Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(3):643-653. doi:10.7150/jca.28542 This issue Cite
Review
Advances in Identification of Susceptibility Gene Defects of Hereditary Colorectal Cancer
Qiang Liu1,2,3, Yue-Qiu Tan2,3 ![]()
1. Hunan Key Laboratory of Translational Radiation Oncology, Hunan cancer Hospital and The Affiliated Cancer of Xiangya School of Medicine, Central South University, Changsha, China
2. Institute of Reproductive and Stem Cell Engineering, School of Basic Medical Science, Central South University, Changsha, China
3. Reproductive and Genetic Hospital of CITIC-Xiangya, Changsha, China
Received 2018-7-15; Accepted 2018-12-8; Published 2019-1-1
Citation:
Liu Q, Tan YQ. Advances in Identification of Susceptibility Gene Defects of Hereditary Colorectal Cancer. J Cancer 2019; 10(3):643-653. doi:10.7150/jca.28542. https://www.jcancer.org/v10p0643.htm
Other stylesAbstract
Colorectal cancer (CRC) is a common malignant tumor of the digestive system worldwide, associated with hereditary genetic features. CRC with a Mendelian genetic predisposition accounts for approximately 5-10% of total CRC cases, mainly caused by a single germline mutation of a CRC susceptibility gene. The main subtypes of hereditary CRC are hereditary non-polyposis colorectal cancer (HNPCC) and familial adenomatous polyposis (FAP). With the rapid development of genetic testing methods, especially next-generation sequencing technology, multiple genes have now been confirmed to be pathogenic, including DNA repair or DNA mismatch repair genes such as APC, MLH1, and MSH2. Since familial CRC patients have poor clinical outcomes, timely clinical diagnosis and mutation screening of susceptibility genes will aid clinicians in establishing appropriate risk assessment and treatment interventions at a personal level. Here, we systematically summarize the susceptibility genes identified to date and the potential pathogenic mechanism of HNPCC and FAP development. Moreover, clinical recommendations for susceptibility gene screening, diagnosis, and treatment of HNPCC and FAP are discussed.
Keywords: hereditary colorectal cancer, hereditary non-polyposis colorectal cancer (HNPCC), familial adenomatous polyposis (FAP), susceptibility genes
Introduction
Colorectal cancer (CRC) is the third most common malignant tumor among all cancer types worldwide, accounting for more than 8% of the annual global cancer-related death rate [1]. The incidence of CRC in men is second only to lung and prostate cancer, while that in women is second only to lung and breast cancer [2]. Although the majority of CRC cases are considered to be sporadic, approximately 30% of CRCs develop from genetic factors [3], and approximately 5% are associated with known susceptibility gene mutations [4]. The most common hereditary CRCs are hereditary nonpolyposis colorectal cancer (HNPCC) and familial adenomatous polyposis (FAP), followed by MutY homolog (MUTYH)-associated polyposis (MAP), Peutz-Jeghers syndrome, juvenile polyposis, Cowden/PTEN hamartoma syndrome, and other subtypes [5, 6]. Harboring susceptibility gene defects increase the possibility of developing CRC owing to genetic instability. The major HNPCC susceptibility genes discovered to date include DNA mismatch repair (MMR) genes such as MLH1, MSH2, MSH6, and PMS2 [7-10], whereas the main FAP susceptibility gene is adenomatous polyposis coli gene (APC) [11]. Moreover, recent progress in sequencing technology has accelerated the discovery of new susceptibility genes involved in CRC [9, 12-14].
At present, approximately 40 susceptibility genes have been identified to be associated with hereditary CRC [15, 16]. In this review, we searched for susceptibility genes of CRC to obtain a documented evidence (clinical, functional, or experimental) of the genes causing HNPCC and FAP in humans using PubMed and other computer-based internet websites. The key words included hereditary colorectal cancer (or CRC), hereditary nonpolyposis colorectal cancer (or HNPCC), Lynch syndrome, familial adenomatous polyposis (or FAP), susceptibility genes, gene mutations or variants, genetics, and genetic detection. For the susceptibility genes in HNPCC, we mainly concentrated on the genes related to MMR. In addition, we focused our attention on recent studies conducted in the last five years and highlighted the susceptibility genes with validated evidence. Other informative websites such as Online Mendelian Inheritance in Man (www.OMIM.org) and Gene Cards (www.genecards.org) were then used to compile an updated list of genes from these major sources. We firmly believe that the detection and mechanism elucidation of CRC susceptibility genes are of great significance toward gaining a comprehensive understanding of the molecular mechanism of hereditary CRC. In turn, these efforts will lead to improved accuracy in the clinical judgment of each case, which will allow for more appropriate monitoring or intervention at a personal level to improve patient care and outcome. Overall, this review summarizes the recent progress in identifying hereditary CRC susceptibility genes and elucidation of the underlying mechanism, along with associated recommendations for targeted clinical guidance and screening.
1. Susceptibility genes and mechanism of HNPCC
HNPCC is the most common form of hereditary CRC susceptibility syndrome, also known as Lynch syndrome [17, 18]. HNPCC is inherited in an autosomal dominant manner with high penetrance [19]. Compared with adenoma polyps, the number of adenomas in HNPCC is smaller (<3) [20]. Patients with HNPCC have a higher risk of developing CRC than the general population; approximately 2-4% of all CRC cases are caused by HNPCC [21]. In addition, HNPCC increases the risk of developing other types of cancers such as endometrial cancer, gastric cancer, ovarian cancer, and small intestine cancer [22]. Breast cancer, prostate cancer, and pancreatic cancer have also been associated with HNPCC [23-25].
The diagnosis of HNPCC is based on the Amsterdam II criteria [26] and revised Bethesda criteria [27]. According to the Amsterdam II criteria, patients diagnosed with Lynch syndrome should meet the following conditions: 1) at least three relatives suffered from HNPCC-related tumors (colorectal, endometrial, gastric, ovarian, ureteral, renal, pelvic, small bowel, hepatobiliary, or skin), and one of the relatives is a first-degree relative of the other two; 2) at least two consecutive generations were affected; 3) at least one HNPCC-related tumor was diagnosed before 50 years old; 4) FAP was ruled out; and 5) tumor tissue was confirmed by a pathology method [26]. According to the revised Bethesda criteria, DNA microsatellite instability (MSI) detection should be performed for patients who meet the following conditions: 1) diagnosed with CRC before the age of 50 years; 2) simultaneous or heterochrony colorectal or other HNPCC-related tumors without limit of the diagnosis age; 3) CRC patients under 60 years old who showed histological features of high DNA microsatellite instability (MSI-H, known as MSI of greater than 30% of unstable MSI loci [28]); 4) CRC and another kind of HNPCC-related tumor were diagnosed in one or more first-degree relatives, one of whom was diagnosed before 50 years old; and 5) two or more first- or second-degree relatives were diagnosed with CRC or HNPCC-related tumors without limit of diagnosis age [27].
