Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(15):2258-2265. doi:10.7150/jca.16966 This issue Cite
Review
TFIIH: New Discoveries Regarding its Mechanisms and Impact on Cancer Treatment
Mario Zurita ![]() , Grisel Cruz-Becerra
, Grisel Cruz-Becerra
Departamento de Genética del Desarrollo, Instituto de Biotecnología, Universidad Nacional Autónoma de México. Av. Universidad 2001, Cuernavaca, Morelos 62250, México.
Received 2016-7-24; Accepted 2016-9-30; Published 2016-11-9
Abstract
The deregulation of gene expression is a characteristic of cancer cells, and malignant cells require very high levels of transcription to maintain their cancerous phenotype and survive. Therefore, components of the basal transcription machinery may be considered as targets to preferentially kill cancerous cells. TFIIH is a multisubunit basal transcription factor that also functions in nucleotide excision repair. The recent discoveries of some small molecules that interfere with TFIIH and that preferentially kill cancer cells have increased researchers' interest to elucidate the complex mechanisms by which TFIIH operates. In this review, we summarize the knowledge generated during the 25 years of TFIIH research, highlighting the recent advances in TFIIH structural and mechanistic analyses that suggest the potential of TFIIH as a target for cancer treatment.
Keywords: Anticancer drug, Cell cycle inhibitors, DNA damage, Natural anticancer compounds, Transcription modulators.
Introduction
Since the discovery of TFIIH in the early 1990s by the laboratory of Jean-Marc Egly [54], TFIIH has been the object of extensive research, with studies ranging from structural analyses of the complex to in vitro and in vivo molecular and biochemical evaluations of its functions in transcription and DNA repair. Ten proteins organized in two subcomplexes form the TFIIH complex. Among these proteins, two DNA helicases/ATPases, one of which is also a DNA translocase, a putative E3 ubiquitin ligase and a cyclin-dependent kinase have been identified [2]. However, most of the TFIIH subunits are considered to be regulatory or structural components [3]. The inhibition of some of the TFIIH enzymatic activities has been demonstrated to be effective in cancer treatment [20,41]. Interestingly, recent advances in the solution of the TFIIH complex organization increase the possibility of designing drugs that interfere with the functions of TFIIH. Thus, this review will describe recent discoveries on how TFIIH operates in transcription and DNA repair and how TFIIH is becoming an important target for cancer therapy.
TFIIH's subunits
TFIIH's subunits are organized into two sub-complexes: the CAK and the core, which interact to form the holo-TFIIH complex that is involved in transcription [1]. The CAK contains the cyclin-dependent kinase Cdk7, Cyclin H and MAT1 subunits, which operate in cell cycle control [2,3]. The core is composed of XPB, a 3´-5´ DNA helicase/ATPase, which is also a DNA translocase, XPD, a 5´-3´ DNA helicase/ATPase, p44, a putative E3 ubiquitin ligase, and the structural subunits p62, p52, p34 and p8 [4]. The core of TFIIH plays a determinant role in Nucleotide Excision Repair (NER) [5]. In addition, it has been suggested that XPD modulates the role of the CAK in cell cycle control in an intermediary XPD-CAK complex [6] (Fig. 1). Further, the XPG protein was demonstrated to interact with XPB, XPD and p62 and copurifies with the holo-TFIIH, suggesting that it could be another component of TFIIH [7-10]. On the other hand, the XPD subunit is also a component of the human MMXD complex, which is required for chromosome segregation and is not related to transcription or DNA repair [11]. However, it is still unknown if the enzymatic activities of XPD helicase/ATPase are necessary for the function of MMXD in chromosome segregation during mitosis. Moreover, the possibility of other subunits of TFIIH, besides XPD, being a part of other protein complexes still is open.
The complex life of TFIIH. (a) The 10-subunit TFIIH participates in transcription by RNA polymerase II. The XPG endonuclease participates in RNAPII transcription by binding to TFIIH. (b) The XPD subunit of TFIIH forms an intermediary complex with the CAK. (c) The CAK subcomplex phosphorylates several cdks that participates in cell cycle regulation. (d) The core subcomplex losses the CAK by the incorporation of XPA to TFIIH when the complex detects a bulky lesion in the DNA. (e) After the recognition of the damaged strand in the DNA the Pre-Incision-Complex is formed by TFIIH and other components of the NER machinery that maintain the reparation bubble in the DNA damaged region, it is stabilized by XPA and RPA and then the XPG and XPF-ERCC1 endonucleases cut the damaged strand. After that a DNA polymerase and a ligase repair the DNA. (f) TFIIH participates in rRNA elongation during RNAPI transcription. However it is unknown whether the CAK subcomplex participates in this mechanism. (g) The XPD subunit is a component of the MMXD complex that is important for the proper segregation of chromosomes during mitosis.
