Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(24):4762-4773. doi:10.7150/jca.25138 This issue Cite
Research Paper
Functional Genome-wide Screening Identifies Targets and Pathways Sensitizing Pancreatic Cancer Cells to Dasatinib
Wenwen Chien1,2 ![]() *, Makoto Sudo1*, Ling-Wen Ding1, Qiao-Yang Sun1, Peer Wuensche1, Kian Leong Lee1, Norimichi Hattori1, Manoj Garg1, Liang Xu1, Yun Zheng3, Sigal Gery2,4, Sarawut Wongphayak1, Henry Yang1, Erkan Baloglu5, Sharon Shacham5, Michael Kauffman5, Seiichi Mori1, H. Phillip Koeffler1,2,6
*, Makoto Sudo1*, Ling-Wen Ding1, Qiao-Yang Sun1, Peer Wuensche1, Kian Leong Lee1, Norimichi Hattori1, Manoj Garg1, Liang Xu1, Yun Zheng3, Sigal Gery2,4, Sarawut Wongphayak1, Henry Yang1, Erkan Baloglu5, Sharon Shacham5, Michael Kauffman5, Seiichi Mori1, H. Phillip Koeffler1,2,6
1. Cancer Science Institute of Singapore, National University of Singapore, Singapore
2. Department of Hematology-Oncology, Cedars-Sinai Medical Center, Los Angeles, CA, USA
3. State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University Cancer Center, Guangzhou, China
4. David Geffen School of Medicine, UCLA, Los Angeles, CA, USA
5. Karyopharm Therapeutics, Boston, MA, USA
6. National University Cancer Institute, National University Hospital, Singapore
*These authors contributed equally to this work
Received 2018-1-25; Accepted 2018-8-1; Published 2018-12-10
Abstract
This study is an unbiased genomic screen to obtain functional targets for increased effectiveness of dasatinib in pancreatic cancer. Dasatinib, a multi-targeted tyrosine kinase inhibitor, is used in clinical trials for treatment of pancreatic cancer; however, intrinsic and acquired resistance often occurs. We used a dasatinib-resistant pancreatic cancer cell line SU8686 to screen for synthetic lethality that synergizes with dasatinib using a pooled human shRNA library followed by next generation sequencing. Novel genes were identified which when silenced produced a prominent inhibitory effect with dasatinib against the pancreatic cancer cells. Several of these genes are involved in the regulation of epigenetics, as well as signaling pathways of the FOXO and hedgehog families. Small molecule inhibitors of either histone deacetylases or nuclear exporter had marked inhibitory effect with dasatinib in pancreatic cancers, suggesting their potential therapeutic effectiveness in this deadly cancer.
Keywords: Pancreatic Cancer, Dasatinib, XPO
Introduction
Pancreatic cancers are increasing in frequency, representing the fourth most common cancer in the United States and globally the seventh most malignancy [1]. Peak incidence of the disease is in the sixth and seventh decades of life. Causes of pancreatic cancer are unknown, but it is associated with chronic pancreatitis and diabetes [2, 3]. Genomic abnormalities include frequent mutations of KRAS, TP53, CDKN2A, and SMAD4 [4-7]. Genetic alteration in a core set of 12 cellular signaling pathways (60-100%) are involved in pancreatic cancer [8]. Pancreatic cancer has a high mortality rate and ninety-five percent of patients die within five years. No biomarkers for early diagnosis are available; and at diagnosis, the disease is usually no longer curable by surgery. Chemotherapy with either gemcitabine or in combination with other drugs (e.g. paclitaxel, erlotinib) has limited activity against this cancer [9, 10]. Multi-targeted kinase inhibitor dasatinib retards growth of human pancreatic adenocarcinoma cells in an orthotopic nude murine model [11] and inhibits metastasis in a murine model of pancreatic ductal adenocarcinoma [12].
We previously identified dasatinib-sensitive and -resistant pancreatic cancer cell lines [13]. In that study, a panel of human pancreatic cancer cell lines was screened with dasatinib plus 65 kinase inhibitors. At the highest concentration of dasatinib (1 μM), 9 cell lines (AsPc1, CFPAC, BxPc3, MiaPaCa2, Panc0203, Panc0327, Panc1, SU8686, PL45) did not achieve an IC50 (defined as dasatinib-resistant). Six cell lines (HPAC1, Panc0403, Panc0504, Panc1005, Panc0813, CaPan2) had IC50 of dasatinib between 20 - 250 nM (defined as dasatinib-sensitive). Here, we used an unbiased screening of dasatinib-resistant pancreatic cancer cells (SU8686) with a genome-wide shRNA library followed by massively parallel sequencing to identify novel targets and signaling pathways that can potentiate synergistic inhibitory effects of dasatinib. We identified inhibitors of the FOXO and hedgehog families, histone deacetylases, as well as XPO1 (nuclear protein exporter) which markedly enhanced ability of dasatinib to suppress cellular proliferation of pancreatic cancer cells.