1.1 Susceptibility genes of HNPCC
HNPCC-related susceptibility genes mainly belong to the families of DNA MMR genes such as MLH1, MSH2, MSH6, and PMS2. To date, more than 400 mutations have been detected in over 748 families [7, 29-32], with MLH1 and MSH2 mutations accounting for up to 90% of the total (Fig. 1A, B). The common susceptibility genes and some of the mutation sites identified in the past five years have been shown in Table 1. MLH1 is located on chromosome 3p21-23 and contains 19 exons. The most common mutations detected in this gene are missense and splice-site mutations [33, 34]. MSH2 is located on chromosome 2p21 and contains 16 exons. The main type of MSH2 mutation detected is frameshift mutations due to small deletions and insertions. Schneider et al. analyzed the mutational profile of MMR genes in 60 unrelated probands diagnosed with colorectal cancer or Lynch syndrome and identified pathogenic or likely pathogenic variants in MLH1 or MSH2 in 21 probands (35%) [34]. Another MMR gene, MSH6, accounts for about 7-10% of all mutations detected in HNPCC [35], with c.39593962delCAAG and c.39843987dupGTCA being the two most common mutations [36, 37]. PMS2 mutations are relatively rare, accounting for only about 5% of all mutations in HNPCC [5, 38, 39]. Recent studies found novel deleterious PMS2 mutations such as c.1492del11 [40]. Importantly, a substantial proportion of CRCs with solitary loss of PMS2 expression are associated with a deleterious MLH1 germline mutation, which supports the screening for MLH1 in patients with tumors of this immunophenotype [41]. Overall, the cumulative risk of developing CRC in MLH1, MSH2, and MSH6 mutation carriers above 70 years old is 41%, 48%, and 12%, respectively [42]. Engel et al. [5] found that the risk of CRC for HNPCC patients with MLH1 and MSH2 deletions was 22-74%, while that for patients with MSH6 or PMS2 mutations was 10-22% and 15-20%, respectively. Moreover, Chan et al. [43] found that MSH1 and MSH2 germline allele-specific hypermethylation and somatic hypermethylation resulted in loss of MSH1 and MSH2 proteins, and patients with these alterations showed early CRC symptoms. In addition, germline deletions of the epithelial cell adhesion molecule (EPCAM) gene result in hypermethylation of the MSH2 promoter, leading to MSH2 silencing. EPCAM is also considered to be a susceptibility gene that induces HNPCC [44], although the identified mutations of EPCAM account for only 1-3% of HNPCC cases [45].
Figure 1
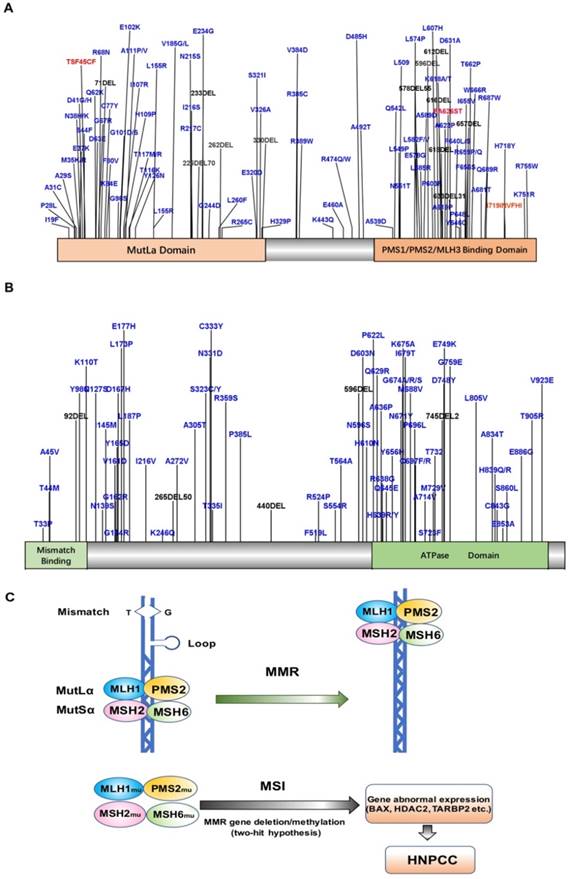
A. Map of MLH1 protein mutation sites B. Map of MSH2 protein mutation sites C. Model of MLH1/MSH2 in HNPCC.
Table 1
Common Susceptibility Genes of HNPCC and FAP and related hot mutation sites identified in the past five years.
| Disease | Gene | OMIM | Transcript | New hot mutation sites |
|---|---|---|---|---|
| HNPCC | BUB1 | 602452 | NM_004336.4 | c.67C>G[53] c.3005C>G[54] |
| EPCAM | 185535 | NM_002354.2 | Exon 8-9 deletion[55] | |
| EXO1 | 606063 | NM_003686.4 | c.2212-1G>C [56] | |
| MLH1 | 120436 | NM_000249.3 | c.-63_-58delins18[57] c.888_890delAGAinsC[34] c.1681dupT[34] c.1863delG[34] c.225delT[34] c.113A>G[41] c.677+3A>G[41] c.1833dup[58] c.2059C > T[59] | |
| MLH3 | 604395 | NM_001040108.1 | c.2152C>T [60] c.2615C>G [60] | |
| MSH2 | 609309 | NM_000251.2 | c.965G>A [53] c.1676_1679 delTAAA [61] c.212-1G>A[62] | |
| MSH3 | 600887 | NM_002439.4 | c.1035del[63] c.2732T>G[63] | |
| MSH6 | 600678 | NM_000179.2 | c.2300_2302delCTC[53] c.431G>T [53] c.3103C>T[64] c.3261dupC[64] | |
| MUTYH | 604933 | NM_001128425.1 | c.1075C>A [53] | |
| PMS2 | 600259 | NM_000535.6 | c.1492del11[40] | |
| POLD2 | 600815 | NM_001127218.1 | c.203G>T [56] | |
| SMAD4 | 600993 | NM_005359.5 | c.1217C>T [53] c.1106A>G [53] c.1573A>G [53] | |
| FAP | APC | 611731 | NM_000038.5 | c.1219delC [65] c.3331G>T [65] c.3418delC [66] c.2971G>T [67] c.510_511insA[68] c.1317delA[69] |
| MSH3 | 617100 | NM_002439.4 | c.1148delA[70] c.2319-1G>A[70] c.2760delC[70] c.3001-2A>C | |
| MUTYH | 604933 | NM_001128425.1 | c.325C>G [65] IVS10-2A>G[71] c.1187G>A[51] c.536A>G[51] c.1214C>T[51] c.1437_1439delGGA[51] | |
| NTHL1 | 602656 | NM_001318193.1 | c.268C>T[72] c.709+1G>A[73] | |
| POLD1 | 612591 | NM_002691 | c.1421T>C[74] | |
| POLE | 615083 | NM_006231 | c.1270C>G[74] | |
| STK11 | 175200 | NM_000455 | c.167 G>C[75] c.1062C>G[75] |
HNPCC: hereditary non-polyposis colorectal cancer; FAP: familial adenomatous polyposis
Apart from MMR genes, other genes have also been detected to be responsible for HNPCC. In particular, BRCA1/2, APC, and MUTYH biallelic mutations have been detected in HNPCC patients [46-48]. In addition, PMS1, MLH3, BRAF, and TGFBR2 have been shown to directly or indirectly affect the development of HNPCC [24]. The chromosome 11q24 region has also been found to be associated with increased susceptibility of CRC. A study by Pinheiro et al. showed a different mutation frequency in MSH3 and TGFBR2 between Lynch syndrome and sporadic MSI CRC regarding tumor location, indicating different pathways of carcinogenesis [49]. Moreover, Rudkjobing et al. [50] found that ALKBH8 in this region may be associated with HNPCC in a Danish family, although mutations in this gene were also detected in unaffected family members, suggesting the need for further investigation. DeRycke et al. [51] found missense mutations in BUB1, MSH2, MSH6, MUTYH, and SMAD4 in HNPCC patients by targeting 36 known CRC susceptibility genes in 1231 CRC patients. However, a recent study by Mur et al. suggested that the contribution of NUDT1 germline mutations to hereditary CRC and to polyposis is negligible as well as OGG1 mutations, indicating that further investigations are still needed to validate this [52].
