Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(12):2163-2172. doi:10.7150/jca.19940 This issue Cite
Research Paper
CENPH Inhibits Rapamycin Sensitivity by Regulating GOLPH3-dependent mTOR Signaling Pathway in Colorectal Cancer
Wei Wu#, Fan Wu#, Zaozao Wang, Jiabo Di, Jie Yang, Pin Gao, Beihai Jiang ![]() , Xiangqian Su
, Xiangqian Su ![]()
Key laboratory of Carcinogenesis and Translational Research (Ministry of Education), Department of Gastrointestinal Surgery IV, Peking University Cancer Hospital & Institute, Beijing 100142, China.
# These authors contributed equally to this work.
Received 2017-3-6; Accepted 2017-4-25; Published 2017-7-15
Citation:
Wu W, Wu F, Wang Z, Di J, Yang J, Gao P, Jiang B, Su X. CENPH Inhibits Rapamycin Sensitivity by Regulating GOLPH3-dependent mTOR Signaling Pathway in Colorectal Cancer. J Cancer 2017; 8(12):2163-2172. doi:10.7150/jca.19940. https://www.jcancer.org/v08p2163.htm
Other stylesAbstract
Background: Centromere protein H (CENPH) is known as a fundamental component of the active centromere complex, and its overexpression is correlated with poor prognosis in various solid tumors. mTOR inhibitor rapamycin has been shown to possess antitumor activity, as well as prevent intestinal tumorigenesis. However, the prognostic value of CENPH in colorectal cancer (CRC) and the role of CENPH in rapamycin sensitivity remain unknown.
Materials and methods: The effect of CENPH on the cell proliferation, clonogenicity, and cell response to rapamycin in CRC were evaluated by MTT and/or colony formation assays. For the underlying mechanisms, the interaction between CENPH and GOLPH3 were detected by co-immunoprecipitation, GST pull-down, and His-tag pull-down assays, as well as the laser scanning confocal microscopy. The status of kinases in mTOR signaling was determined by Western blot. Finally, the clinical significance of CENPH was analyzed using public CRC datasets with CENPH transcripts and clinical information.
Results: CENPH inhibited CRC malignant phenotypes, conferred reduced sensitivity to rapamycin, and attenuated both mTORC1 and mTORC2 in mTOR signaling pathway through the interaction with golgi phosphoprotein 3 (GOLPH3), which has been identified as a potential oncogene and modulates the response to rapamycin. Moreover, elevated levels of CENPH were detected in CRC tissues, compared with normal colorectal tissues. High levels of CENPH expression gradually decreased according to CRC tumor stages. Patients with high CENPH expression had favorable survival.
Conclusions: Our results suggest that CENPH inhibits rapamycin sensitivity by regulating GOLPH3 dependent mTOR pathway. High CENPH expression is associated with better prognosis in CRC patients. Taken together, CENPH may serve as a potential predictor for rapamycin sensitivity and therapeutic target for CRC patients.
Keywords: CENPH, GOLPH3, Colorectal cancer (CRC), Rapamycin, mTOR, Prognosis.
Introduction
Colorectal cancer (CRC) is one of the most common forms of cancer and the second leading cause of cancer deaths worldwide [1, 2]. Due to the application of adjuvant radiation and chemotherapy treatments as well as the development of surgical techniques, the 5-year survival rate of CRC has been improved during the past two decades [3]. Like other solid cancers, the development of CRC is a multistep progression involving the deregulation of oncogenes and tumor suppressor genes, which influence various intracellular and extracellular signal transductions responsible for cellular processes including proliferation, apoptosis, invasion, and metastasis [4].
PI3K/AKT signaling pathway plays critical roles in orchestrating multiple cellular functions [5-8]. Mammalian target of rapamycin (mTOR), as downstream effector of PI3K/AKT pathway, consists of two functionally distinct multi-protein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), which were involved in protein synthesis, cell growth, autophagy, metabolism, and survival [5-8]. Since the PI3K/AKT/mTOR pathway is upregulated in almost all cancers, anticancer drugs including LY294002, MK2206, everolimus, rapamycin, and AZD8055 have been developed to target crucial kinases associated with this pathway [7, 8]. Among them, rapamycin inhibits mTOR activity by binding to the protein raptor, resulting in decreased cell proliferation and prevention in intestinal tumorigenesis [9-10]. However, there are no conclusive protein indicators to predict the response to rapamycin nowadays, except golgi phosphoprotein 3 (GOLPH3), which is identified as a potent oncogene and a potential positive predictor for rapamycin sensitivity involving in mTOR signaling regulation [9, 11].
Centromere protein H (CENPH) is a fundamental element of the active centromere complex, which is responsible for linking centromeres to spindle microtubules [12, 13]. CENPH is overexpressed in most CRC, and high levels of CENPH leads to chromosome missegregation and aneuploidy in diploid cell lines [14]. Consequently, continuous chromosome missegregation results in chromosomal instability (CIN), which plays a critical role in the development and progression of cancer [14]. CENPH cooperates with CENPI or CSPP1 on kinetochores and contributes to kinetochore microtubules dynamics for accurate chromosome segregation [15, 16]. However, the role of CENPH in mTOR signaling pathway and rapamycin sensitivity remains less characterized. With regard to the clinical significance of CENPH, previous studies demonstrated that the expression levels of CENPH is elevated in a series of cancers including colorectal cancer, gastric cancer, renal cell carcinoma, hepatocellular carcinoma, et al [14, 17-25]. However, the prognostic value of CENPH in CRC patients remains largely unclear.
In this study, we investigated the role CENPH played in cancer cell response to rapamycin and the underlying mechanism. Moreover, we determine the clinical significance of CENPH using the public CRC datasets. Our results suggest that high levels of CENPH impaired CRC malignant phenotypes and rapamycin sensitivity through cooperating with GOLPH3 on the modulation of mTOR signaling pathway. Taken together, these findings indicate that CENPH may become a valuable predictor for rapamycin sensitivity.
Materials and Methods
Ethics statement
The study was approved and supervised by the Research Ethics Committee of Peking University Cancer Hospital and Institute, Beijing, China (No. 2014KT97).
Cell lines and cell culture
All human CRC cell lines (RKO, LoVo, HCT116, SW480) were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cells were maintained in RPMI-1640 or DMEM medium (HyClone) supplemented with 10% FBS (PAA), penicillin 100 units/mL, and streptomycin 100μg/mL in a 5% CO2 humidified atmosphere.
