have a distended abdomen, but no ascites was found in the abdomen.
A dual reporter system may greatly enhance the value of mouse cancer models as shown by Dai et al. [22]. In the present study, we used the same previously well-established vector [22]. The LMB-H226-GL cells highly coexpressed Luc/GFP. We used luciferase to quantitate in vivo tumor growth. The light emission of the luciferase system can be detected in the imager after i.p. injection of the substrate luciferin into the mouse. The signal quickly reached its peak three minutes after injection, and the peak was maintained for at least 30 minutes. As for the GFP reporter, it may be particularly useful to isolate mesothelioma cells from mice by flow cytometry after forming tumors as previously shown [22] and to investigate the molecular oncogenesis of mesothelioma by analyzing the changes in gene expression profiles.
At the end of the experiment, the mice were euthanized, and tumors were immediately removed and measured by weight (Fig. 3B) and volume (Fig. 3C). We compared the photon intensity with tumor weight and volume, and found that comparing the bioluminescence (photons/sec) to tumor weight formed a better correlation than comparing the bioluminescence to tumor size (correlation coefficients were 0.77 and 0.57, respectively).
Necropsy and histopathology
At necropsy, white mesothelioma tumors were attached to and sometimes invaded the peritoneal wall (Fig. 4A). Similar masses were present on the visceral peritoneum of the pancreas, liver, diaphragm and gonadal fat pads.
Necropsy and histological observance. A. Mesothelioma in peritoneal cavity. Arrow indicates the tumor mass. B. H&E staining (20x). C. Mesothelin IHC (20x) with membrane staining. D. GFP IHC (20x) with cytoplasmic staining.
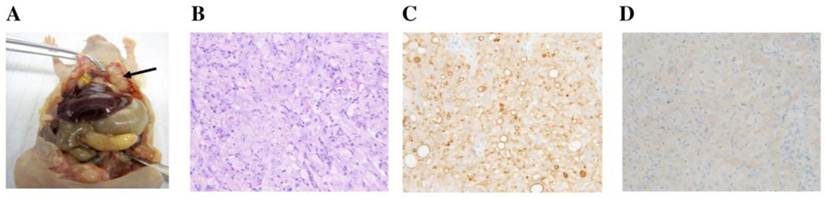
Histopathological evaluation confirmed the mesothelioma attributes of the injected cell line. On H&E, mesotheliomas in the peritoneal cavity were composed of sheets of cells with eosinophilic cytoplasm and large atypical nuclei with prominent nucleoli (Fig. 4B). Immunohistochemistry using the anti-mesothelin antibody showed strong membrane and some cytoplasmic staining (Fig. 4C). With the anti-GFP antibody, there was mild to moderate cytoplasmic staining (Fig. 4D).
Immunotoxin treatment
As a proof-of-concept experiment, we tested in vivo anti-tumor activity using SS1P in the established mesothelioma model. The treatment was designed based on clinical regimens. One treatment started on day one (the day when the mice received cells was set as day1), which may represent those patients who receive SS1P immediately after surgery to remove the visible tumor tissues, but may have remnant tumor cells that may cause relapse. Another treatment began on day 9, which may represent the early stages of MPM. A dramatic decrease of bioluminescence can be seen in both treatments two days after the first SS1P injection (Fig. 5).
SS1P treatment. Each mouse was i.p. injected with 10 million cells. At day 1 (treatd1) or day 9 (treatd9), the mice were given the first SS1P treatment by i.p. injection at a dose of 0.4 mg/kg body weight. The treatments were performed every other day as indicated by arrows. *Indicates the significant (p < 0.05) difference between treatment and control by using the one-way ANOVA statistical test (GraphPad Prism 5.03).
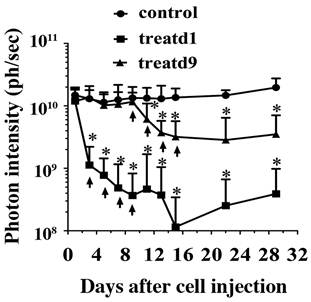
There was a significant difference between the treatment and control groups. Lungs were examined microscopically from the subset of mice. Interestingly, 50% of the control group (2/4) and none of the treated groups (0/6) had mesothelioma micrometastases. Neither of the treatment groups completely eradicated the tumor cells. We speculated that this might be due to an acquired apoptotic drug resistance to SS1P after treatment; however, we did not find changes in sensitivity to SS1P using the surviving tumor cells grown as monolayers for in vitro cytotoxicity assays on culture dishes (data not shown).
Discussion
We generated a Luc/GFP-expressing mesothelioma cell line, LMB-H226-GL. The LMB-H226-GL line is able to form solid tumors seeded in the peritoneal cavity of nude mice, which exhibited similar characteristics of human MM. The growth and spread of tumors can be noninvasively monitored by in vivo bioluminescent imaging.