1.2 Mechanism of action of HNPCC susceptibility genes
The main cause of HNPCC is mutation of MMR genes [76], which have been implicated in a variety of cellular functions essential for maintaining the basic integrity of genetic material and regulation of the cell cycle, including repair of DNA mis-synthesis or DNA double-strand breaks, and in resistance to DNA recombination and DNA destabilization [77]. A heterodimeric MutSα loop consisting of MSH2 and MSH6 (or MSH2 and MSH3) forms a sliding clamp structure that surrounds DNA, which is responsible for recognizing DNA mismatches and recruiting MLH1-PMS1, MLH1-MLH2, or MLH1-MLH3 heterodimers [78]. In particular, the heterodimer consisting of MLH1 and PMS2 is responsible for recruiting the remaining proteins required for MMR [79, 80]. Thus, when MLH1 and MLH2 are mutated, the MMR function is completely lost, and when MSH6 is mutated, the basal function of MMR is lost. However, mutated PMS2, MLH2, and MSH3 rarely affect MMR function [81]. HNPCC patients typically harbor germline mutations in MMR genes. MSI is a condition of genetic hypermutability (predisposition to mutation) that results from impaired DNA repair, the presence of which represents phenotypic evidence of MMR dysfunction [82]. Therefore, when the normal allele in the targeted organ undergoes a somatic mutation or deletion, the associated MMR protein will lose its function, and DNA repair cannot be completed. Subsequently, the wrong nucleotide insertion and/or deletion in the genome will gradually accumulate, and, in turn, increase the state of MSI, leading to MSI-H. MSI-H tumors are often accompanied by methylation of the MLH1 promoter, resulting in epigenetic silencing of the gene and sustained loss of MMR protein. This lack of repair decreases the rate of apoptosis of cancer cells to increase their survival, which, in turn, is more likely to translate into the invasion and development of tissue-specific cancers (Figure 1C) [83].
Thus, MMR deficiency, a feature of advanced adenomas, is considered to reflect the early stage of transformation from adenoma to cancer. However, MMR deficiency has also been detected in a small subset of low-grade adenomas associated with HNPCC. Moreover, novel MMR deficiency can be identified in non-crypt foci with MSI, suggesting that in some adenomas, the development of MMR dysfunction follows the progression of MSI and that other HNPCC-associated adenomas may develop independently of MMR deficiency [84]. Furthermore, mutations in genes that cause loss of MMR function do not necessarily increase HNPCC susceptibility. For example, BRAF (V600E) is a common mutation that promotes the hypermethylation-induced silencing of the MLH1 promoter and is also one of the hallmarks of sporadic CRC with MSI; however, BRAF mutations have neither been detected in HNPCC patients with MLH1 and MSH2 germline mutations, nor in tumors of MSH6 germline mutations and MMR-negative families, indicating that BRAF is not involved in the process of HNPCC transformation into hereditary CRC [85].
2. FAP susceptibility genes and mechanism of action
FAP is a common autosomal dominant genetic disorder that typically manifests in adolescence and is classified as a hereditary CRC syndrome caused by endodermal mutations of APC genes [86]. FAP accounts for about 1% of all cases of CRC and the clinical manifestation is multiple adenomatous polyps in the large intestine [87]. Specifically, FAP is characterized by the appearance of hundreds to thousands of adenomatous polyps from late childhood or early adolescence, with most of the adenomas originating from dysplastic aberrant crypt foci, which are considered to be precursor lesions of adenomas or cancers [88]. FAP is also usually accompanied by some other symptoms such as inflammation, osteoma, epidermoid cyst, upper air-intestinal polyps, and congenital hypertrophy of the retinal pigment epithelium [89]. Without intervention, FAP will inevitably transform into CRC after the age of 35 years [90]. Therefore, improving the early diagnosis rate of FAP is of great significance for clinical decision-making, prognosis, and outcome.
FAP can be generally divided into two subcategories according to the number of colorectal adenomas or polyps and the onset age: classical FAP (CFAP) and attenuated FAP (AFAP) [91, 92]. The diagnosis of CFAP is usually based on the presence of more than 100 colorectal adenomatous polyps with an earlier age of onset than polyposis and CRC, whereas AFAP is characterized by 10-100 colorectal adenomatous polyps with an age of onset later than polyposis and CRC. FAP also includes a special subclass of recessive dominant disorders that are mainly caused by mutations in MUTYH, known as MUTYH-associated FAP (MFAP). Patients with MFAP generally have greater than 20 colorectal adenomatous polyps, an increased incidence of extraintestinal malignancies, and, rarely, exhibit sebaceous gland tumors, with an age of onset concentrated at 40-70 [93].
2.1 FAP-associated susceptibility genes
APC is one of the currently recognized FAP susceptibility genes, which is located on chromosome 5q21 and encodes a 2843-amino acid protein [94-96]. APC is an important component protein in the Wnt signaling pathway (Figure 2A). FAP-associated APC gene mutations are dominated by germline mutations, including small deletions, nonsense or frameshift mutations, and genomic rearrangements caused by splice-site mutations and intron deletions, followed by the formation of truncated APC proteins and loss of APC function, ultimately leading to the development of FAP [97, 98]. A mutation cluster region (MCR) in exon 15 of APC [99], covering mainly the β-catenin-binding functional domain of APC, has been identified as a site with higher mutation rates, including gene deletions occurring in c.39273931delAAAGA and c.31833187delACAAA, and site mutations occurring in exon 15, c.1744G>A, or c.1744G>T [97, 100] (Figure 2A). Besides, according to a study by Kojima et al., APC and STK11 mutations found in duodenal adenomas/adenocarcinomas highlight the importance of proteins encoded by these genes in tumor development [75]. Mutations in MUTYH have also been associated with FAP in special cases of recessive genetic FAP [101]. MUTYH, located on chromosome 1p32, is commonly found as biallelic mutations and is less commonly associated with genomic deletion or duplication. The main mutation sites are located in exon 7 (c.536A>G) and exon 13 (c.1187G>A) [98, 102].
The role of mutations in the clip excision repair gene Nth-like DNA glycosylase 1 (NTHL1) in hereditary CRC diseases has attracted increasing attention in recent years. Weren et al. [72] reported three families carrying the NTHL1 c.268C>T mutation, in which all affected members presented the phenotype of a base excision repair-associated adenomatous polyposis and CRC inherited in a recessive manner. Using whole-genome sequencing or other validated methods, POLE and POLD1 mutations were suggested to affect the occurrence of inherited polymerase proofreading-associated polyposis or even CRC [74, 103-105], and hot mutation sites such as c.1270C>G in POLE or c.1433G>A in POLD1 were found. CD36 encodes a scavenger receptor involved in fatty acid processing in the intestine. A recent study found a substantially lower age of polyposis diagnosis for patients in the severe FAP group and higher age for patients in the attenuated FAP group with CD36 rs1761667 and rs1984112 SNPs, respectively [106]. However, whether different CD36 mutations have distinct consequences on FAP need to be further investigated.
Figure 2
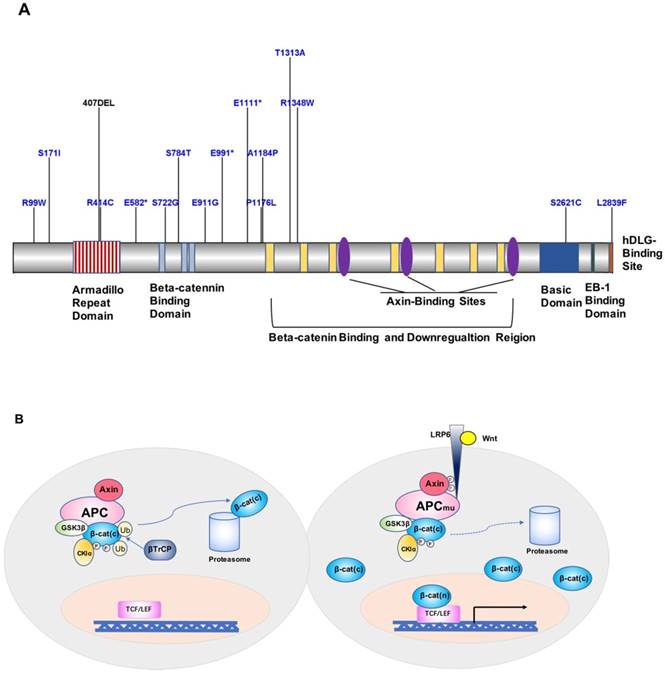
A. Map of APC protein mutation sites. B. Model of the contribution of the APC gene to FAP development and progression.
In addition, some rare cases of dominant polyposis have also been associated with genetic mutations; for example, mutations in SMAD4 and BMPR1A were detected in juvenile polyposis patients [106-108]. Moreover, mutations in LKB1/SKT11 were associated with Peutz-Jeghers syndrome; GREM1 mutations were detected in mixed-type (serrated type and juvenile type) polyposis patients [109]; and Cowden/PTEN hamartoma syndrome was associated with mutations at the G129E site of PTEN [110]. Besides, exome sequencing by Adam et al. in 102 unrelated individuals with unexplained adenomatous colorectal polyposis identified biallelic MSH3 germline mutations as a recessive subtype of colorectal adenomatous polyposis [70]. However, elucidating the mechanisms by which these genes increase the susceptibility to CRC requires more research and data support.