Quantitative Real-time PCR
RNA samples were extracted using Trizol reagent (Invitrogen). The isolated total RNA was reverse-transcribed into cDNA using the reverse transcription kit (Promega, Madison, WI, USA). For quantitative Real-time PCR, we used the SYBR Green PCR Master Mix system (Toyobo Co. Ltd., Osaka, Japan). The primer sequences of Real-time PCR were: CENPH forward, 5'- TCATACGACAAAACCTACAGATGGA-3'; reverse, 5'- TGTCAACATTCTTCTCAAGCTGC-3'. Primers for GAPDH have been described before [26]. The quantitation of CENPH mRNA relative to an internal control GAPDH was performed by the ΔCt method as previously described [26].
Antibodies and Western blot
Cells were harvested and total protein (100 μg) isolated from cells were separated by electrophoresis in 8%-12% SDS-PAGE and transferred onto a nitrocellulose membrane. The protein levels were detected using primary antibody against CENPH (1:100, Cat#HPA036494, Sigma-Aldrich, USA), AKT (1:1000, Cat#9272, Cell Signaling Technology, USA), p-AKT (1:1000, Cat#9271, Cell Signaling Technology, USA), mTOR Substrates Antibody Sampler Kit (1:1000, Cat#9862, Cell Signaling Technology, USA), p70S6K (1:1000, Cat#2708, Cell Signaling Technology, USA), p-p70S6K (1:200, Cat#9234, Cell Signaling Technology, USA), GOLPH3 (1:1000, Cat#19112-1-AP, Proteintech), and β-actin (1:3000. Cat#A1978, Sigma-Aldrich, USA). The signal was detected by the ECL Western blot detection kit (Amersham). Densitometric analysis of the Western blot protein was performed using the ImageJ software.
Plasmids and siRNA transfections
Full-length human CENPH cDNA were cloned into pCMV-3Tag-1A to constructed pCMV-3Tag-1A-CENPH. The small inhibitor RNAs (siRNA) ON-target plus SMARTpool CENPH (human: #L-003254-00-0005) were from Dharmacon. The GOLPH3-targeting siRNA and the negative control siRNA were purchased from Shanghai Gene Pharma (Shanghai, China). Primer sequences of siRNAs have been described previously [26]. RKO and LoVo cells were transiently transfected with plasmids or siRNAs using Lipofectamine™ 2000 (Invitrogen) according to the manufacturer's instructions, and incubated for another 24 hours before further treatment.
MTT proliferation assay
The MTT assay was performed to determine the cell proliferation and cell sensitivity to rapamycin as previously described [26, 27]. Briefly, cells (0.5×104 cells/well) transfected with plasmids or siRNAs were seeded in 96-well plates and incubated overnight. For cell proliferation assay, numbers of live cells were determined at 0h, 24h, 48h, 72h, 96h, for RKO cells, and 0h, 24h, 48h, 72h, for LoVo cells using the MTT reagent by reading at 570 nm. To determine cell sensitivity to rapamycin, on the following day, different concentrations of rapamycin (0, 5, 20, 40 or 50μM) (Cat#S1039, Selleckchem) were added in the culture medium, and then incubated for another 48h. All treatments were performed in triplicate. The inhibition of cell proliferation at 48 hours was calculated by the fomula: [(OD value of control - OD value of the sample)/OD value of control] × 100%. The experiment was repeated three times. All the graphs were generated by GraphPad Prism 5 software.
Colony formation assay
Cells transfected with plasmids or siRNAs were seeded 250 cells/well into 6-well plates and incubated at 37°C for 14 days. Colonies were fixed and stained with 0.01% crystal violet, photographed and counted. All relevant assays were performed independently at least three times.
Co-immunoprecipitation (co-IP) assay
Cells transfected with pCMV-3Tag-1A-CENPH and pCMV-Myc-GOLPH3 [25] were harvested and subjected to co-IP assays. Protein lysates were incubated with 1μg of anti-Flag antibody (Cat#F1804, Sigma-Aldrich, USA) or anti-c-Myc antibody (Cat#631206, Clontech, USA) for 4 h at 4°C under rotation, and then incubated with rProtein A sepharose bead (GE healthcare) for 2 h. The precipitates were washed three times with lysis buffer, and then subjected to Western blot using anti-Flag (1:1000) and anti-c-Myc antibodies (1:500).
GST pull-down and His-tag pull-down assays
Human full-length CENPH and GOLPH3 cDNA was cloned into pET-28a(+) and pGEX-4T-1 vector, respectively. The recombinant His-CENPH and GST-GOLPH3 protein were expressed and purified according to manufacturer's instructions (Amersham). GST pull-down and His-tag pull-down assays were performed as described previously [27, 28]. Briefly, E. coli cell lysates containing GST and His-tag fusion protein were prepared in lysis buffer (1% Triton X-100, 1 mmol/L PMSF, and 1 mg/mL lysozyme in PBS) followed by sonication. After centrifugation, supernatants were incubated with 10μl glutathione sepharose 4B beads (GE Healthcare) and Ni-NTA agarose (QIAGEN) for 4 h at 4°C, respectively, for GST pull-down and His-tag pull-down assays. The pellets were washed with PBS extensively. The bound proteins were resolved by 12% SDS-PAGE and detected by western blot analysis with anti-GST and anti-His tag antibodies (1:1000, Beijing Zhongshan Golden Bridge Biotechnology Co Ltd, China).
Confocal microscopy
Human full-length CENPH and GOLPH3 cDNA was cloned into pDsRed-Express-C1 and pEGFP-C1 vector, respectively. Cells grown in 35-mm glass bottom dishes were cotransfected with pDsRed-Express-C1-CENPH and pEGFP-C1-GOLPH3. After transfection for 48 h, the cells were stained with Hoechst 33342 at 37℃for 10 minutes. A Leica SP5 confocal system (Leica Microsystems, Dresden, Germany) was used to observe the localization of CENPH and GOLPH3.
Analysis of gene expression datasets
The gene expression profiles of human CRC for this study were downloaded from Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/). CRC tumor expression data for CENPH mRNA and clinical information were extracted from the public CRC datasets. Differences in CENPH mRNA expression between normal biopsies and tumor tissues were evaluated using paired-sample t-tests. One-way ANOVA method was performed to analyze the differences in expression levels of CENPH between the TNM stages (I-IV). Patients from each dataset were classified into CENPH high and low-expression groups, according to the levels of CENPH transcript level. Chi-square test was performed to analyze the association between CENPH expression and clinicopathological variables. Survival curves were evaluated using the Kaplan-Meier method and compared using the log-rank test. Disease-free survival (DFS) was calculated as the time from primary surgery to the first of either recurrence or relapse, second cancer, or death. Disease-specific survival (DSS) was estimated as the time from primary surgery to cancer related death. Overall survival (OS) was defined as the time from primary surgery to death or date of last follow-up. Univariate and multivariate Cox proportional hazard regression models were used to evaluate the predictors for survival. All statistical analyses were performed using SPSS software 13.0 (SPSS Inc., Chicago, IL, USA). A two-sided P value of < 0.05 was considered statistically significant.