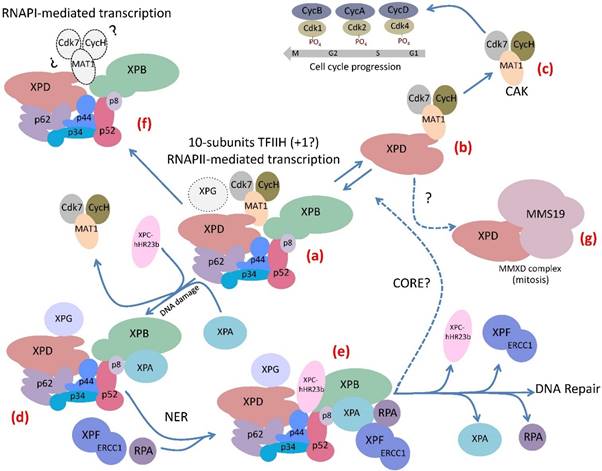
Several protein-protein interactions among the components of TFIIH have been identified by different approaches. Although the crystallographic structure of some subunits and the global architecture of TFIIH has been determined using high-resolution cryo-electron microscopy [10,12,13], determining the atomic structure of this complex is a great challenge. The interaction network in the 10-subunit complex has been recently described using chemical cross-linking combined with mass spectrophotometry and previous high-resolution electron microscopy data. This integrative analysis showed that the p34/p44 heterodimer represents a central "submodule" that connects the other core subunits. The VWA domains of p34 and p44 were shown to contact each other at multiple sites. In addition, these domains interact with p62 and p52. In turn, p62 interacts with XPD. Furthermore, XPD and XPB contact each other and both interact with p44, as well as with MAT1, which anchors Cdk7 and CycH [10]. In addition to the previously reported interactions between XPB and p52 and between p8 and p52, this analysis shows that the p8/p52 heterodimer directly interacts with XPB and p8 does not contact XPD, as was previously suggested [5,10,14]. This detailed model of the TFIIH architecture could be useful to search for drugs that affect the interface between the different subunits through in silico modeling approaches.
TFIIH-related human diseases
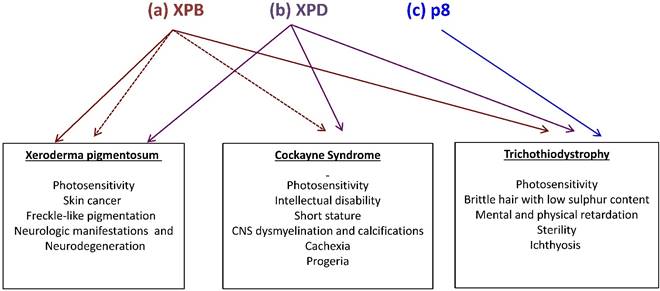
There is a relationship between mutations in some of the TFIIH subunits and three human syndromes associated with DNA repair and transcriptional deficiencies. Most of the TFIIH-affected patients show mutations in the XPB and XPD subunits, which are linked to xeroderma pigmentosum (XP), Cockayne syndrome (CS) and trichothiodystrophy (TTD) [7]. However, the different mutations described in the p8 subunit in humans have only been related to TTD-A [5]. Photosensitivity is a clinical feature among all these diseases, other manifestations are shared in a less extent and some others are syndrome-specific (Fig. 2). For example, XP patients affected in XPD or XPB can develop skin cancer, but TTD patients with different mutations in the same genes are not cancer prone. On the other hand, anomalies in the hair, nails and skin are hallmarks in TTD patients. In agreement, recent studies demonstrated a correlation between skin defects and alterations in the levels of collagen types I and VI in the extracellular matrix (ECM) of primary dermal fibroblast and skin from TTD patients [15,16]. Orioli and colleagues demonstrated the transcript down-regulation of the COL6A1 gene (which encodes for the collagen type VI alpha1 subunit) in skin fibroblasts from XPD-affected TTD patients [16]. On the other hand, transcript overexpression and high protein levels of the active form of the matrix metalloproteinase 1 (which degrades the interstitial collagen from the ECM) and concomitant low levels of collagen type I were detected in primary dermal fibroblast and skin from several different TFIIH-affected TTD patients, when compared with fibroblast from TFIIH-affected XP patients or healthy normal donors [15]. In addition, differential alterations in the TFIIH assembly at the RARB2 promoter, TFIIH enzymatic activities and chromatin remodeling were revealed by the analysis of the transcriptional role of TFIIH in several cell lines bearing mutations in XPB, XPD or p8. Interestingly the strongest defects were observed in cell lines carrying point mutations in the N-terminal domain of XPB found in TTD or XP/CS patients, while point mutations of the p8 show no effect on any of the steps analyzed [17]. Strikingly, the study of the phenotypes of these patients in relation to the identification of the molecular nature of the mutations that they carry in these genes have been very useful to understand how TFIIH works and reciprocally, to understand these syndromes. An enigmatic question that rises from the analysis of the manifestations in the patients afflicted in XPB, XPD and p8 is why these individuals only present very specific phenotypes even that TFIIH is required for three fundamental cellular functions? Although we still do not have a conclusive answer for this question, we know that the described mutations from these patients are not null and only partially affect the TFIIH functions, making these defects in TFIIH compatible with life [4,18,19].