Materials and Methods
shRNA library screen
We used MISSION LentiPlex Human Pooled shRNA Library SHP001 containing more than 75,000 shRNA constructs targeting 15,000+ human genes (Sigma, St. Louis, MO) (Supplementary Figure 1). At day 0, human pancreatic cancer cell line SU8686 was seeded in 150 mm tissue culture dishes at a density that by the next day the cells were between 30 to 40% confluent. At day 1, the cells were infected with lentivirus for 24 hr. Viral titer of 0.3 MOI (multiplicity of infection) was used to obtain a single viral infection per cell. At day 2, the virus was removed and replaced with regular culture medium. At day 3, cells were incubated under selection with puromycin (5 μg/ml, 48h). At day 5, culture medium containing puromycin was removed and replenished with fresh medium without puromycin. At day 7, cells reached confluence and were passaged at 1:4 for subsequent dasatinib treatment and 1:8 for controls. At day 8, dasatinib (10 ng/ml) was added to the cells. After 5 days of incubation, both control and dasatinib-treated plates reached confluence, and cells were harvested for genomic DNA using QiaAMP DNA micro kit (Qiagen, Valencia, CA). Hairpins from genomic DNA were retrieved with PCR (forward primer: ATCTTGTGGAAAGGACGAAAC; reverse primer: ACCTCGAGACAAATGGGCAGTA) and 114 bp products were purified for deep sequencing with SOLEXA (Illumina, San Diego, CA). Sequencing and data analyses were performed at the Core Facility in National University of Singapore. (Supplementary Figure S2).
Databases
Public databases used included RIGER (http://www.broadinstitute.org/cancer/software/GENE-E/), PANTHER (protein analysis through evolutionary relationships) classification system (http://www.pantherdb.org/), Oncomine (http://www.webcitation.org/), and Pathway interaction database (PID) (http://pid.nci.nih.gov/). For RIGER analysis, Second Best Rank feature was used; shRNAs were selected when at least two different shRNAs had the same effect.
Real-time PCR
Total RNA was isolated with TRIZOL (Life Technologies, Carlsbad, CA) and cDNA was synthesized using RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Carlsbad, CA) according to manufacturers' instructions. Detection of gene expression was performed on Applied Biosystems 7500 Fast real-time PCR system (Life Technologies, Carlsbad, CA). Expression levels of mRNA were normalized to β-actin and expression of MAX in SU8686 was designated at 1.
MTT assay
Human pancreatic cancer cells (3x103 per well in 96 well plates) were incubated with drugs for 72h, and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Sigma-Aldrich, St. Louis, MO) was used to assess cell viability. For drug combination, drugs were added simultaneously to cells and incubated for 72h and assayed with MTT. Lipofectamine RNAiMAX (Life Technologies, Carlsbad, CA) was used for siRNA transfection. Forty-eight hours after transfection, cells were incubated with drugs for 72h. Combination index (CI) was calculated using Calcusyn [14]. Values of CI indicate synergistic (<1), additive (=1), or antagonistic (>1) effect. Calculation of IC50 was based on sigmoidal dose-response curve fitting, using Graphpad prism (La Jolla, CA). Hypoxic experiments were carried out in temperature-controlled hypoxia chamber at 2% O2, 5% CO2, 93% N2. Data represent means ± standard deviations of either two or three independent experiments with quadruplicate wells per experiment.
Colony formation assay
For clonal growth on plastic, cells were seeded at a density that resulted in 30-40% confluence in 6-well plates on the following day. For colony formation in soft agar, 3,000 cells were seeded in the top layer of 0.25% agar with a bottom layer of 0.5% agar. After addition of drugs, the plates were incubated for 14 days. Cells were stained with 0.1% crystal violet and photographed. Number of colonies was counted with ImageJ (http://imagej.nih.gov). Data represent the average of two independent experiments each done with triplicate wells.
Animal studies
All animal studies were in accordance with protocols approved by Institutional Animal Care and Use Committee (IACUC) at National University of Singapore. The human pancreatic cancer cells (MiaPaCa2, 107 cells) were injected subcutaneously into six-week-old NSG mice. Mice were randomly divided into four groups: 1. Oral treatment with KPT-330 (selinexor) (Karyopharm Therapeutics, Newton, MA) (10mg/kg); 2. Intraperitoneal injection with dasatinib (10mg/kg); 3. Treatment with both KPT-330 and dasatinib; 4. Diluent control. Each mouse carried 1 tumor with 6 mice per group. The mice received the drug/diluent control three times per week for 4 weeks. During the study, mice were supplemented with Nutri-cal (Tomlyn, Fort Worth, TX). Tumor size and body weight were measured weekly. Tumor volume was calculated using the formula: volume (mm3) = (long diameter) x (short diameter)2 /2. At the end of experiments, the tumors were carefully dissected and weighed. Blood chemistries (ALB, GLU, TP, CREA, BUN, AST and ALT) and complete blood cell counts were measured. Organ weights (liver, kidney, spleen) were measured. Portions of the tumors were used for immunohistochemistry (IHC) to examine levels of Ki-67 (ab15580, Abcam, Cambridge, UK) and phosphorylated ERK1/2 (#4376, Cell Signaling, Danvers, MA). IHC was performed as previously described [15].