2.2 Mechanism of FAP susceptibility genes
Loss of APC protein function is currently considered to be the key factor in increasing FAP susceptibility [94]. Wild-type APC protein and glycogen synthesis kinase 3β (GSK-3β) combine with axin to form a so-called “destruction complex”, promoting β-catenin degradation and preventing its accumulation in the cytoplasm [111, 112]. Upon mutations of APC, the Wnt signaling pathway is activated, and β-catenin is transferred to the nucleus and activates the transcription factor TCF/LEF, which promotes oncogene expression [62]. In addition, APC protein binds to the cellular structure microfilament proteins F-actin and tubulin, which affects intercellular adhesion and plays a tumor-suppressor role in digestive organs such as the stomach, colon, and pancreas [113, 114]. Moreover, APC mutations result in increased chromosomal instability, causing reduced fidelity of segregated chromosomes and increased chromosome number (aneuploidy) imbalance or loss of heterozygosity rates, thereby further exacerbating mutations in APC (Figure 2B). According to Knudson's two-hit hypothesis, additional somatic APC mutations will occur in the diseased tissue and further promote tumor formation [115]. Indeed, Wang et al. [112] found that in FAP patients of a Chinese family, the c.1744-1G>A site mutation in APC occurred at the wild-type splice acceptor site at the boundary of exons 14 and 15, resulting in aberrant splicing of the gene to form a differential spliceosome, thus leading to the disease phenotype.
In contrast to the pathogenic mechanism of APC mutation, MUTYH mutations mainly cause MUTYH-FAP susceptibility by affecting the base repair mechanism [116]. MUTYH encodes a 535-amino acid protein and is involved in base-repair mechanisms by binding to MSH2/MSH6 to form covalent intermediates with DNA substrates, recognizing dA:dG and dA:dC mismatches generated during DNA replication, so as to clear erroneous adenines from newly generated daughter strands [117]. When MUTYH is mutated in both alleles, its glucokinase activity is lost. Experiments with Escherichia coli showed that mutation of Y82C (hY165C) severely impaired the catalytic activity of the MUTYH glycokinase, resulting in decreased mismatch specificity and adenine flip ability. Further, mutation of G253D (hG382D) reduced the rate of adenine removal catalyzed by MUTYH protein by 85%, along with the level of MUTYH expression, thereby exacerbating the loss of MUTYH function, leading to inability of repairing incorrect bases and contributing to recessively inherited FAP [118].
3. Genetic testing of hereditary CRC susceptibility genes
There are five main genetic methods available for the detection of hereditary CRC susceptibility genes: (1) judgment according to the clinical history, (2) clinical prediction models, (3) direct sequencing, (4) MSI or MMR immunohistochemistry (IHC), and (5) high-throughput sequencing. The main advantage of focusing on clinical history is that it is an inexpensive approach that can be used for individuals with or without a personal history of cancer, while the disadvantages are its poor sensitivity and specificity, inconsistent use, and difficulty in applying complex standards [119]. The use of clinical prediction models (such as PREMM5) is a more cost-effective method that can also be used for individuals with or without a personal history of cancer, and the model does not take into account MSI/MMR IHC results; however, the main disadvantage of this approach is that the model performance for the group of patients unaffected by cancer is unclear [120]. Direct sequencing is the most accurate and direct mutation detection method, which mainly involves the use of traditional Sanger sequencing. This method is of relatively low cost, but the workload is large, making it difficult to process large amounts of data. Therefore, this method is mostly suitable in validation studies of a small number of genes [53]. Universal testing with MSI and/or MMR IHC has the main advantage of cost-effectiveness and high sensitivity for HNPCC screening, although the disadvantage is that it cannot be used for individuals without a cancer history, and the results are susceptible to individual effects of non-HNPCC forms of MSI-H/MMR-D; for example, MSH6 levels are artificially reduced in patients with rectal cancer receiving chemotherapy, which would bias the MMR immunohistochemistry results [119, 121]. Finally, high-throughput sequencing technology is suitable for patients with large errors in MSI and/or MMR immunohistochemistry [122] and can be targeted according to the specific research need by selecting the whole genome or key genomic regions for sequencing analysis. This technology can generate a large amount of data in each region of interest with high accuracy, providing great convenience for mutation identification. For example, the combination of exome profiling with high-throughput sequencing has proven to be the most rapid and effective method for identifying the genetic causes of inherited diseases [123].
With the development of next-generation sequencing technology, large-scale series of products can now reduce the cost of sequencing and experimental reactions by simplifying the sample pool, which can improve the ability of differential information capture and analysis through specific targeted region design; this approach has also been shown to specifically improve the efficiency and accuracy of the detection of hereditary CRC susceptibility genes [124-126]. Moreover, combining high-throughput sequencing technology with other new types of gene test services such as RNA sequencing, multi-gene sequencing panels, and epigenetic high-throughput sequencing will provide great convenience and more options for clinical practice and fundamental scientific research [123, 127, 128]. In practical applications, the most appropriate detection method should be selected according to the requirements of test time, sample characteristics, and the medical cost tolerance of patients.
4. Genetic counseling and fertility guidance for hereditary CRC
Familial CRC is characterized by late onset and rapid progression. Thus, the lesion is typically not detected early enough with traditional surgical detection methods. Accordingly, genetic testing is helpful for the early screening and assessment of cancer risk in patients and their families, facilitating early diagnosis and treatment, allowing the clinicians to provide timely guidance for fertility, and ultimately reducing cancer incidence and mortality [129, 130]. Given the wide variety of germline mutations in CRC patients as reviewed above, many of which lack obvious clinical features of genetic risk, it is recommended that all individuals diagnosed with CRC (especially those diagnosed before the age of 50) should undergo multigene panel testing [120]. In particular, genetic counseling and testing should be performed in HNPCC or FAP probands and their relatives.
Newly diagnosed HNPCC patients can undergo immunohistochemical and MSI tests for the presence of MLH1, MSH2, MSH6, and PMS2 proteins. Approximately 15% of sporadic CRCs also show MSI resulting from hypermethylation of the MLH1 promoter [131]. The BRAF (V600E) mutation is also quite common in sporadic MSI-H tumors (63.5%) [132], but is rare in HNPCC. Therefore, MLH1 hypermethylation and BRAF mutation analysis can be used to distinguish HNPCC-associated CRC from sporadic CRC [133]. Patients with MLH1-deleted HNPCC should be further tested for BRAF mutation status along with assessment of MLH1 promoter methylation levels. For HNPCC patients with a history of tumors, genetic evaluation should be performed when there is evidence of MMR defects, to confirm the lack of BRH mutation or MLH1 hypermethylation.
Patients with classical FAP and mild-phenotype FAP should firstly be tested for mutations of APC, and their children should undergo preventive genetic testing as of the age of 10 years. In FAP patients with a recessive mode of inheritance, if there is no APC mutation, the entire MUTYH needs to be tested. The currently emerging family multigene panel test based on next-generation sequencing technology includes many cancer susceptibility genes and different cancer risk profiles, which is practical for HNPCC probands who do not meet the Amsterdam criteria or Bethesda guidelines and for patients with other specific syndromes (such as germline breast cancer).