Mutations in the XPB, XPD and p8 subunits of TFIIH are associated with three human syndromes: xeroderma pigmentosum (XP), Cokayne syndrome (CS) and trichothiodystrophy (TTD). (a) Mutations in XPB may generate TTD, XP (brown continuous arrow) or combinations of XP with CS (brown fragmented arrows). (b) Mutations in XPD may generate any of the three syndromes (purple continuous arrow) (c) Mutations in p8 (blue) on the other hand, are only linked to TTD. Syndrome-associated features are enlisted.
TFIIH's functions
Cell Cycle. As mentioned before components of TFIIH participates at least in three functions in the cell. The CAK has a central role in the activities of several Cdks that control cell cycle [2]. Cdk7 phosphorylates Cdk4 controlling the transition G1 to S phase. Also it phosphorylates Cdk2 to promote the entrance for S to G2 phases and Cdk1 to conduct G2 to mitosis [3] (Fig. 1). CAK subunits as the case of the rest of TFIIH are expressed constitutively during G1 in all cells, it is always active and seems to be very promiscuous in the phosphorylation of the Cdks that control the cell cycle which are modulated by specific cyclins. Therefore, the CAK is more like a supported factor for the control of the cell cycle than a regulator. It will be interesting to use the new Cdk7 inhibitor TZH1 [20], which is very specific to see how the cell cycle is affected when the catalytic activity of Cdk7 is inhibited.
Nucleotide excision repair. NER removes helix-distorting DNA lesions that include cyclobutane pyrimidine-pyrimidine dimers and pyrimidine-pyrimidone (6-4) photoproducts. Two pathways recognize this type of DNA damage: transcription coupled-NER (TC-NER) in the actively transcribed DNA strands and global genome-NER (GG-NER) in the non-transcribed DNA sequences. In TC-NER, the RNA polymerase, which is stalled in the presence of a bulky lesion, recognizes the DNA damage. In GG-NER, depending on the DNA-damaging agent, the protein complex RAD23B-Centrin 2 or XPE recognizes the bulky DNA lesion. In any case, the DNA damage recognition step is followed by the recruitment of the NER machinery, including TFIIH, which unwinds the DNA by the ATPase and helicase activities of XPB and XPD, respectively, the XPA and RPA proteins that stabilize the DNA repair bubble, and the endonucleases XPF-ERCC1 and XPG, which remove a segment of 23-27 nucleotides from the damaged DNA strand, which is ultimately refilled by the DNA replication machinery [21-23] (Fig. 1). It was initially proposed that after TFIIH recruitment, the CAK subcomplex dissociates from the core in an XPA-dependent manner [24]. New data suggest that the holo-TFIIH scans the DNA through its XPB and XPD helicase/ATPase activities, which are inhibited by bulky DNA lesions. Then, TFIIH is stalled in the damaged DNA region, followed by the ejection of CAK from the core [25]. However, in vivo experiments are required to confirm this model.
Transcription. TFIIH participates in transcription mediated by RNA polymerases I and II; however, our understanding of the role of TFIIH in RNAPI transcription is still obscure. Since the first report by Grummt and Elgy's groups in 2002, which demonstrated that TFIIH is dispensable for the initiation of RNAPI transcription but required for rRNA elongation [26], only a few studies about the role of TFIIH in RNAPI-mediated transcription have been published. For example, it has recently been reported that TFIIH binds to RNAPI after the initiation of transcription. In agreement, the ATPase activity of XPB is required during rRNA elongation, and CS-related mutations in XPB and XPD disturb the interaction between TFIIH and rDNA, which affects elongation but not transcription initiation [27,28]. In addition, it has been suggested that the NER-related proteins CSB and CSA form a complex with TFIIH in the nucleolus, which is necessary for rDNA transcription [29,30].