Fig 1
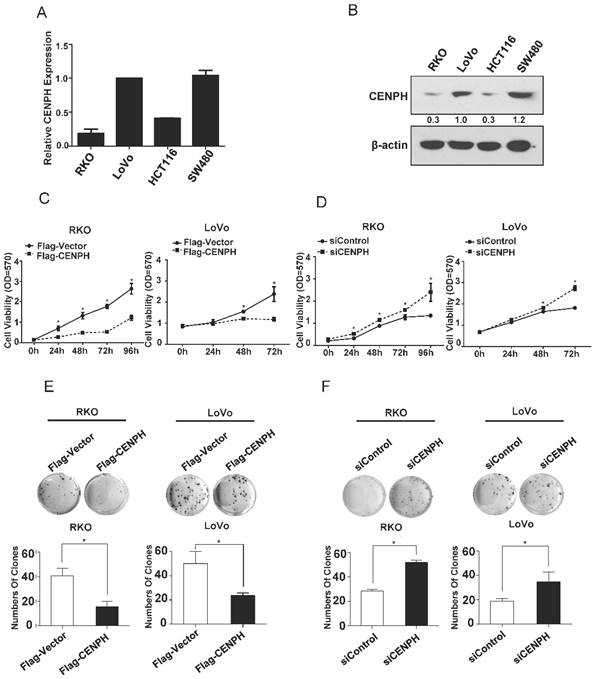
CENPH inhibits CRC cellular malignant phenotypes. (A) The expression levels of CENPH in 4 CRC cell lines were analyzed by Real-time PCR. (B) The levels of CENPH in CRC cell lines were determined by Western blot. Ratios of CENPH/β-actin shown under the representative blots were normalized to CENPH in LoVo cells. (C) Overexpression of CENPH reduced cell proliferation in RKO and LoVo cell lines. (D) Knockdown of CENPH promoted CRC cell growth. (E) Overexpression of CENPH suppressed colony formation in both RKO and LoVo cell lines. (F) Depletion of CENPH enhanced colony formation in CRC cells. *, P < 0.05.
Results
CENPH inhibited CRC malignant phenotypes
We first assessed CENPH mRNA and protein levels in four CRC cell lines (RKO, LoVo, HCT116 and SW480) by Real-time PCR and Western blot, respectively (Fig. 1A, B). CENPH was highly expressed in LoVo and SW480 cells at both mRNA and protein levels. In contrast, RKO and HCT116 cells exhibited low expression of CENPH. Thus, LoVo and RKO cells with high and low expression levels of CENPH, respectively, were chosen for further studies in vitro. Moreover, to facilitate the experiments, we used plasmid encoding full length of CENPH and CENPH siRNAs to overexpress and knockdown CENPH levels in both RKO and LoVo cells. The efficiency of CENPH overexpression and knockdown was confirmed in both cell lines by Real-time PCR and Western blot (Fig. S1).
To evaluate the roles of CENPH in the viability of CRC cells, MTT and colony formation assays were performed. The results showed that overexpression of CENPH impaired cellular malignant phenotypes including cell proliferation and colony formation (Fig. 1C, E). However, depletion of CENPH significantly enhanced malignant phenotypes (Fig. 1D, F). These findings suggest that CENPH inhibits the development of CRC.
Fig 2
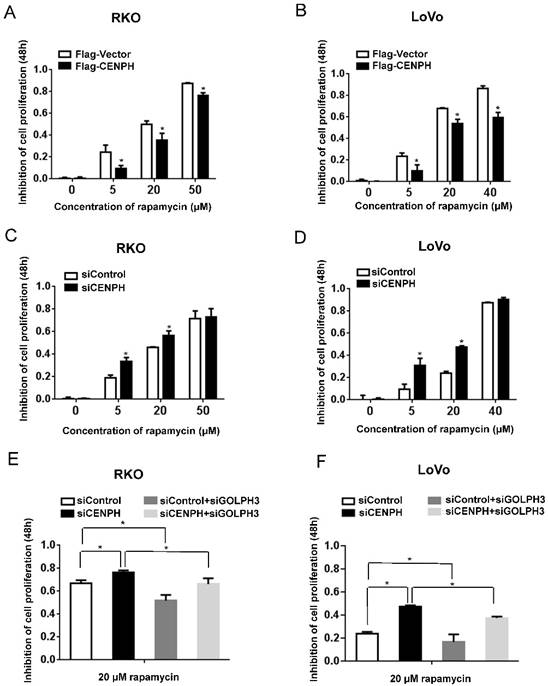
CENPH modulates rapamycin sensitivity in CRC cells. Cells were treated with increasing concentration of rapamycin, and cell viability was determined by MTT assays. (A, B) Overexpression of CENPH in RKO and LoVo cells significantly decreased inhibition of cell proliferation by rapamycin treatment. (C, D) Depletion of CENPH in RKO and LoVo cells promoted rapamycin-induced inhibition of cell growth. (E, F) GOLPH3 is required for the induction of rapamycin cytotoxicity by CENPH depletion in RKO and LoVo cells. *, P < 0.05.
CENPH modulates rapamycin sensitivity through GOLPH3
The effects of CENPH on rapamycin cytotoxicity to CRC cells were determined by MTT assays. Although the mRNA and protein levels of CENPH and GOLPH3 altered minimally in response to low doses of rapamycin in LoVo cells (Fig. S2), overexpression of CENPH significantly decreased rapamycin-induced inhibition of cell proliferation in a rapamycin-dose-dependent manner in RKO and LoVo cells (Fig. 2A, B). Furthermore, knockdown of CENPH significantly promoted rapamycin-induced cytotoxicity (Fig. 2C, D). The data confirmed the roles of CENPH on tumor cell sensitivity to rapamycin.
GOLPH3 confers increased sensitivity to rapamycin in cancer [9]. To further determine whether CENPH regulates rapamycin sensitivity via GOLPH3 in CRC cells, we knocked down both CENPH and GOLPH3 by siRNAs. As expected, CENPH depletion enhanced rapamycin-induced cytotoxicity. However, knockdown of GOLPH3 significantly attenuated the elevated rapamycin-induced inhibition of cell growth detected with CENPH depletion (Fig. 2E, F). Taken together, the results suggest that CENPH functions partially depend on the expression of GOLPH3.