SS1P was previously tested in ecotopic (subcutaneous) mouse models [23, 24]. These models used a transfected non-mesothelioma cell line (A431) that highly expresses mesothelin (~1×106 sites per cell), as inoculums. Regardless, these may not be ideal models given that both the genetic background and antigen (mesothelin) expression levels are different from those of native mesothelioma cells. In addition, ectopic models use manual callipering for tumor measurement, which may introduce manual deviation depending on the operator. On the other hand, LMB-H226-GL tumor cells express Luc/GFP at a very high level allowing for in vivo human mesothelioma tumors to be measured directly. This can be accomplished according to either luciferase or GFP fluorescence using an in vivo bioluminescence imaging system. As such, we believe that this new model introduces an improved method for the evaluation of anti-mesothelioma therapeutics.
Advances in antibody-based therapies represent a promising new approach to treating solid cancers, but a major challenge involves delivering a sufficient amount of antibody and immunoconjugate to the tumor mass [25]. For an anti-cancer antibody agent to be successful, it must satisfy two requirements: the agent must be effective in the tumor microenvironment, and the agent must reach the tumor cells in optimal quantities. In the present study, the SS1P treatment alone exhibited significant anti-mesothelioma activity. However, it was not able to completely eradicate tumors. We found that SS1P bound to the remaining tumor cells and killed them with similar activity when tested in vitro as tumor monolayers (data not shown). In a recent study, we examined how the tumor microenvironment affected the penetration and killing activity of SS1P in a three-dimensional (3D) spheroid model cultured in vitro using NCI-H226 and two primary cell lines isolated from the ascites of MM patients [26]. Mesothelioma cells grown as monolayers or as spheroids expressed comparable levels of mesothelin; however, spheroids were at least 100 times less affected by SS1P. We examined the time course of SS1P penetration within spheroids. The penetration was limited after 4 hours. Interestingly, we found a significant increase in the number of tight junctions in spheroids. Expression of E-Cadherin, a protein involved in the assembly and sealing of tight junctions and highly expressed in MM, was found significantly increased in spheroids as compared to monolayers. Moreover, we found that siRNA silencing and antibody inhibition targeting E-Cadherin could enhance SS1P therapy in vitro. Further studies will be needed to understand the molecular mechanisms underlying drug resistance and for innovative combinations of SS1P with agents that can increase the penetration of drugs and allow for sufficient amounts of SS1P to reach the tumor cells.
Acknowledgements
This work was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, in part by a Mesothelioma Applied Research Foundation Grant in Honor of Craig Kozicki to MH, and in part by the National Cancer Institute, NIH under contract No. HHSN261288800001E.
We thank Ira Pastan (NCI, NIH) for helpful discussions, Betty Ortiz-Conde (Gene Expression Laboratory, SAIC-Frederick, Inc.) for producing lentivirus, Bih-Rong Wei (NCI, NIH) for technical assistance in in vivo tumor imaging, Barbara Taylor (NCI Flow Cytometry Core, NIH) for technical assistance in flow cytometry, and Donna Butcher (Pathology/Histotechnology Laboratory, SAIC-Frederick) for immunohistochemistry technical assistance. We also thank the NIH Fellows Editorial Board for their review of the manuscript, Yen Phung (NCI, NIH) and Anna Mazzuca (NCI, NIH) for editorial assistance. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
1. Robinson BWS, Lake RA. Advances in malignant mesothelioma. N Engl J Med. 2005;353:1591-603
2. Ceresoli GL, Zucali PA, Gianoncelli L, Lorenzi E, Santoro A. Second-line treatment for malignant pleural mesothelioma. Cancer Treat Rev. 2010;36:24-32
3. Chang K, Pastan I, Willingham MC. Isolation and characterization of a monoclonal antibody, K1, reactive with ovarian cancers and normal mesothelium. Int J Cancer. 1992;50:373-81
4. Chang K, Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci USA. 1996;93:136-40