However, more extensive studies have been performed to elucidate the role of TFIIH in RNAPII transcription. During the Pre-Initiation Complex (PIC) assembly, TFIIH is recruited by TFIIE and binds to RNAPII [31]. While the enzymatic activities of XPD are dispensable for RNAPII transcription, the ATPase activity of XPB is required for the unwinding of 11 bp in the promoter during the formation of the open complex [32,33]. This configuration is stabilized by the RNAPII cleft and jaw domains [13,34]. Interestingly, XPB does not directly bind at the site of DNA unwinding. Instead, XPB tracks the DNA in a 5´-3´ direction and translocates the downstream DNA into the cleft region of the RNAPII, which generates the DNA unwinding and allows the recognition of the transcription initiation start site in a process that requires the continuous hydrolysis of ATP by XPB [33]. In addition, the current PIC structure model shows that XPB binds downstream of the initiator site and contacts both DNA strands in the promoter [13]. Then, the function of XPB in transcription relies on its translocase activity, and this mechanism is a rate-limiting step in the initiation of transcription. The other major function of TFIIH in RNAPII transcription takes place through its kinase activity. The kinase module phosphorylates serines 5 and 7 (S5 and S7) of the heptapeptide repeat (consensus sequence: Y1S2P3T4S5P6S7) in the CTD of the largest subunit of RNAPII. It has been established that S5 phosphorylation is fundamental for transcription initiation and promoter clearance [35]. However, the in vitro inhibition of the kinase activity of Cdk7 by the Cdk7-specific inhibitor THZ1 (see ahead) had no effect on transcription initiation and promoter realese, but preferentially affected the pausing of the RNAPII after the synthesis of 30-50 nucleotides [36]. Furthermore, the phosphorylation of S5 by Cdk7 is important for the recruitment of the pausing factors, the Negative Elongation Factor (NELF) and the DRB-sensitive factor (DSIF), as well as the capping enzyme that guanylates the 5´end of the nascent transcript, a process that requires pausing [37]. However, more recent in vivo experiments in budding yeast show that by using an engineered Kin28 (Cdk7 in yeast) that can be specifically and irreversible inhibited, affect promoter scape as well as RNAPII elongation [38]. Intriguingly, in the absence of S5 phosphorylation of the CTD, the RNAPII is paused after the transcription initiation site [38]. Then these recent reports, one in vitro [36], and the other in vivo [38] using the budding yeast about the role of the Cdk7 on RNAP II transcription initiation and elongation partially disagree. It will be relevant to perform similar experiments as the reported in yeast, but in human cells. On the other hand, it has been demonstrated that mutations in Kin28 reduce the dissociation of the mediator from the PIC, which affects the escape of the RNAPII to initiate mRNA elongation [39]. Therefore, all of these new data and models on how TFIIH operates in transcription in vitro must be integrated with the in vivo results.
TFIIH as a target for cancer therapy
It is well established that cancer cells require high levels of transcription to survive and to maintain their malignant phenotype [40]. Therefore, the components of the basal transcription machinery could be considered good targets to reduce global transcription, and they might have stronger effects in cancer cells when compared with non-cancerous cells (reviewed in 39).
A very promising drug for the treatment of many cancer types is triptolide (TPL). TPL is a diterpen triepoxide purified from the plant Tripterygium wilfordii. Extracts from this plant have been used for centuries in traditional Chinese medicine, and preclinical studies have shown high effectiveness against cancer due to its very strong anti-proliferative effect and apoptosis induction. TPL binds covalently to the ATPase domain of XPB, inhibiting both RNAPII transcription and NER [41]. Although other TPL targets have been proposed, structural and biochemical data show that it is very specific for XPB, and intriguingly, the treatment of Drosophila imaginal discs with TPL phenocopies the defects observed in different TFIIH mutant subunits in the fly [41-43]. RNA-seq and DNA microarray analyses from TPL-treated cancerous cells have shown the down-regulation of nearly all the RNAPII-dependent transcripts [44,45]; however, comparative studies at this level between cancer cells and their progenitors are required. TPL is not highly permeable to the cell membrane, and it is hepatotoxic [46]. To overcome these inconveniences in cancer therapy, more permeable and potentially less toxic TPL derivatives have been developed, although it will be important to demonstrate that they are as effective as TPL to kill cancer cells [41].