CENPH interacts with GOLPH3 in vivo and in vitro
To investigate the association between CENPH and GOLPH3, co-IP experiments were performed to determine whether CENPH interacts with GOLPH3 in vivo. RKO cells were transiently transfected with pCMV-3Tag-1A-CENPH and pCMV-Myc-GOLPH3, the lysates were immunoprecipitated with the indicated antibodies, and the immune complexes were analyzed with Western blot. Myc-GOLPH3 was detected in immunoprecipitates of Flag-CENPH (Fig. 3A). Similarly, Flag-CENPH was detected in immune complexes of Myc-GOLPH3 by reciprocal IP (Fig. 3B). Thus, these data suggested that CENPH could interact with GOLPH3 in vivo.
We further examined whether CENPH directly interacts with GOLPH3 in vitro by GST pull-down and His-tag pull-down assays. For GST pull-down assays, CENPH was bacterially expressed as His-tag fusion protein, whereas GOLPH3 was bacterially expressed as GST fusion protein. The results showed that His-CENPH recombinant protein bound to GST-GOLPH3, but not to the GST alone (Fig. 3C). Likewise, His-tag pull-down assays showed GST-GOLPH3 was pulled down by His-CENPH, while there was no protein pulled down with GST control (Fig. 3D), suggesting that CENPH and GOLPH3 could directly interact in vitro.
Fig 3
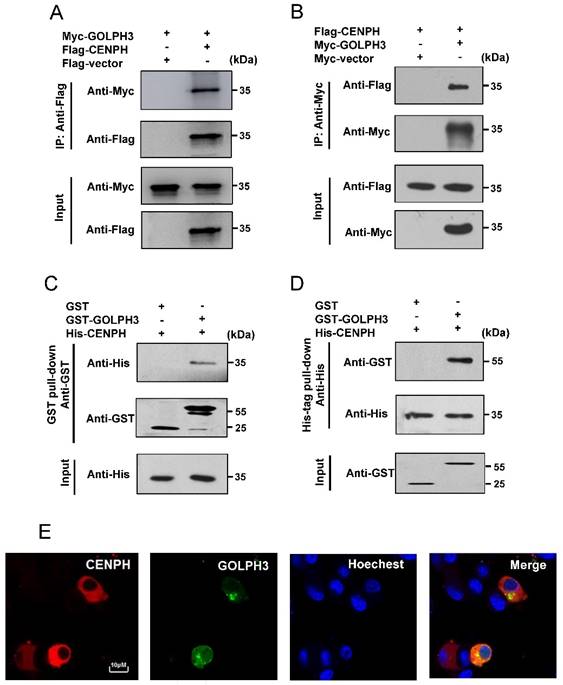
CENPH interacts with GOLPH3. (A, B) Exogenous CENPH interacts with GOLPH3. The indicated plasmids were expressed in cells and proteins were affinity purified. CENPH and GOLPH3 protein levels were assessed against indicated antibodies, respectively. (C, D) His-CENPH directly interacted with GST-GOLPH3, but not GST, as determined by the in vitro GST pull-down and His-tag pull-down assays, respectively. (E) Colocalization of CENPH and GOLPH3 in CRC cells. Cells cotransfected with pDsRed-Express-C1-CENPH and pEGFP-C1-GOLPH3 were observed under a laser scanning confocal microscope. Colocalization was shown by merge (yellow or orange). Nuclei were counterstained with Hoechest 33342 (blue).
Intracellular colocalization of CENPH and GOLPH3
The expression profile of CENPH and GOLPH3 was observed with a laser scanning confocal microscopy. The expression of DsRed-CENPH fusion protein was distributed in the cytoplasm (Fig. 3E, CENPH). It has been well described that GOLPH3 was located to the Golgi apparatus [9]. Consistently, GFP-GOLPH3 fusion protein was mainly located around the nucleus where Golgi network is usually located (Fig. 3E, GOLPH3). The red and green fluorescences overlapped in the cytoplasm or around the nucleus, resulting in yellow or orange color in merged images (Fig. 3E, Merge), suggesting the detection of an intense colocalization of CENPH and GOLPH3.
CENPH regulates mTOR signaling pathway via GOLPH3
Since CENPH regulated cancer cell sensitivity to rapamycin, which inhibits the kinase activity of mTORC1, we assessed whether CENPH could modulate mTOR signaling in CRC cells. Firstly, GSEA was performed using the gene expression profiles from GEO dataset GSE38832. mTOR signaling pathway was identified to be significantly associated with CENPH expression in GSE38832 (Fig. S3). To confirm the GSEA analysis of CENPH, the status of molecules associated with mTOR signaling was determined. The results showed that overexpression of CENPH reduced phosphorylation of pAKT and pS6K, which are the direct substrates of mTORC2 and mTORC1, respectively (Fig. 4A). In addition, CENPH overexpression decreased GOLPH3 mRNA and protein levels in RKO and LoVo cells (Fig. 4A, Fig. S1A, B). In contrast, the depletion of CENPH expression by transfection of siRNAs resulted in elevated levels of pAKT and pS6K in CRC cells (Fig. 4B). Meanwhile, CENPH knockdown upregulated the GOLPH3 mRNA and protein levels in CRC cells (Fig. 4B, Fig. S1C, D). Furthermore, due to the role of GOLPH3 in activating mTOR signaling [9], and the binding of CENPH with GOLPH3, we investigated whether GOLPH3 is required for CENPH-mediated mTOR signaling. The phosphorylation of AKT and S6K was examined with reduced expression of GOLPH3 and CENPH by siRNAs. These results suggested that knockdown of GOLPH3 attenuated the induction of phosphorylation of pAKT and pS6K upon CENPH suppression (Fig. 4C).
Fig 4
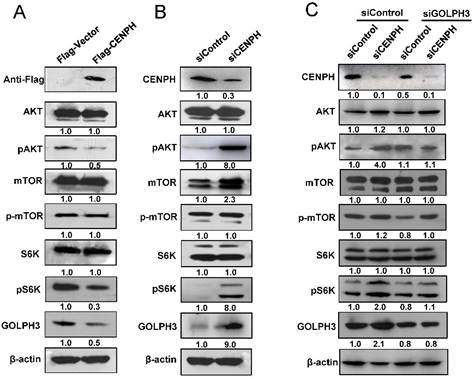
CENPH modulates mTOR signaling pathway though GOLPH3. Western blot was performed with indicated antibodies to determine the status of several mTOR substrates. (A) Overexpression of CENPH in LoVo cells reduced phosphorylation of pAKT and pS6K. (B) Knockdown of CENPH in LoVo cells increased the levels of pAKT and pS6K. (C) Depletion of GOLPH3 partially reversed the activation of pAKT and pS6K induced by CENPH knockdown. Ratios of the detected target/β-actin shown under the representative blots were normalized to that of controls in CRC cells.