In cases in which the pathogenesis can be identified, we recommend that genetic counseling be performed for both the patients and their families. Moreover, fertility guidance can be offered as needed during the counseling. For family members carrying the same pathogenic genes, the risk of cancer should be assessed, so that early intervention can be initiated as required. Genetic counseling is also recommended for patients with FAP or HNPCC without identified pathogenic genes because the family members of these patients will also be susceptible to the disease. With the continuing advance of techniques and the deepening study into the etiology of FAP or HNPCC, new potential causes and mechanisms are also expected to be discovered in the future.
Summary and perspectives
The defect in one of the susceptibility genes of HNPCC and FAP is one of the root causes of hereditary CRC. Therefore, testing for HNPCC and FAP susceptibility gene mutations is of great significance for the diagnosis, prognosis, and timely intervention of familial CRC patients and their high-risk relatives. At the same time, identification of these genetic defects can be used as targets for individualized treatment. For example, regular aspirin use was shown to significantly reduce the incidence of HNPCC and colon cancer [134]. In FAP patients with APC germline mutations, use of the non-steroidal anti-inflammatory drug sulindac and the COX inhibitor celecoxib reduced the adenoma burden in the rectal mucosa preserved in patients after colectomy [135]. Therefore, further study of HNPCC and FAP susceptibility gene defects will surely open up new directions for the treatment of hereditary CRC.
Acknowledgements
This study was supported in part by grants from the National Natural Science Foundation of China (81471432 and 81771645 to Y.T.), and the project of Hunan provincial health department (C2015-53 to Q.L.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: a cancer journal for clinicians. 2018;68:7-30
2. Gupta S, Provenzale D, Regenbogen SE, Hampel H, Slavin TP Jr, Hall MJ. et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 3.2017. Journal of the National Comprehensive Cancer Network: JNCCN. 2017;15:1465-75
3. Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology. 2010;138:2044-58
4. Jones N, Vogt S, Nielsen M, Christian D, Wark PA, Eccles D. et al. Increased colorectal cancer incidence in obligate carriers of heterozygous mutations in MUTYH. Gastroenterology. 2009;137:489-94 94 e1; quiz 725-6
5. Ma H, Brosens LAA, Offerhaus GJA, Giardiello FM, de Leng WWJ, Montgomery EA. Pathology and genetics of hereditary colorectal cancer. Pathology. 2018;50:49-59
6. Stoffel EM, Mangu PB, Gruber SB, Hamilton SR, Kalady MF, Lau MW. et al. Hereditary colorectal cancer syndromes: American Society of Clinical Oncology Clinical Practice Guideline endorsement of the familial risk-colorectal cancer: European Society for Medical Oncology Clinical Practice Guidelines. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2015;33:209-17
7. Peltomaki P, Vasen H. Mutations associated with HNPCC predisposition - Update of ICG-HNPCC/INSiGHT mutation database. Disease markers. 2004;20:269-76
8. Espenschied CR, LaDuca H, Li S, McFarland R, Gau CL, Hampel H. Multigene Panel Testing Provides a New Perspective on Lynch Syndrome. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2017;35:2568-75
9. Hansen MF, Johansen J, Sylvander AE, Bjornevoll I, Talseth-Palmer BA, Lavik LAS. et al. Use of multigene-panel identifies pathogenic variants in several CRC-predisposing genes in patients previously tested for Lynch Syndrome. Clinical genetics. 2017;92:405-14
10. Zelga P, Przybylowska-Sygut K, Zelga M, Dziki A, Majsterek I. The 116G > A MSH6 and IVS1-1121C > T PMS2 Genes Polymorphisms Modulate the Risk of the Sporadic Colorectal Cancer Development in Polish Population. Pathology oncology research: POR. 2018;24:231-5
11. Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A. et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665-9
12. Gylfe AE, Katainen R, Kondelin J, Tanskanen T, Cajuso T, Hanninen U. et al. Eleven candidate susceptibility genes for common familial colorectal cancer. PLoS genetics. 2013;9:e1003876
13. Smith CG, Naven M, Harris R, Colley J, West H, Li N. et al. Exome resequencing identifies potential tumor-suppressor genes that predispose to colorectal cancer. Human mutation. 2013;34:1026-34
14. Esteban-Jurado C, Vila-Casadesus M, Garre P, Lozano JJ, Pristoupilova A, Beltran S. et al. Whole-exome sequencing identifies rare pathogenic variants in new predisposition genes for familial colorectal cancer. Genetics in medicine: official journal of the American College of Medical Genetics. 2015;17:131-42
15. Stoffel EM, Boland CR. Genetics and Genetic Testing in Hereditary Colorectal Cancer. Gastroenterology. 2015;149:1191-203 e2
16. Chubb D, Broderick P, Frampton M, Kinnersley B, Sherborne A, Penegar S. et al. Genetic diagnosis of high-penetrance susceptibility for colorectal cancer (CRC) is achievable for a high proportion of familial CRC by exome sequencing. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2015;33:426-32
17. Carethers JM, Stoffel EM. Lynch syndrome and Lynch syndrome mimics: The growing complex landscape of hereditary colon cancer. World journal of gastroenterology. 2015;21:9253-61
18. Chen E, Xu X, Liu T. Hereditary Nonpolyposis Colorectal Cancer and Cancer Syndromes: Recent Basic and Clinical Discoveries. Journal of oncology. 2018;2018:3979135
19. Kanth P, Grimmett J, Champine M, Burt R, Samadder NJ. Hereditary Colorectal Polyposis and Cancer Syndromes: A Primer on Diagnosis and Management. The American journal of gastroenterology. 2017;112:1509-25
20. Brosens LA, Offerhaus GJ, Giardiello FM. Hereditary Colorectal Cancer: Genetics and Screening. The Surgical clinics of North America. 2015;95:1067-80
21. Connell LC, Mota JM, Braghiroli MI, Hoff PM. The Rising Incidence of Younger Patients With Colorectal Cancer: Questions About Screening, Biology, and Treatment. Current treatment options in oncology. 2017;18:23
22. Yu H, Hemminki A, Sundquist K, Hemminki K. Familial Associations of Colorectal Cancer with Other Cancers. Scientific reports. 2017;7:5243
23. Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P. et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:5783-8
24. Sijmons RH, Hofstra RM. Review: Clinical aspects of hereditary DNA Mismatch repair gene mutations. DNA repair. 2016;38:155-62
25. Bujanda L, Herreros-Villanueva M. Pancreatic Cancer in Lynch Syndrome Patients. Journal of Cancer. 2017;8:3667-74
26. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453-6
27. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J. et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. Journal of the National Cancer Institute. 2004;96:261-8
28. Gelsomino F, Barbolini M, Spallanzani A, Pugliese G, Cascinu S. The evolving role of microsatellite instability in colorectal cancer: A review. Cancer treatment reviews. 2016;51:19-26
29. Ponti G, Castellsague E, Ruini C, Percesepe A, Tomasi A. Mismatch repair genes founder mutations and cancer susceptibility in Lynch syndrome. Clinical genetics. 2015;87:507-16