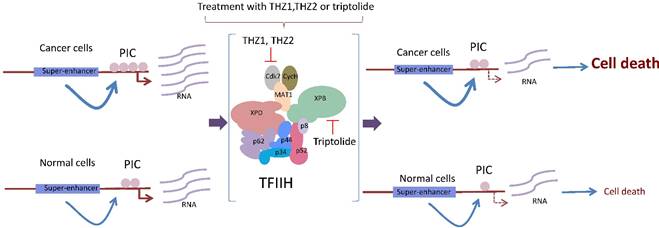
Based on structural analysis predictions, other drugs have been developed to specifically target the enzymatic functions of TFIIH. For example, the antitumor drug BS-181 is a pyrazolo [1,5-a] pyrimidine-derived compound that inhibits the phosphorylation of Cdk7 targets, inducing cell cycle arrest and apoptosis [45]. More recently, it was shown that THZ1 is a phenylaminopyrimidine with a cysteine reactive acrylamide moiety that covalently binds to the cysteine 312 located outside of the catalytic domain, making THZ1 very specific for Cdk7 [20], although it inhibits Cdk12 at very high concentrations and after long incubation times [36]. Approximately 1000 cancerous cell lines tested were very sensitive to nanomolar concentrations of THZ1, particularly T acute lymphoblastic leukemia cells [20]. Intriguingly, gene-specific effects in transcription were observed in different cell lines after the THZ1 treatment [20,47,48]. For example, the expression of RUNX1, a transcription factor associated with leukemia and whose expression is modulated by a super-enhancer, was highly sensitive to THZ1 [20] (Fig. 3). Similarly, THZ1 affects the expression of genes controlled by super-enhancers, particularly Myc, as well as lineage-specific transcription factors required by the maintenance of the cancerous phenotype in lung cancer cells [47,49] (Fig. 3). However, a new study showed a high potential for THZ1 in the treatment of triple negative breast cancer (TNBC) tumors [50]. TNBC cells were substantially more sensitive to THZ1 when compared to ER/Pr+ (positive for the estrogen receptor) tumor cells. Interestingly, some sets of genes that encode for signal transduction and transcription factors related to breast cancer, such as TGFβ, STAT, EGFR, and WNT, among other genes, were affected; these genes were named the Achilles cluster of TNBC, and their expression is also regulated by super-enhancers [50]. Therefore, it seems that genes modulated by super-enhancers, which are large regions of DNA that accumulate large quantities of transcription factors [48] and are directly involved in the cancerous phenotype, may be more sensitive to THZ1 [20,47,49] (Fig. 3). A new THZ1 analog named THZ2 is more stable than THZ1 and has an improved effect on Cdk7 inhibition [50].
The cancerous cell requires high levels of global transcription to maintain its malignant phenotype. Highly transcribed genes in cancer cells, such as oncogenes, are modulated by super enhancers. These super-enhancers allow the incorporation of the basal transcription machinery at higher frequency in the cancerous cell. The incubation of these cells with either THZ1, THZ2 or triptolide affects the functions of TFIIH by reducing the rate of transcription initiation and elongation, which has a more deleterious effect in malignant than in normal cells.
Another recently identified TFIIH inhibitor is spironolactone (SP), which induces the proteasome-mediated degradation of the XPB subunit and enhances the effect of platinum derivatives in cancer cells [51]. The SP-induced degradation of XPB clearly affects both NER and transcription. The effect of SP in tumor cells from different origins should be analyzed to determine whether it also preferentially kills malignant cells.
Conclusions and Perspectives
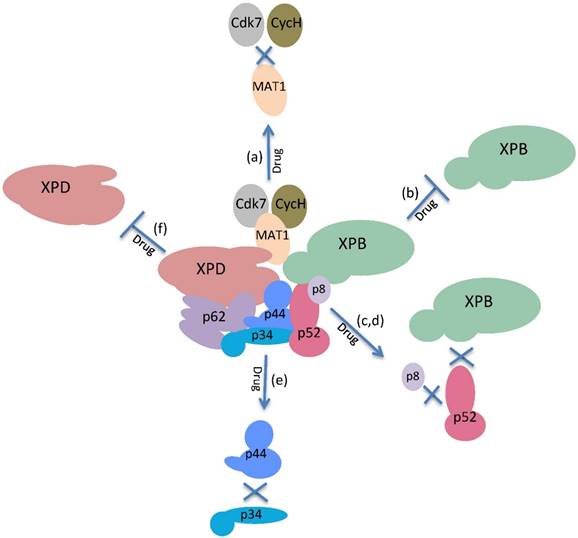
Twenty-five years after the discovery of TFIIH, we have a more comprehensive view of the mechanisms of different functions of TFIIH; however, there are still many questions that must be answered. Although we understand when the enzymatic activities of TFIIH are required and the identities of some of their substrates, it is highly possible that other targets will be discovered. For example, Cdk7 may phosphorylate other transcription factors. TFIIH exhibits crosstalk with the mediator [52] which may be modulated by TFIIH and vice versa; however, this intercommunication is not clear. The role of the other subunits of the core that lack enzymatic activities may be modulators of TFIIH activity, as p52 regulates the ATPase activity of XPB. In addition, p38 and p62 have protein domains that suggest dynamic interactions with the other subunits of the core, and we believe that the complex is not static and exhibits strong internal dynamics. However, important questions remain; for example: Is p44 really a ubiquitin ligase, and if so, what are its targets? Is p8 a real stabilizer of the complex, or does it have a regulatory role in the XPB ATPase activity? Is the CAK subcomplex required for rDNA transcription even if the RNAPI lacks a CTD domain such as the one present in RNAPII? In addition, the fact that two promising drugs for the treatment of cancer exhibit target subunits of TFIIH suggests that new small molecules may be found that inhibit TFIIH but recognize other subunits besides XPB and Cdk7. Furthermore, because we understand in detail the regions of each of the 10 subunits of TFIIH that interact, it is possible to search for small molecules that interrupt the interface of these interactions and affect the assembly of TFIIH and therefore, its functions (Fig. 4). In fact, a new antibiotic that inhibits the interaction between a mediator subunit with a transcriptional activator and that preferentially kills a pathogen fungus has recently been identified [53]. A similar approach may be used to look for new inhibitors of TFIIH. On the other hand, a challenging problem for the development and use of drugs that target nuclear factors is to increase the permeability of these compounds in the nucleus. The treatment with TPL or THZ1 cause an important reduction in transcription in cancer cell lines, showing that an important amount of these molecules permeates the nuclear membrane [4,44,45], however more precise experiments with animal models are needed. In addition, new methods to increase the efficiency to deliver new drugs to the nucleus still have to be developed.