Table 1
Correlations between CENPH expression and clinicopathological characteristics in CRC patients from dataset GSE39582.
| Variables | Cases | CENPH expression | ||
|---|---|---|---|---|
| Low | High | P | ||
| Age | ||||
| <60 yr | 292 | 210 | 82 | 0.301 |
| ≥60 yr | 292 | 221 | 71 | |
| Gender | ||||
| Female | 322 | 157 | 165 | 0.477 |
| Male | 263 | 136 | 127 | |
| Tumor location | ||||
| Distal | 197 | 171 | 180 | 0.477 |
| Proximal | 293 | 120 | 112 | |
| TNM stage | ||||
| Ⅰ | 38 | 16 | 22 | 0.080 |
| Ⅱ | 272 | 125 | 147 | |
| Ⅲ | 209 | 117 | 92 | |
| Ⅳ | 60 | 34 | 26 | |
| Depth of Invasion | ||||
| T1/T2 | 57 | 22 | 35 | 0.076 |
| T3/T4 | 498 | 254 | 244 | |
| Lymph node metastasis | ||||
| Negative | 168 | 62 | 104 | <0.001 |
| Positive | 391 | 216 | 175 | |
| Distance metastasis | ||||
| Negative | 452 | 227 | 255 | 0.203 |
| Positive | 61 | 27 | 34 | |
| Survival | ||||
| Alive | 288 | 183 | 105 | 0.134 |
| Dead | 291 | 202 | 89 | |
| Recurrence | ||||
| Positive | 179 | 98 | 81 | 0.112 |
| Negative | 395 | 188 | 207 | |
P values in bold were statistically significant.
High CENPH expression indicated favorable prognosis in patients with CRC
To determine mRNA levels of CENPH expression in CRC tissues, public available datasets were used. The results showed that CENPH transcripts was significantly increased in CRC tumors compared with matched normal tissues from 2 independent datasets GSE9348 and GSE4183 (P < 0.001 and P = 0.005, respectively, Fig. 5A). The development of CRC can be divided into four stages (stage I, II, III, and IV) according to depth of tumor invasion (T), metastasis in reginal lymph nodes (N), and distant metastasis (M) features. To evaluate the effects of CENPH in CRC progression, we assessed the levels of CENPH according to the stages in the datasets GSE38832 and GSE39582. One-way ANOVA showed CENPH mRNA level was decreased gradually in respect to tumor stages. There was significantly different in the levels of CENPH among different stages (P = 0.043, and P = 0.041 for GSE38832 and GSE39582, respectively, Fig. 5B).
Patients in each dataset were divided into CENPH high and low expression groups, according to CENPH transcript levels. The correlation between CENPH expression and clinicopathological features in GSE39582 dataset is shown in Table 1. The results revealed that CENPH mRNA expression was significantly associated with lymph node metastasis (P < 0.001). However, no significant associations were detected between CENPH expression and other clinicopathological variables including age, gender, tumor location, tumor depth of invasion, distant metastasis, survival or recurrence (P > 0.05, Table 1).
Kaplan-Meier analysis was performed to investigate the prognostic relevance of CENPH in CRC. According to datasets of GSE38832, which included disease-free survival (DFS) and disease-specific survival (DSS) information, patients with high levels of CENPH expression had marginally significantly better DFS and DSS than those with low levels of CENPH expression (DFS, P = 0.050; DSS, P = 0.050; Fig. 5C, D). Similarly, in another CRC dataset GSE39582, patients with CENPH-high tumors had improved DFS and overall survival (OS), compared to those with CENPH-low tumors (DFS, P = 0.096; OS, P = 0.047; Fig. 5E, F). These results suggest that high levels of CENPH mRNA expression could predict favorable prognosis for patients with CRC.
Table 2
Univariate and multivariate analysis of CENPH in CRC patients from GSE39582 with respect to OS.
| Variables | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| HR | 95%CI | P | HR | 95%CI | P | |
| Age (≥60 yr vs <60 yr) | 1.405 | 0.944-1.982 | 0.054 | |||
| Gender (Male vs Female) | 1.308 | 0.980-1.744 | 0.068 | |||
| Tumor location (Distal vs Proximal) | 0.923 | 0.691-1.232 | 0.585 | |||
| Depth of invasion (T3/T4 vs T1/ T2) | 2.367 | 1.211-4.626 | 0.012 | 1.980 | 1.000-3.919 | 0.050 |
| Lymph node metastasis (Positive/ Negative) | 2.491 | 1.436-4.323 | 0.001 | 1.479 | 1.037-2.111 | 0.031 |
| Distant Metastasis (Positive/ Negative) | 5.176 | 0.044-2.330 | <0.001 | 4.558 | 2.976-6.982 | <0.001 |
| CENPH Expression (High vs Low) | 0.752 | 0.567-0.988 | 0.048 | 0.765 | 0.562-1.042 | 0.089 |
| GOLPH3 Expression (High vs Low) | 0.888 | 0.670-1.178 | 0.411 |
HR hazard ratio, CI confidence interval, P values in bold were statistically significant.
Moreover, Cox proportional hazard regression analysis was performed to analyze the prognostic value of CENPH expression. In univariate analysis, patients whose tumors have high CENPH expression had a longer period of OS (HR 0.752; 95% CI 0.567-0.988; P = 0.048) than those whose tumors have low CENPH expression. All variables that indicated prognostic significance in the univariate analysis were involved in the multivariate analysis. The results revealed that patients with high levels of CENPH expression and superficial tumor invasion had marginally significantly longer periods of OS than those with low levels of CENPH expression (HR 0.765; 95% CI 0.562-1.042; P = 0.089, and HR 1.980; 95% CI 1.000-3.919; P = 0.050, respectively, Table 2). As expected, positive lymph node metastasis and distant metastasis were found to be independent predictors for poor OS (HR 1.479; 95% CI 1.037-2.111; P = 0.031, and HR 4.558; 95% CI 2.976-6.982; P < 0.001, respectively, Table 2). However, there was no significant association between other clinocophathological features and OS in multivariate analysis (Table 2).
Discussion
In this study, we demonstrated that CENPH inhibited CRC malignant phenotypes, conferred impaired sensitivity to rapamycin, and regulated mTOR signaling through the interaction with GOLPH3. Moreover, high levels of CENPH expression were associated with favorable prognosis in patients with CRC. These finding raised the possibility that the expression levels of CENPH may serve as a predictor of sensitivity to mTOR inhibitor, rapamycin.