30. Aggarwal N, Donald ND, Malik S, Selvendran SS, McPhail MJ, Monahan KJ. The Association of Low-Penetrance Variants in DNA Repair Genes with Colorectal Cancer: A Systematic Review and Meta-Analysis. Clinical and translational gastroenterology. 2017;8:e109
31. McGee SF, AlGhareeb W, Ahmad CH, Armstrong D, Babak S, Berry S. et al. Eastern Canadian Colorectal Cancer Consensus Conference 2017. Current oncology. 2018;25:262-74
32. Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K. et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA oncology. 2017;3:464-71
33. Takehara Y, Nagasaka T, Nyuya A, Haruma T, Haraga J, Mori Y. et al. Accuracy of four mononucleotide-repeat markers for the identification of DNA mismatch-repair deficiency in solid tumors. Journal of translational medicine. 2018;16:5
34. Schneider NB, Pastor T, Paula AE, Achatz MI, Santos ARD, Vianna FSL. et al. Germline MLH1, MSH2 and MSH6 variants in Brazilian patients with colorectal cancer and clinical features suggestive of Lynch Syndrome. Cancer medicine. 2018;7:2078-88
35. Stoffel EM, Koeppe E, Everett J, Ulintz P, Kiel M, Osborne J. et al. Germline Genetic Features of Young Individuals With Colorectal Cancer. Gastroenterology. 2018;154:897-905 e1
36. Boland PM, Yurgelun MB, Boland CR. Recent progress in Lynch syndrome and other familial colorectal cancer syndromes. CA: a cancer journal for clinicians. 2018;68:217-31
37. Raskin L, Schwenter F, Freytsis M, Tischkowitz M, Wong N, Chong G. et al. Characterization of two Ashkenazi Jewish founder mutations in MSH6 gene causing Lynch syndrome. Clinical genetics. 2011;79:512-22
38. Papadopoulos N, Lindblom A. Molecular basis of HNPCC: mutations of MMR genes. Human mutation. 1997;10:89-99
39. Gonzalez-Acosta M, Del Valle J, Navarro M, Thompson BA, Iglesias S, Sanjuan X. et al. Elucidating the clinical significance of two PMS2 missense variants coexisting in a family fulfilling hereditary cancer criteria. Familial cancer. 2017;16:501-7
40. Nomura S, Fujimoto Y, Yamamoto N, Sato Y, Ashihara Y, Kita M. et al. A case of early onset rectal cancer of Lynch syndrome with a novel deleterious PMS2 mutation. Japanese journal of clinical oncology. 2015;45:987-92
41. Rosty C, Clendenning M, Walsh MD, Eriksen SV, Southey MC, Winship IM. et al. Germline mutations in PMS2 and MLH1 in individuals with solitary loss of PMS2 expression in colorectal carcinomas from the Colon Cancer Family Registry Cohort. BMJ open. 2016;6:e010293
42. Bonadona V, Bonaiti B, Olschwang S, Grandjouan S, Huiart L, Longy M. et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. Jama. 2011;305:2304-10
43. Chan TL, Yuen ST, Kong CK, Chan YW, Chan AS, Ng WF. et al. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nature genetics. 2006;38:1178-83
44. Boesch M, Spizzo G, Seeber A. Concise Review: Aggressive Colorectal Cancer: Role of Epithelial Cell Adhesion Molecule in Cancer Stem Cells and Epithelial-to-Mesenchymal Transition. Stem cells translational medicine. 2018;7:495-501
45. Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M. et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3' exons of TACSTD1. Nature genetics. 2009;41:112-7
46. Yurgelun MB, Allen B, Kaldate RR, Bowles KR, Judkins T, Kaushik P. et al. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients With Suspected Lynch Syndrome. Gastroenterology. 2015;149:604-13 e20
47. Yurgelun MB, Kulke MH, Fuchs CS, Allen BA, Uno H, Hornick JL. et al. Cancer Susceptibility Gene Mutations in Individuals With Colorectal Cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2017;35:1086-95
48. Ricci MT, Miccoli S, Turchetti D, Bondavalli D, Viel A, Quaia M. et al. Type and frequency of MUTYH variants in Italian patients with suspected MAP: a retrospective multicenter study. Journal of human genetics. 2017;62:309-15
49. Pinheiro M, Pinto C, Peixoto A, Veiga I, Lopes P, Henrique R. et al. Target gene mutational pattern in Lynch syndrome colorectal carcinomas according to tumour location and germline mutation. British journal of cancer. 2015;113:686-92
50. Rudkjobing LA, Eiberg H, Mikkelsen HB, Binderup ML, Bisgaard ML. The analysis of a large Danish family supports the presence of a susceptibility locus for adenoma and colorectal cancer on chromosome 11q24. Familial cancer. 2015;14:393-400
51. DeRycke MS, Gunawardena S, Balcom JR, Pickart AM, Waltman LA, French AJ. et al. Targeted sequencing of 36 known or putative colorectal cancer susceptibility genes. Molecular genetics & genomic medicine. 2017;5:553-69
52. Mur P, Jemth AS, Bevc L, Amaral N, Navarro M, Valdes-Mas R. et al. Germline variation in the oxidative DNA repair genes NUDT1 and OGG1 is not associated with hereditary colorectal cancer or polyposis. Human mutation. 2018
53. DeRycke MS, Gunawardena S, Balcom JR, Pickart AM, Waltman LA, French AJ. et al. Targeted sequencing of 36 known or putative colorectal cancer susceptibility genes. Molecular genetics & genomic medicine. 2017;5:553-69
54. Vargas-Parra GM, Gonzalez-Acosta M, Thompson BA, Gomez C, Fernandez A, Damaso E. et al. Elucidating the molecular basis of MSH2-deficient tumors by combined germline and somatic analysis. International journal of cancer. 2017;141:1365-80
55. Mur P, Pineda M, Romero A, Del Valle J, Borras E, Canal A. et al. Identification of a founder EPCAM deletion in Spanish Lynch syndrome families. Clin Genet. 2014;85:260-6
56. Talseth-Palmer BA, Bauer DC, Sjursen W, Evans TJ, McPhillips M, Proietto A. et al. Targeted next-generation sequencing of 22 mismatch repair genes identifies Lynch syndrome families. Cancer medicine. 2016;5:929-41
57. Morak M, Ibisler A, Keller G, Jessen E, Laner A, Gonzales-Fassrainer D. et al. Comprehensive analysis of the MLH1 promoter region in 480 patients with colorectal cancer and 1150 controls reveals new variants including one with a heritable constitutional MLH1 epimutation. Journal of medical genetics. 2018;55:240-8
58. Akizawa Y, Yamamoto T, Tamura K, Kanno T, Takahashi N, Ohki T. et al. A novel MLH1 mutation in a Japanese family with Lynch syndrome associated with small bowel cancer. Human genome variation. 2018;5:13
59. von Salome J, Liu T, Keihas M, Morak M, Holinski-Feder E, Berry IR. et al. Haplotype analysis suggest that the MLH1 c.2059C > T mutation is a Swedish founder mutation. Familial cancer. 2017
60. Lu JY, Sheng JQ. Advances in the study of Lynch syndrome in China. World journal of gastroenterology. 2015;21:6861-71
61. Wu B, Ji W, Liang S. et al. A novel heterozygous germline deletion in MSH2 gene in a five generation chinese family with Lynch syndrome. Oncotarget. 2017;8:55194-203
62. Liccardo R, De Rosa M, Izzo P, Duraturo F. Novel MSH2 splice-site mutation in a young patient with Lynch syndrome. Molecular medicine reports. 2018;17:6942-6
63. Morak M, Kasbauer S, Kerscher M, Laner A, Nissen AM, Benet-Pages A. et al. Loss of MSH2 and MSH6 due to heterozygous germline defects in MSH3 and MSH6. Familial cancer. 2017;16:491-500
64. Ling C, Yang W, Sun H, Ge M, Ji Y, Han S. et al. Rare compound heterozygous mutations in gene MSH6 cause constitutive mismatch repair deficiency syndrome. Clinical case reports. 2018;6:1448-51
65. Marabelli M, Molinaro V, Abou Khouzam R, Berrino E, Panero M, Balsamo A. et al. Colorectal Adenomatous Polyposis: Heterogeneity of Susceptibility Gene Mutations and Phenotypes in a Cohort of Italian Patients. Genetic testing and molecular biomarkers. 2016;20:777-85