The enzymatic activities and physical interactions of the subunits of TFIIH could be targets for drugs in cancer treatment. Possible targets for the inactivation of the functions of TFIIH: (a) Disruption of the interactions of the MAT1 subunit with cdk7 and Cyc H. (b) Inhibition of the DNA translocase and ATPase activities of XPB. (c,d) Disruption of the interaction of p52 with p8 or XPB. (e) Disruption of the interactions of p34 with p44. (f) Inhibition of the DNA helicase/ATPase activities of XPD.
In conclusion, further studies of TFIIH should explore how the complex works at high resolution in transcription and DNA repair, how defects in TFIIH affect development, and why it is an important target for cancer therapy.
Abbreviations
RNAPII: RNA polymerase II; RNAPI: RNA polymerase I; NER: nucleotide excision repair; XP: xeroderma pigmentosum; CS: Cockayne syndrome; TTD: trichothiodystropy; TTD-A: trichothiodystropy complementation group A; ECM: extracellular matrix; TC-NER: transcription coupled-NER; GG-NER: global genome-NER; PIC: pre-initiation complex; NELF: negative elongation factor; DSIF: DRB-sensitive factor; TPL: triptolide; TNBC: triple negative breast cancer; SP: spironolactone; CNS: central nervous system.
Acknowledgements
This work was supported by the grants from CONACyT 219673 and DGAPA UNAM number IN200315.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zurita M, Merino C. The transcriptional complexity of the TFIIH complex. TRENDS Genet. 2003;19:578-84
2. Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J. Cell Sci. 2005;118:5171-80
3. Schachter MM, Merrick KA, Larochelle S, Hirschi A, Zhang C, Shokat KM. et al. A Cdk7-Cdk4 T-Loop Phosphorylation Cascade Promotes G1 Progression. Mol. Cell. 2013;50:250-60
4. Egly J-M, Coin F. A history of TFIIH: Two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst). 2011;10:714-21
5. Compe E, Egly J-M. TFIIH: when transcription met DNA repair. Nat. Rev. Mol. Cell Biol. 2012;13:476-476
6. Li X, Urwyler O, Suter B. Drosophila Xpd regulates Cdk7 localization, mitotic kinase activity, spindle dynamics, and chromosome segregation. PLoS Genet. 2010;6:10.1371 /journal.pgen.1000876
7. Schärer OD. The molecular basis for different disease states caused by mutations in TFIIH and XPG. DNA Repair (Amst). 2008;7:339-44
8. Narita T, Narita K, Takedachi A, Saijo M, Tanaka K. Regulation of Transcription Elongation by the XPG-TFIIH Complex Is Implicated in Cockayne Syndrome. Mol. Cell. Biol. 2015;35:3178-88
9. Ito S, Kuraoka I, Chymkowitch P, Compe E, Takedachi A, Ishigami C. et al. XPG Stabilizes TFIIH, Allowing Transactivation of Nuclear Receptors: Implications for Cockayne Syndrome in XP-G/CS Patients. Mol. Cell. 2007;26:231-43
10. Luo J, Cimermancic P, Viswanath S, Ebmeier CC, Kim B, Dehecq M. et al. Architecture of the Human and Yeast General Transcription and DNA Repair Factor TFIIH. Mol. Cell. 2015;59:794-806
11. Ito S, Tan LJ, Andoh D, Narita T, Seki M, Hirano Y. et al. MMXD, a TFIIH-Independent XPD-MMS19 Protein Complex Involved in Chromosome Segregation. Mol. Cell. Elsevier. 2010;39:632-40
12. Grünberg S, Hahn S. Structural insights into transcription initiation by RNA polymerase II. Trends Biochem. Sci. 2013;38:603-11
13. Louder R, He Y, Lopez-Blanco J, Fang J, Chacon P, Nogales E. Structure of promoter-bound TFIID and model of human pre-initiation complex assembly. Nature. 2016;531:604-9
14. Coin F, De Santis LP, Nardo T, Zlobinskaya O, Stefanini M, Egly JM. p8/TTD-A as a repair-specific TFIIH subunit. Mol. Cell. 2006;21:215-26
15. Arseni L, Lanzafame M, Compe E, Fortugno P, Afonso-Barroso A, Peverali FA. et al. TFIIH-dependent MMP-1 overexpression in trichothiodystrophy leads to extracellular matrix alterations in patient skin. Proc. Natl. Acad. Sci. U. S. A. 2015;112:1499-504