Fig 5
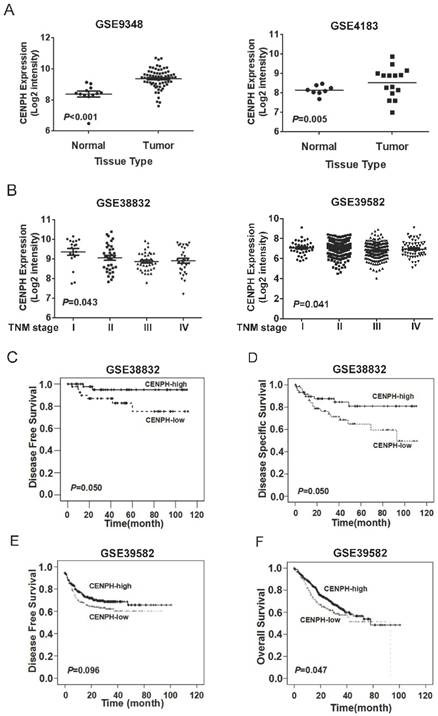
Expression of CENPH in human CRC tissues. (A) CENPH mRNA level was significantly higher in CRC tissues than in adjacent normal CRC tissues from GEO datasets. (B) CENPH mRNA level was decreased gradually according to tumor stage. (C-F) Kaplan-Meier survival curves revealed that high CENPH expression significantly correlated with favorable survival by analyzing GEO datasets GSE38832 (C, D) and GSE39582 (E, F), respectively. The log-rank test was used to calculate P values.
Centromere protein H (CENPH), as a crucial component of the active kinetochore complex, was localized in the inner plate together with CENPA and CENPC [12, 13, 29]. It has been well established that CENPH plays a basic role in linking centromeres to the spindle microtubule [12, 13, 29]. Consequently, CENPH contributes to the function of maintaining centromere structure, kinetochore formation, and sister chromatid separation [12, 13, 29]. Since kinetochore proteins were involved in cancer development and progression, previous studies demonstrated that overexpression of CENPH promoted cell growth and proliferation in gastric cancer, renal cell carcinoma, and tongue cancer [17, 18, 23]. However, inconsistent with previous studies, our results suggested that CENPH abrogated cellular malignant phenotypes including cell growth and colony formation in CRC cells. The completely different function of CENPH in cancer is likely tissue-specific consequence.
The overexpression of CENPH has been validated in in several types of tumors, including colorectal cancer, gastric cancer, esophageal carcinoma, oral squamous cell carcinomas, tongue cancer, hepatocellular carcinoma, nonsmall cell lung cancer, breast cancer, renal cell carcinoma, and nasopharyngeal carcinoma [14, 17-25]. Although elevated CENPH levels indicates poor prognosis in most types of cancers, the clinical significance of CENPH expression in CRC remains largely unknown. In this study, the analysis of public datasets demonstrated that the levels of CENPH transcript were significantly higher in CRC tissues than those in matched normal tissues. High levels of CENPH transcripts decreased gradually according to tumor stages. However, inconsistent with studies in other types of cancer, our data indicated that patients with high CENPH expression had favorable survival, suggesting better clinical outcome.
It has been well established that mTOR pathway plays an essential role in the development and progression of CRC [30, 31]. As mTOR inhibitors, temsirolimus and everolimus had been approved by FDA for the treatment of patients with metastatic renal cell carcinoma [32, 33]. Although rapamycin (sirolimus) was usually used as immunosuppressant for organ transplantation [34], the role of rapamycin in inhibiting the protein kinase activity of mTOR and anitproliferative ability makes it a potential anticancer agent [30, 31]. Preclinical studies have suggested that the combination of rapamycin with a VEGF antibody or trastuzumab exhibited a synergistic antitumor effect in a pancreatic or breast cancer model, respectively [35, 36]. However, many clinical trials did not show the desired pharmacological properties [37]. A phase I study indicated that a combination of sunitinib and rapamycin is unacceptable due to irresistible toxicities in patients with refractory solid malignancies including CRC [34]. Therefore, it is a long way to go before it could be used in the clinic. In this study, the effects of CENPH on rapamycin cytotoxicity to CRC cells were evaluated. The results suggested that change of CENPH expression level significantly regulated rapamycin-induced cytotoxicity.
In this study, CENPH could not only interact with GOLPH3, but also regulate the levels of GOLPH3. GOLPH3 was first identified as a Golgi member protein involved in Golgi trafficking and morphology [38]. It has been reported that GOLPH3, as a potent oncogene, activates mTOR signaling by phosphorylating both mTORC1 and mTORC2 specific substrates. Consequently, GOLPH3 enhances tumor cell sensitivity to rapamycin in vivo [9]. However, Zhu et al. demonstrated that GOLPH3 overexpression is associated with poor response to neoadjuvant chemoradiotherapy in rectal cancer [39]. Given the roles of GOLPH3 in cancer, and the association between CENPH and GOLPH3, we investigated the impact of CENPH on mTOR signaling pathway. The results indicated that the alteration of CENPH expression level could regulate mTOR signaling cascade. In addition, GOLPH3 is required for the function of CENPH. The PI3K/AKT/mTOR signaling pathway is activated in most cancers, representing focused issue for cancer drug development. The role of CENPH in the modulation of mTOR signaling and rapamycin sensitivity raised the possibility that the levels of CENPH together with GOLPH3 may predict sensitivity to mTOR inhibitors.
However, there are some limitations of our study. Although in this study we demonstrated the association between CENPH and GOLPH3, as well as their effects on mTOR signaling, the predictive values of CENPH and GOLPH3 as indicators for rapamycin sensitivity remains to be elucidated by randomized clinical cohorts for patients with or without rapamycin treatment. Moreover, to confirm the clinical significance of CENPH expression in patients with CRC, replication cohorts containing detailed clinicopathological parameters are required.
In conclusion, we demonstrated that CENPH regulates CRC cell proliferation and cell sensitivity to rapamycin. CENPH binds to GOLPH3 and modulates both mTORC1 and mTORC2 in mTOR signaling pathway via GOLPH3. Moreover, CRC patients with high CENPH expression indicates improved survival. These findings suggest that CENPH may serve as a potential predictor for rapamycin sensitivity and a therapeutic target for CRC patients.
Supplementary Material
Supplementary figures and methods.
Abbreviations
CRC: Colorectal cancer; CENPH: Centromere protein H; mTOR: Mammalian target of rapamycin; mTORC1: mTOR complex 1; mTORC2: mTOR complex 2; FBS: Fetal bovine serum; co-IP: Co-immunoprecipitation; DFS: Disease-free survival; DSS: Disease-specific survival; OS: overall survival.
Acknowledgements
This study was supported by the grants from National Natural Science Foundation of China (81272766, 81450028, 81672439, 81470129, 81402346, 81502137), Beijing Natural Science Foundation (7162039, 7132054, 7172042), the Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (XM201309, ZYLX201701), Peking University (PKU) 985 Special Funding for Collaborative Research with PKU Hospitals (To XS and FB), Beijing Health System High-level Health Technical Personnel Training Program (2015-3-074), Science Foundation of Peking University Cancer Hospital.