66. Zhang Z, Liang S, Wang D, Liang S, Li Y, Wang B. et al. A novel pathogenic single nucleotide germline deletion in APC gene in a four generation Chinese family with familial adenomatous polyposis. Scientific reports. 2017;7:12357
67. Li H, Zhang L, Jiang Q, Shi Z, Tong H. Identification a nonsense mutation of APC gene in Chinese patients with familial adenomatous polyposis. Experimental and therapeutic medicine. 2017;13:1495-9
68. D'Elia G, Caliendo G, Casamassimi A, Cioffi M, Molinari AM, Vietri MT. APC and MUTYH Analysis in FAP Patients: A Novel Mutation in APC Gene and Genotype-Phenotype Correlation. Genes. 2018:9
69. Pang M, Liu Y, Hou X, Yang J, He X, Hou N. et al. A novel APC mutation identified in a large Chinese family with familial adenomatous polyposis and a brief literature review. Molecular medicine reports. 2018;18:1423-32
70. Adam R, Spier I, Zhao B, Kloth M, Marquez J, Hinrichsen I. et al. Exome Sequencing Identifies Biallelic MSH3 Germline Mutations as a Recessive Subtype of Colorectal Adenomatous Polyposis. American journal of human genetics. 2016;99:337-51
71. Taki K, Sato Y, Nomura S, Ashihara Y, Kita M, Tajima I. et al. Mutation analysis of MUTYH in Japanese colorectal adenomatous polyposis patients. Familial cancer. 2016;15:261-5
72. Weren RD, Ligtenberg MJ, Kets CM, de Voer RM, Verwiel ET, Spruijt L. et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet. 2015;47:668-71
73. Broderick P, Dobbins SE, Chubb D, Kinnersley B, Dunlop MG, Tomlinson I. et al. Validation of Recently Proposed Colorectal Cancer Susceptibility Gene Variants in an Analysis of Families and Patients-a Systematic Review. Gastroenterology. 2017;152:75-7.e4
74. Valle L, Hernandez-Illan E, Bellido F, Aiza G, Castillejo A, Castillejo MI. et al. New insights into POLE and POLD1 germline mutations in familial colorectal cancer and polyposis. Human molecular genetics. 2014;23:3506-12
75. Kojima Y, Ohtsuka K, Ohnishi H, Abe N, Furuse J, Watanabe T. et al. APC:T1556fs and STK11 mutations in duodenal adenomas and adenocarcinomas. Surgery today. 2018;48:765-72
76. Wielders EA, Hettinger J, Dekker R, Kets CM, Ligtenberg MJ, Mensenkamp AR. et al. Functional analysis of MSH2 unclassified variants found in suspected Lynch syndrome patients reveals pathogenicity due to attenuated mismatch repair. Journal of medical genetics. 2014 jmedgenet-2013-101987
77. Li G-M. Mechanisms and functions of DNA mismatch repair. Cell research. 2008;18:85
78. Honda M, Okuno Y, Hengel SR, Martin-Lopez JV, Cook CP, Amunugama R. et al. Mismatch repair protein hMSH2-hMSH6 recognizes mismatches and forms sliding clamps within a D-loop recombination intermediate. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E316-25
79. Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese LS. Structure of the human MutSα DNA lesion recognition complex. Molecular cell. 2007;26:579-92
80. Schmidt TT, Hombauer H. Visualization of mismatch repair complexes using fluorescence microscopy. DNA repair. 2016;38:58-67
81. Kolodner RD. A personal historical view of DNA mismatch repair with an emphasis on eukaryotic DNA mismatch repair. DNA repair. 2016;38:3-13
82. Gupta R, Sinha S, Paul RN. The impact of microsatellite stability status in colorectal cancer. Current problems in cancer. 2018
83. Sijmons RH, Hofstra RM. Clinical aspects of hereditary DNA Mismatch repair gene mutations. DNA repair. 2016;38:155-62
84. Giuffre G, Müller A, Brodegger T, Bocker-Edmonston T, Gebert J, Kloor M. et al. Microsatellite analysis of hereditary nonpolyposis colorectal cancer-associated colorectal adenomas by laser-assisted microdissection: correlation with mismatch repair protein expression provides new insights in early steps of tumorigenesis. The Journal of Molecular Diagnostics. 2005;7:160-70
85. Domingo E, Niessen RC, Oliveira C, Alhopuro P, Moutinho C, Espín E. et al. BRAF-V600E is not involved in the colorectal tumorigenesis of HNPCC in patients with functional MLH1 and MSH2 genes. Oncogene. 2005;24:3995-8
86. Leoz ML, Carballal S, Moreira L, Ocana T, Balaguer F. The genetic basis of familial adenomatous polyposis and its implications for clinical practice and risk management. The application of clinical genetics. 2015;8:95-107
87. Rustgi AK. The genetics of hereditary colon cancer. Genes & development. 2007;21:2525-38
88. Nucci MR, Robinson CR, Longo P, Campbell P, Hamilton SR. Phenotypic and genotypic characteristics of aberrant crypt foci in human colorectal mucosa. Human pathology. 1997;28:1396-407
89. Gerdehsang PS, Ranji N, Gorji M, Pakizehkar S, Kiani AA, Veysi S. New Mutations in APC Gene Among Familial Adenomatous Polyposis (FAP) Patients in Iran. International Journal of Human Genetics. 2017;17:145-50
90. Petersen GM, Slack J, Nakamura Y. Screening guidelines and premorbid diagnosis of familial adenomatous polyposis using linkage. Gastroenterology. 1991;100:1658-64
91. Byrne RM, Tsikitis VL. Colorectal polyposis and inherited colorectal cancer syndromes. Annals of gastroenterology. 2018;31:24-34
92. Kinney AY, Hicken B, Simonsen SE, Venne V, Lowstuter K, Balzotti J. et al. Colorectal cancer surveillance behaviors among members of typical and attenuated FAP families. The American journal of gastroenterology. 2007;102:153-62
93. Aretz S. The differential diagnosis and surveillance of hereditary gastrointestinal polyposis syndromes. Deutsches Arzteblatt International. 2010;107:163
94. Nallamilli BR, Hegde M. Detecting APC Gene Mutations in Familial Adenomatous Polyposis (FAP). Current protocols in human genetics. 2017;92:10 8 1- 8 6
95. Zhang M, Yang D, Gold B. The Adenomatous Polyposis Coli (APC) mutation spectra in different anatomical regions of the large intestine in colorectal cancer. Mutation research. 2018;810:1-5
96. Zhang L, Shay JW. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. Journal of the National Cancer Institute. 2017:109
97. Plawski A, Slomski R. APC gene mutations causing familial adenomatous polyposis in Polish patients. Journal of applied genetics. 2008;49:407-14
98. Aretz S, Genuardi M, Hes FJ. Clinical utility gene card for: MUTYH-associated polyposis (MAP), autosomal recessive colorectal adenomatous polyposis, multiple colorectal adenomas, multiple adenomatous polyps (MAP)-update 2012. European journal of human genetics. 2013:21
99. Bertario L, Russo A, Sala P, Varesco L, Giarola M, Mondini P. et al. Multiple approach to the exploration of genotype-phenotype correlations in familial adenomatous polyposis. Journal of Clinical Oncology. 2003;21:1698-707
100. Aretz S, Uhlhaas S, Sun Y, Pagenstecher C, Mangold E, Caspari R. et al. Familial adenomatous polyposis: aberrant splicing due to missense or silent mutations in the APC gene. Human mutation. 2004;24:370-80
101. Yanus GA, Akhapkina TA, Ivantsov AO, Preobrazhenskaya EV, Aleksakhina SN, Bizin IV. et al. Spectrum of APC and MUTYH germ-line mutations in Russian patients with colorectal malignancies. Clinical genetics. 2018;93:1015-21
102. Wang L, Baudhuin LM, Boardman LA, Steenblock KJ, Petersen GM, Halling KC. et al. MYH mutations in patients with attenuated and classic polyposis and with young-onset colorectal cancer without polyps. Gastroenterology. 2004;127:9-16