16. Orioli D, Compe E, Nardo T, Mura M, Giraudon C, Botta E. et al. XPD mutations in trichothiodystrophy hamper collagen VI expression and reveal a role of TFIIH in transcription derepression. Hum. Mol. Genet. 2013;22:1061-73
17. Singh A, Compe E, Le May N, Egly JM. TFIIH subunit alterations causing xeroderma pigmentosum and trichothiodystrophy specifically disturb several steps during transcription. Am. J. Hum. Genet. 2015;96:194-207
18. Singh A. et al. TFIIH Subunit Alterations Causing Xeroderma Pigmentosum and Trichothiodystrophy Specifically Disturb Several Steps during Transcription. 2015.
19. Theil AF, Nonnekens J, Steurer B, Mari PO, de Wit J, Lemaitre C. et al. Disruption of TTDA Results in Complete Nucleotide Excision Repair Deficiency and Embryonic Lethality. PLoS Genet. 2013;9:e1003431. doi:10.1371/journal.pgen.1003431
20. Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB. et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. Nature Publishing Group. 2014;511:616-20
21. Liu X. In vitro chromatin templates to study nucleotide excision repair. DNA Repair (Amst). 2015;36:68-76
22. Schärer OD. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb Perspect Biol. 2013;5:doi 10.1101/cshperspect.a012609
23. Oksenych V, Coin F. The long unwinding road: XPB and XPD helicases in damaged DNA opening. Cell Cycle. 2010;9:90-6
24. Coin F, Oksenych V, Mocquet V, Groh S, Blattner C, Egly JM. Nucleotide Excision Repair Driven by the Dissociation of CAK from TFIIH. Mol. Cell. 2008;31:9-20
25. Li CL, Golebiowski FM, Onishi Y, Samara NL, Sugasawa K, Yang W. Tripartite DNA Lesion Recognition and Verification by XPC, TFIIH, and XPA in Nucleotide Excision Repair. Mol. Cell. 2015;59:1025-34
26. Iben S, Tschochner H, Bier M, Hoogstraten D, Hozak P, Egly JM. et al. TFIIH plays an essential role in RNA polymerase I transcription. Cell. 2002;109:297-306
27. Assfalg R, Lebedev A, Gonzalez OG, Schelling A, Koch S, Iben S. TFIIH is an elongation factor of RNA polymerase I. Nucleic Acids Res. 2012;40:650-9
28. Nonnekens J, Perez-Fernandez J, Theil AF, Gadal O, Bonnart C, Giglia-Mari G. Mutations in TFIIH causing trichothiodystrophy are responsible for defects in ribosomal RNA production and processing. Hum. Mol. Genet. 2013;22:2881-93
29. Bradsher J, Auriol J, De Santis LP, Iben S, Vonesch JL, Grummt I. et al. CSB is a component of RNA pol I transcription. Mol. Cell. 2002;10:819-29
30. Koch S, Gonzalez OG, Assfalg R, Schelling A, Schafer P, Scharffetter-Kochanek K. et al. Cockayne syndrome protein A is a transcription factor of RNA polymerase I and stimulates ribosomal biogenesis and growth. Cell Cycle. 2014;13:2029-37
31. He Y, Fang J, Taatjes DJ, Nogales E. Structural visualization of key steps in human transcription initiation. Nature. 2013;495:481-6
32. Kuper J, Braun C, Elias A, Michels G, Sauer F, Schmitt DR. et al. In TFIIH, XPD Helicase Is Exclusively Devoted to DNA Repair. PLoS Biol. 2014;12:doi 10. 1371/journal.pbio.1001954
33. Fishburn J, Tomko E, Galburt E, Hahn S. Double-stranded DNA translocase activity of transcription factor TFIIH and the mechanism of RNA polymerase II open complex formation. Proc. Natl. Acad. Sci. U. S. A. 2015;112:3961-6
34. Jeronimo C, Collin P, Robert F. The RNA Polymerase II CTD: The Increasing Complexity of a Low-Complexity Protein Domain. J. Mol. Biol. 2015 doi.org/10.1016/j.jmb.2016.02.006
35. Akhtar MS, Heidemann M, Tietjen JR, Zhang DW, Chapman RD, Eick D. et al. TFIIH Kinase Places Bivalent Marks on the Carboxy-Terminal Domain of RNA Polymerase II. Mol. Cell. 2009;34:387-93