Competing interests
All authors declare that they have no competing interests.
References
1. Siegel R, Desantis C, Jemal A. Colorectal cancer statistics2014. CA Cancer J Clin. 2014;64(2):104-17
2. DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, Alteri R, Robbins AS, Jemal A. Cancer treatmentand survivorship statistics, 2014. CA Cancer J Clin. 2014;64(4):252-71
3. Tong L, Ahn C, Symanski E, Lai D, Du XL. Effects of newly developed chemotherapy regimens, comorbidities, chemotherapy-related toxicities on the changing patterns of the leading causes of death in elderly patients with colorectal cancer. Ann Oncol. 2014;25(6):1234-42
4. Yu L, Lu Y, Han X, Zhao W, Li J, Mao J, Wang B, Shen J, Fan S, Wang L, Wang M, Li L, Tang J, Song B. MicroRNA -140-5p inhibits colorectal cancer invasion and metastasis by targeting ADAMTS5 and IGFBP5. Stem Cell Res Ther. 2016;7(1):180
5. Li X, Wu C, Chen N, Gu H, Yen A, Cao L, Wang E, Wang L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget. 2016;7(22):33440-50
6. Mayer IA, Arteaga CL. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu Rev Med. 2016;67:11-28
7. Polivka J Jr, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther. 2014;142(2):164-75
8. Phyu SM, Tseng CC, Fleming IN, Smith TA. Probing the PI3K/Akt/mTor pathway using 31P-NMR spectroscopy: routes to glycogen synthase kinase 3. Sci Rep. 2016;6:36544
9. Scott KL, Kabbarah O, Liang MC, Ivanova E, Anagnostou V, Wu J, Dhakal S, Wu M, Chen S, Feinberg T, Huang J, Saci A, Widlund HR, Fisher DE, Xiao Y, Rimm DL, Protopopov A, Wong KK, Chin L. GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature. 2009;459(7250):1085-90
10. Faller WJ, Jackson TJ, Knight JR, Ridgway RA, Jamieson T, Karim SA, Jones C, Radulescu S, Huels DJ, Myant KB, Dudek KM, Casey HA, Scopelliti A, Cordero JB, Vidal M, Pende M, Ryazanov AG, Sonenberg N, Meyuhas O, Hall MN, Bushell M, Willis AE, Sansom OJ. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature. 2015;517(7535):497-500
11. Peng J, Fang Y, Tao Y, Li K, Su T, Nong Y, Xie F, Lai M. Mechanisms of GOLPH3 associated with the progression of gastric cancer: a preliminary study. PLoS One. 2014;9(10):e107362
12. Fukagawa T, Mikami Y, Nishihashi A, Regnier V, Haraguchi T, Hiraoka Y, Sugata N, Todokoro K, Brown W, Ikemura T. CENP-H, a constitutive centromere component, is required for centromere targeting of CENP-C in vertebrate cells. EMBO J. 2001;20(16):4603-17
13. Westermann S, Cheeseman IM, Anderson S, Yates JR 3rd, Drubin DG, Barnes G. Architecture of the budding yeast kinetochore reveals a conserved molecular core. J Cell Biol. 2003;163(2):215-22
14. Tomonaga T, Matsushita K, Ishibashi M, Nezu M, Shimada H, Ochiai T, Yoda K, Nomura F. Centromere Protein H Is Up-regulated in Primary Human Colorectal Cancer and Its Overexpression Induces Aneuploidy. Cancer Res. 2005;65(11):4683-9
15. Zhu L, Wang Z, Wang W, Wang C, Hua S, Su Z, Brako L, Garcia-Barrio M, Ye M, Wei X, Zou H, Ding X, Liu L, Liu X, Yao X. Mitotic Protein CSPP1 Interacts with CENP-H Protein to Coordinate Accurate Chromosome Oscillation in Mitosis. J Biol Chem. 2015;290(45):27053-66
16. Amaro AC, Samora CP, Holtackers R, Wang E, Kingston IJ, Alonso M, Lampson M, McAinsh AD, Meraldi P. Molecular control of kinetochore-microtubule dynamics and chromosome oscillations. Nat Cell Biol. 2010;12(4):319-29
17. He WL, Li YH, Yang DJ, Song W, Chen XL, Liu FK, Wang Z, Li W, Chen W, Chen CY, He YL, Zhan WH. Combined evaluation of centromere protein H and Ki-67 as prognostic biomarker for patients with gastric carcinoma. Eur J Surg Oncol. 2013;39(2):141-9
18. Guo XZ, Zhang G, Wang JY, Liu WL, Wang F, Dong JQ, Xu LH, Cao JY, Song LB, Zeng MS. Prognostic relevance of Centromere protein H expression in esophageal carcinoma. BMC Cancer. 2008;8:233
19. Shigeishi H, Higashikawa K, Ono S, Mizuta K, Ninomiya Y, Yoneda S, Taki M, Kamata N. Increased expression of CENP-H gene in human oral squamous cell carcinomas harboring high-proliferative activity. Oncol Rep. 2006;16(5):1071-5
20. Liao WT, Yu CP, Wu DH, Zhang L, Xu LH, Weng GX, Zeng MS, Song LB, Li JS. Upregulation of CENP-H in tongue cancer correlates with poor prognosis and progression. J Exp Clin Cancer Res. 2009;28:74
21. Lu G, Shan T, He S, Ren M, Zhu M, Hu Y, Lu X, Zhang D. Overexpression of CENP-H as a novel prognostic biomarker for human hepatocellular carcinoma progression and patient survival. Oncol Rep. 2013;30(5):2238-44
22. Liao WT, Wang X, Xu LH, Kong QL, Yu CP, Li MZ, Shi L, Zeng MS, Song LB. Centromere protein H is a novel prognostic marker for human nonsmall cell lung cancer progression and overall patient survival. Cancer. 2009;115(7):1507-17
23. Liao WT, Feng Y, Li ML, Liu GL, Li MZ, Zeng MS, Song LB. Overexpression of centromere protein H is significantly associated with breast cancer progression and overall patient survival. Chin J Cancer. 2011;30(9):627-37
24. Wu X, Lin Y, Shi L, Huang Y, Lai C, Wang Y, Zhang M, Wang S, Heng B, Yu G, Du X, Fang L, Fu Y, Chen J, Guo Z, Su Z, Wu S. Upregulation of centromere protein H is associated with progression of renal cell carcinoma. J Mol Histol. 2015;46(4-5):377-85
25. Liao WT, Song LB, Zhang HZ, Zhang X, Zhang L, Liu WL, Feng Y, Guo BH, Mai HQ, Cao SM, Li MZ, Qin HD, Zeng YX, Zeng MS. Centromere Protein H Is a Novel Prognostic Marker for Nasopharyngeal Carcinoma Progression and Overall Patient Survival. Clin Cancer Res. 2007;13(2 Pt 1):508-14