5. Hassan R, Bera T, Pastan I. Mesothelin: a new target for immunotherapy. Clin Cancer Res. 2004;10:3937-42
6. Ho M, Hassan R, Zhang J, Wang Q, Onda M, Bera T, Pastan I. Humoral immune response to mesothelin in mesothelioma and ovarian cancer patients. Clin Cancer Res. 2005;11:3814-20
7. Hassan R, Ho M. Mesothelin targeted cancer immunotherapy. Eur J Cancer. 2008;44:46-53
8. Ordonez NG. Application of mesothelin immunostaining in tumor diagnosis. Am J Surg Pathol. 2003;27:1418-28
9. Ho M, Bera TK, Willingham MC, Onda M, Hassan R, FitzGerald D, Pastan I. Mesothelin expression in human lung cancer. Clin Cancer Res. 2007;13:1571-5
10. Yu L, Feng M, Kim H, Phung Y, Kleiner DE, Gores GJ, Qian M, Wang XW, Ho M. Mesothelin as a potential therapeutic target in human cholangiocarcinoma. J Cancer. 2010;1:141-9
11. Robinson BWS, Creaney J, Lake R, Nowak A, Musk AW, Klerk N, Winzell P, Hellstrom KE, Hellstrom I. Mesothelin-family proteins and diagnosis of mesothelioma. Lancet. 2003;362:1612-16
12. Ho M, Onda M, Wang Q, Hassan R, Pastan I, Lively M. Mesothelin is shed from tumor cells. Cancer Epidemiol Biomarkers Prev. 2006;15:1751
13. Chowdhury PS, Pastan I. Improving antibody affinity by mimicking somatic hypermutation in vitro. Nat Biotechnol. 1999;17:568-72
14. Hassan R, Bullock S, Premkumar A, Kreitman RJ, Kindler H, Willingham MC, Pastan I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 2007;13:5144-9
15. Robert J. Kreitman, Raffit Hassan, David J. FitzGerald, Pastan I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res. 2009;15:5274-9
16. Hassan R, Ebel W, Routhier EL, Patel R, Kline JB, Zhang J, Chao Q, Jacob S, Turchin H, Gibbs L, Phillips MD, Mudali S, Donahue CI, Jaffee EM, Moreno M, Pastan I, Sass PM, Nicolaides NC, Grasso L. Preclinical evaluation of MORAb-009, a chimeric antibody targeting tumor-associated mesothelin. Cancer Immun. 2007;7:20-30
17. Ho M, Feng M, Fisher RJ, Rader C, Pastan I. A novel high-affinity human monoclonal antibody to mesothelin. Int J Cancer. 2010 [Epub ahead of print]
18. Bera TK, Pastan I. Mesothelin is not required for normal mouse development or reproduction. Mol Cell Biol. 2000;20:2902-6
19. Rump A, Morikawa Y, Tanaka M, Minami S, Umesaki N, Takeuchi M, Miyajima A. Binding of ovarian cancer antigen CA125/MUC16 to mesothelin mediates cell adhesion. J Biol Chem. 2004;279:9190-8
20. Kaneko O, Gong L, Zhang J, Hansen J, Hassan R, Lee B, Ho M. A binding domain on mesothelin for CA125/MUC16. J Biol Chem. 2009;284:3739-49
21. Yanagihara K, Tsumuraya M, Takigahira M, Mihara K, Kubo T, Ohuchi K, Seyama T. An orthotopic implantation mouse model of human malignant pleural mesothelioma for in vivo photon counting analysis and evaluation of the effect of S-1 therapy. Int J Cancer. 2010;126:2835-46
22. Day CP, Carter J, Bonomi C, Esposito D, Crise B, Ortiz-Conde B, Hollingshead M, Merlino G. Lentivirus-mediated bifunctional cell labeling for in vivo melanoma study. Pigment Cell Melanoma Res. 2009;22:283-95
23. Zhang Y, Xiang L, Hassan R, Paik CH, Carrasquillo JA, Jang BS, Le N, Ho M, Pastan I. Synergistic antitumor activity of taxol and immunotoxin SS1P in tumor-bearing mice. Clin Cancer Res. 2006;12:4695-701
24. Hassan R, Broaddus VC, Wilson S, Liewehr DJ, Zhang J. Anti-mesothelin immunotoxin SS1P in combination with gemcitabine results in increased activity against mesothelin-expressing tumor xenografts. Clin Cancer Res. 2007;13:7166-71
25. Jain RK. Transport of molecules, particles, and cells in solid tumors. Annu Rev Biomed Eng. 1999;1:241-63
26. Xiang X, Phung Y, Feng M, Nagashima K, Zhang J, Broaddus VC, Hassan R, FitzGerald D, Ho M. The development and characterization of a human mesothelioma in vitro 3D model to investigate immunotoxin therapy. PLoS One. 2011;6:e14640
Author contact
![]() Corresponding author: Dr. Mitchell Ho, Antibody Therapy Unit, Laboratory of Molecular Biology, National Cancer Institute, 37 Convent Drive, Room 5002C, Bethesda, MD 20892-4264. Tel: (301) 451-8727; Fax: (301) 402-1344; E-mail: hominih.gov
Corresponding author: Dr. Mitchell Ho, Antibody Therapy Unit, Laboratory of Molecular Biology, National Cancer Institute, 37 Convent Drive, Room 5002C, Bethesda, MD 20892-4264. Tel: (301) 451-8727; Fax: (301) 402-1344; E-mail: hominih.gov