36. Nilson K a, Guo J, Turek ME, Brogie JE, Delaney E, Luse DS. et al. THZ1 Reveals Roles for Cdk7 in Co-transcriptional Capping and Pausing. Mol. Cell. 2015;59:576-87
37. Komarnitsky P, Cho EJ, Buratowski S. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 2000;14:2452-60
38. Rodriguez-Molina JB, Tseng SC, Simonett SP, Taunton J, Ansari AZ, Tseng SC. et al. Engineered Covalent Inactivation of TFIIH-Kinase Reveals an Elongation Checkpoint and Results in Widespread mRNA Stabilization. Mol. Cell. 2016;63:433-44
39. Wong KH, Jin Y, Struhl K. TFIIH Phosphorylation of the Pol II CTD Stimulates Mediator Dissociation from the Preinitiation Complex and Promoter Escape. Mol. Cell. 2014;54:601-12
40. Villicaña C, Cruz G, Zurita M. The basal transcription machinery as a target for cancer therapy. Cancer Cell Int. 2014;14:18
41. Titov D V, Gilman B, He Q-L, Bhat S, Low W-K, Dang Y. et al. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat. Chem. Biol. 2011;7:182-8
42. Villicaña C, Cruz G, Zurita M. The genetic depletion or the triptolide inhibition of TFIIH in p53-deficient cells induces a JNK-dependent cell death in Drosophila. J. Cell Sci. 2013;126:2502-15
43. Yi J-M, Huan X-J, Song S-S, Zhou H, Wang Y-Q, Miao Z-H. Triptolide Induces Cell Killing in Multidrug-Resistant Tumor Cells via CDK7/Rpb1 rather than XPB or p44. Mol. Cancer Ther. 2016 doi:10.1158/1535-7163.MCT-15-0753
44. Chen F, Gao X, Shilatifard A, Shilatifard A. Stably paused genes revealed through inhibition of transcription initiation by the TFIIH inhibitor triptolide. Genes Dev. 2015;29:39-47
45. Wang B, Chen J. Selective CDK7 inhibition with BS-181 suppresses cell proliferation and induces cell cycle arrest and apoptosis in gastric cancer. Drug Des Devel Ther. 2016:1181-9
46. Qu L, Qu F, Jia Z, Wang C, Wu C, Zhang J. Integrated targeted sphingolipidomics and transcriptomics reveal abnormal sphingolipid metabolism as a novel mechanism of the hepatotoxicity and nephrotoxicity of triptolide. J. Ethnopharmacol. 2015;170:28-38
47. Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM. et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159:1126-39
48. Pott S, Lieb JD. What are super-enhancers? Nat. Genet. 2014;47:8-12
49. Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T. et al. Targeting Transcriptional Addictions in Small Cell Lung Cancer with a Covalent CDK7 Inhibitor. Cancer Cell. 2014;26:909-22
50. Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S. et al. CDK7-Dependent Transcriptional Addiction in Triple-Negative Breast Cancer. Cell. 2015;163:174-86
51. Alekseev S, Ayadi M, Brino L, Egly JM, Larsen AK, Coin F. A small molecule screen identifies an inhibitor of DNA repair inducing the degradation of TFIIH and the Chemosensitization of tumor cells to platinum. Chem. Biol. 2014;21:398-407
52. Kumafuji M, Umemura H, Furumoto T, Fukasawa R, Tanaka A, Ohkuma Y. Mediator MED18 subunit plays a negative role in transcription via the CDK/cyclin module. Genes to Cells. 2014;19:582-93
53. Nishikawa JL, Boeszoermenyi A, Vale-Silva LA, Torelli R, Posteraro B, Sohn Y-J. et al. Inhibiting fungal multidrug resistance by disrupting an activator-Mediator interaction. Nature. 2016;530:485-9
54. Gerard M, Fischer L, Moncollin V, Chipoulet JM, Chambon P, Egly JM. Purification and interaction properties of the human RNA polymerase B(II) general transcription factor BTF2. J Biol Chem. 1991;266:20940-5
Author contact
![]() Corresponding author: Mario Zurita, Departamento de Genética del Desarrollo, Instituto de Biotecnología/UNAM. Tel. 52 555 6227657 Email: mariozunam.mx.
Corresponding author: Mario Zurita, Departamento de Genética del Desarrollo, Instituto de Biotecnología/UNAM. Tel. 52 555 6227657 Email: mariozunam.mx.