26. Wang Z, Jiang B, Chen L, Di J, Cui M, Liu M, Ma Y, Yang H, Xing J, Zhang C, Yao Z, Zhang N, Dong B, Ji J, Su X. GOLPH3 predicts survival of colorectal cancer patients treated with 5-fluorouracil-based adjuvant chemotherapy. J Transl Med. 2014;12:15
27. Wang A, Cui M, Qu H, Di J, Wang Z, Xing J, Wu F, Wu W, Wang X, Shen L, Jiang B, Su X. Induction of anti-EGFR immune response with mimotopes identified from a phage display peptide library by panitumumab. Oncotarget. 2016;7(46):75293-306
28. Li X, Chen C, Wang F, Huang W, Liang Z, Xiao Y, Wei K, Wan Z, Hu X, Xiang S1, Ding X, Zhang J. KCTD1 Suppresses Canonical Wnt Signaling Pathway by Enhancing b-catenin Degradation. PLoS One. 2014;9(4):e94343
29. Sugata N, Li S, Earnshaw WC, Yen TJ, Yoda K, Masumoto H, Munekata E, Warburton PE, Todokoro K. Human CENP-H multimers colocalize with CENP-A and CENP-C at active centromere-kinetochore complexes. Hum Mol Genet. 2000;9(19):2919-26
30. Wen X, Chen Y. Expression of set is downregulated by rapamycin in human colorectal cancer cells. Biomed Rep. 2013;1(5):727-30
31. Thoreen CC, Sabatini DM. Rapamycin inhibits mTORC1, but not completely. Autophagy. 2009;5(5):725-6
32. Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, Kovacevic Z, Lesovoy V, Schmidt-Wolf IG, Barbarash O, Gokmen E, O'Toole T, Lustgarten S, Moore L, Motzer RJ; Global ARCC Trial. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356(22):2271-81
33. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA, Hollaender N, Kay A, Ravaud A; RECORD-1 Study Group. Phase 3 trial of everolimus for metastatic renal cell carcinoma: final results and analysis of prognostic factors. Cancer. 2010;116(18):4256-65
34. Li J, Kluger H, Devine L, Lee JJ, Kelly WK, Rink L, Saif MW. Phase I study of safety and tolerability of sunitinib in combination with sirolimus in patients with refractory solid malignancies and determination of VEGF (VEGF-A) and soluble VEGF-R2 (sVEGFR2) in plasma. Cancer Chemother Pharmacol. 2016;77(6):1193-200
35. Stephan S, Datta K, Wang E, Li J, Brekken RA, Parangi S, Thorpe PE, Mukhopadhyay D. Effect of rapamycin alone and in combination with antiangiogenesis therapy in an orthotopic model of human pancreatic cancer. Clin Cancer Res. 2004;10(20):6993-7000
36. Lu CH, Wyszomierski SL, Tseng LM, Sun MH, Lan KH, Neal CL, Mills GB, Hortobagyi GN, Esteva FJ, Yu D. Preclinical testing of clinically applicable strategies for overcoming trastuzumab resistance caused by PTEN deficiency. Clin Cancer Res. 2007;13(19):5883-8
37. Meng LH, Zheng XF. Toward rapamycin analog (rapalog)-based precision cancer therapy. Acta Pharmacol Sin. 2015;36(10):1163-9
38. Scott KL, Chin L. Signaling from the Golgi: mechanisms and models for Golgi phosphoprotein 3-mediated oncogenesis. Clin Cancer Res. 2010;16(8):2229-34
39. Zhu K, Zhao Q, Yue J, Shi P, Yan H, Xu X, Wang R. GOLPH3 overexpression correlates with poor response to neoadjuvant therapy and prognosis in locally advanced rectal cancer. Oncotarget. 2016;7(42):68328-38
Author contact
![]() Corresponding authors: Beihai Jiang, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Department of Gastrointestinal Surgery IV, Peking University Cancer Hospital & Institute, 52 Fucheng Road, Haidian District, Beijing 100142, China. Tel: 8610-88196836; Fax: 8610-88122437; E-mail: beihai_jiangedu.cn and Xiangqian Su, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Department of Gastrointestinal Surgery IV, Peking University Cancer Hospital & Institute, 52 Fucheng Road, Haidian District, Beijing 100142, China. Tel: 8610-88196579; Fax: 8610-88122437; E-mail: suxiangqianedu.cn
Corresponding authors: Beihai Jiang, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Department of Gastrointestinal Surgery IV, Peking University Cancer Hospital & Institute, 52 Fucheng Road, Haidian District, Beijing 100142, China. Tel: 8610-88196836; Fax: 8610-88122437; E-mail: beihai_jiangedu.cn and Xiangqian Su, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Department of Gastrointestinal Surgery IV, Peking University Cancer Hospital & Institute, 52 Fucheng Road, Haidian District, Beijing 100142, China. Tel: 8610-88196579; Fax: 8610-88122437; E-mail: suxiangqianedu.cn
Citation styles
APA
Wu, W., Wu, F., Wang, Z., Di, J., Yang, J., Gao, P., Jiang, B., Su, X. (2017). CENPH Inhibits Rapamycin Sensitivity by Regulating GOLPH3-dependent mTOR Signaling Pathway in Colorectal Cancer. Journal of Cancer, 8(12), 2163-2172. https://doi.org/10.7150/jca.19940.
ACS
Wu, W.; Wu, F.; Wang, Z.; Di, J.; Yang, J.; Gao, P.; Jiang, B.; Su, X. CENPH Inhibits Rapamycin Sensitivity by Regulating GOLPH3-dependent mTOR Signaling Pathway in Colorectal Cancer. J. Cancer 2017, 8 (12), 2163-2172. DOI: 10.7150/jca.19940.
NLM
Wu W, Wu F, Wang Z, Di J, Yang J, Gao P, Jiang B, Su X. CENPH Inhibits Rapamycin Sensitivity by Regulating GOLPH3-dependent mTOR Signaling Pathway in Colorectal Cancer. J Cancer 2017; 8(12):2163-2172. doi:10.7150/jca.19940. https://www.jcancer.org/v08p2163.htm
CSE
Wu W, Wu F, Wang Z, Di J, Yang J, Gao P, Jiang B, Su X. 2017. CENPH Inhibits Rapamycin Sensitivity by Regulating GOLPH3-dependent mTOR Signaling Pathway in Colorectal Cancer. J Cancer. 8(12):2163-2172.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.