103. Esteban-Jurado C, Garre P, Vila M, Lozano JJ, Pristoupilova A, Beltran S. et al. New genes emerging for colorectal cancer predisposition. World journal of gastroenterology. 2014;20:1961-71
104. Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P. et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45:136-44
105. Rohlin A, Eiengard F, Lundstam U, Zagoras T, Nilsson S, Edsjo A. et al. GREM1 and POLE variants in hereditary colorectal cancer syndromes. Genes, chromosomes & cancer. 2016;55:95-106
106. Holmes M, Connor T, Oldmeadow C, Pockney PG, Scott RJ, Talseth-Palmer BA. CD36 - a plausible modifier of disease phenotype in familial adenomatous polyposis. Hereditary cancer in clinical practice. 2018;16:14
107. Marmol I, Sanchez-de-Diego C, Pradilla Dieste A, Cerrada E, Rodriguez Yoldi MJ. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. International journal of molecular sciences. 2017:18
108. Nejadtaghi M, Jafari H, Farrokhi E, Samani KG. Familial Colorectal Cancer Type X (FCCTX) and the correlation with various genes-A systematic review. Current problems in cancer. 2017;41:388-97
109. Tomlinson I. The Mendelian colorectal cancer syndromes. Annals of clinical biochemistry. 2015;52:690-2
110. Leslie NR, Longy M. Inherited PTEN mutations and the prediction of phenotype. Seminars in cell & developmental biology. 2016;52:30-8
111. Baldwin AT, Phillips BT. The tumor suppressor APC differentially regulates multiple β-catenins through the function of axin and CKIα during C. elegans asymmetric stem cell divisions. J Cell Sci. 2014;127:2771-81
112. Wang D, Liang S, Zhang Z, Zhao G, Hu Y, Liang S. et al. A novel pathogenic splice acceptor site germline mutation in intron 14 of the APC gene in a Chinese family with familial adenomatous polyposis. Oncotarget. 2017;8:21327
113. Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:3046-50
114. Nagase H, Nakamura Y. Mutations of the APC (adenomatous polyposis coli) gene. Hum Mutat. 1993;2:425-34
115. Knudson AG. Two genetic hits (more or less) to cancer. Nature Reviews Cancer. 2001;1:157-62
116. Talseth-Palmer BA. The genetic basis of colonic adenomatous polyposis syndromes. Hereditary cancer in clinical practice. 2017;15:5
117. Gu Y, Parker A, Wilson TM, Bai H, Chang D-Y, Lu A-L. Human MutY homolog, a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins human MutS homolog 2/human MutS homolog 6. Journal of Biological Chemistry. 2002;277:11135-42
118. Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT. et al. Inherited variants of MYH associated with somatic G:C->T:A mutations in colorectal tumors. Nat Genet. 2002;30:227-32
119. Joly MO, Attignon V, Saurin JC, Desseigne F, Leroux D, Martin-Denavit T. et al. Somatic MMR Gene Mutations as a Cause for MSI-H Sebaceous Neoplasms in Muir-Torre Syndrome-Like Patients. Human mutation. 2015;36:292-5
120. Boland PM, Yurgelun MB, Boland CR. Recent progress in Lynch syndrome and other familial colorectal cancer syndromes. CA: a cancer journal for clinicians. 2018
121. Goldstein JB, Wu W, Borras E, Masand G, Cuddy A, Mork ME. et al. Can Microsatellite Status of Colorectal Cancer Be Reliably Assessed after Neoadjuvant Therapy? Clinical Cancer Research. 2017;23:5246-54
122. Nowak JA, Yurgelun MB, Bruce JL, Rojas-Rudilla V, Hall DL, Shivdasani P. et al. Detection of mismatch repair deficiency and microsatellite instability in colorectal adenocarcinoma by targeted next-generation sequencing. The Journal of Molecular Diagnostics. 2017;19:84-91
123. Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nature reviews Genetics. 2011;13:36-46
124. Aloraifi F, Boland MR, Green AJ, Geraghty JG. Gene analysis techniques and susceptibility gene discovery in non-BRCA1/BRCA2 familial breast cancer. Surgical oncology. 2015;24:100-9
125. Kayser K, Degenhardt F, Holzapfel S, Horpaopan S, Peters S, Spier I. et al. Copy number variation analysis and targeted NGS in 77 families with suspected Lynch syndrome reveals novel potential causative genes. International journal of cancer. 2018
126. Maida M, Macaluso FS, Ianiro G, Mangiola F, Sinagra E, Hold G. et al. Screening of colorectal cancer: present and future. Expert review of anticancer therapy. 2017;17:1131-46
127. Binder H, Hopp L, Schweiger MR, Hoffmann S, Juhling F, Kerick M. et al. Genomic and transcriptomic heterogeneity of colorectal tumours arising in Lynch syndrome. The Journal of pathology. 2017;243:242-54
128. Wu H, Zhang XY, Hu Z, Hou Q, Zhang H, Li Y. et al. Evolution and heterogeneity of non-hereditary colorectal cancer revealed by single-cell exome sequencing. Oncogene. 2017;36:2857-67
129. Seppala T, Pylvanainen K, Evans DG, Jarvinen H, Renkonen-Sinisalo L, Bernstein I. et al. Colorectal cancer incidence in path_MLH1 carriers subjected to different follow-up protocols: a Prospective Lynch Syndrome Database report. Hereditary cancer in clinical practice. 2017;15:18
130. Guglielmo A, Staropoli N, Giancotti M, Mauro M. Personalized medicine in colorectal cancer diagnosis and treatment: a systematic review of health economic evaluations. Cost effectiveness and resource allocation: C/E. 2018;16:2
131. Young J, Simms LA, Biden KG, Wynter C, Whitehall V, Karamatic R. et al. Features of colorectal cancers with high-level microsatellite instability occurring in familial and sporadic settings: parallel pathways of tumorigenesis. The American journal of pathology. 2001;159:2107-16
132. Parsons MT, Buchanan DD, Thompson B, Young JP, Spurdle AB. Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: a literature review assessing utility of tumour features for MMR variant classification. Journal of medical genetics. 2012;49:151-7
133. Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA. et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nature genetics. 2006;38:787-93
134. Vasen HF, Blanco I, Aktan-Collan K, Gopie JP, Alonso A, Aretz S. et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut. 2013 gutjnl-2012-304356
135. Kim B, Giardiello FM. Chemoprevention in familial adenomatous polyposis. Best practice & research Clinical gastroenterology. 2011;25:607-22
Author contact
![]() Corresponding author: Dr. Yue-Qiu Tan, Institute of Reproduction and Stem Cell Engineering, School of Basic Medical Science, Central South University, Changsha, Hunan, 410078, China. E-mail: tanyueqiuedu.cn; Tel: 86-731-82355301
Corresponding author: Dr. Yue-Qiu Tan, Institute of Reproduction and Stem Cell Engineering, School of Basic Medical Science, Central South University, Changsha, Hunan, 410078, China. E-mail: tanyueqiuedu.cn; Tel: 86-731-82355301
Citation styles
APA
Liu, Q., Tan, Y.Q. (2019). Advances in Identification of Susceptibility Gene Defects of Hereditary Colorectal Cancer. Journal of Cancer, 10(3), 643-653. https://doi.org/10.7150/jca.28542.
ACS
Liu, Q.; Tan, Y.Q. Advances in Identification of Susceptibility Gene Defects of Hereditary Colorectal Cancer. J. Cancer 2019, 10 (3), 643-653. DOI: 10.7150/jca.28542.
NLM
Liu Q, Tan YQ. Advances in Identification of Susceptibility Gene Defects of Hereditary Colorectal Cancer. J Cancer 2019; 10(3):643-653. doi:10.7150/jca.28542. https://www.jcancer.org/v10p0643.htm
CSE
Liu Q, Tan YQ. 2019. Advances in Identification of Susceptibility Gene Defects of Hereditary Colorectal Cancer. J Cancer. 10(3):643-653.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.