Western blot analysis
Protein lysates were extracted from cells with RIPA lysis buffer (ThermoScientific, Rockford, IL) and resolved with SDS-PAGE. Following transfer to PVDF membrane, antibodies from Cell Signaling (Danvers, MA), Santa Cruz (Santa Cruz, CA), or Sigma-Aldrich (St. Louis, MO) were used to examine protein expression.
Results
Genome wide screening for modulators of dasatinib response
A pooled library containing ~75,000 shRNAs targeting ~15,000 genes was used to screen for synthetic lethal siRNAs in the dasatinib-resistant human pancreatic cancer cell line SU8686. By employing the screening strategy (Supplementary Figure S1), we identified genes that when suppressed, potentiated the activity of dasatinib to induce cell death of SU8686 pancreatic cancer cells. RIGER analysis showed 729 depleted shRNAs targeting 410 genes with a high hairpin score (> 0.2) at a significant level (p < 0.05) for dasatinib treated cells (Supplementary Table S1). These 410 genes may be targets for overcoming dasatinib resistance in pancreatic cancer cells. Conversely, 616 amplified hairpins targeting 350 genes in dasatinib treated cells had enhanced shRNA suggesting that these silenced genes may be pro-proliferative in the presence of dasatinib.
The 410 genes from the primary screen were categorized by PANTHER (protein analysis through evolutionary relationships) classification system. Twelve biological processes were enriched in relation to dasatinib resistance (Supplementary Figure S3). Not surprisingly, “metabolic process” was over-represented (182 genes) since metabolic phenomena related to chemo-resistance is well established. Genes attributed to the “apoptotic process” (16 genes) and “response to stimulus” (46 genes) were likewise represented. Pathway analysis of these 410 genes using PID (pathway interaction database) indicated multiple pathways were related to dasatinib-resistance (Table 1). Notably XPO1 (nuclear protein exporter) was involved in several pathways including signaling through FOXO, hedgehog, and canonical NF-κB as well as signaling events mediated by HDAC class 1 (Table 1). XPO1 and other key components of these pathways may be targeted for potential synergistic effects with dasatinib.
Potential targeted pathways*
| Pathway Name | Genes depleted in experimentals | p-value |
|---|---|---|
| FAS (CD95) signaling pathway | FADD, FAIM2, FASLG, MAPK10, PDK1, PDPK1 | 3.5E-05 |
| mTOR signaling pathway | KRAS, MAP2K1, PDK1, PDPK1, RAF1, SGK1, YWHAZ | 1.3E-04 |
| ATM pathway | ATM, BLM, CDC25C, FAM175A, TOP3A | 2.0E-04 |
| GMCSF-mediated signaling events | KRAS, MAP2K1, PIM1, RAF1, YWHAZ | 2.7E-04 |
| Trk receptor signaling mediated by PI3K and PLC-gamma | KRAS, PDK1, PDPK1, TRPV1, YWHAZ | 3.0E-04 |
| CXCR3-mediated signaling events | KRAS, MAP2K1, PDK1, PDPK1, RAF1 | 0.001 |
| TCR signaling in naïve CD4+ T cells | CSK, DBNL, HLA-DRA, KRAS, PDK1, PDPK1 | 0.001 |
| Signaling events mediated by HDAC Class I | LOC729458, MAX, MBD3L2, SAP18, WDR77, XPO1 | 0.001 |
| Hedgehog signaling events mediated by Gli proteins | MAP2K1, MTSS1, SAP18, SPOP, XPO1 | 0.001 |
| FoxO family signaling | FASLG, MAPK10, SGK1, XPO1, YWHAZ | 0.001 |
| Ras signaling in the CD4+ TCR pathway | KRAS, MAP2K1, RAF1 | 0.001 |
| IGF1 pathway | PDK1, PDPK1, RAF1, YWHAZ | 0.001 |
| ErbB1 downstream signaling | KRAS, MAP2K1, PDK1, PDPK1, RAF1, SH2D2A, YWHAZ | 0.002 |
| Trk receptor signaling mediated by the MAPK pathway | KRAS, MAP2K1, RAF1, TRPV1 | 0.002 |
| Class I PI3K signaling events mediated by Akt | PDK1, PDPK1, RAF1, YWHAZ | 0.003 |
| PLK3 signaling events | ATM, CDC25C | 0.004 |
| PDGFR-beta signaling pathway | CSK, KRAS, MAP2K1, MAPK10, RAF1, SIPA1, YWHAZ | 0.004 |
| BCR signaling pathway | CSK, MAP2K1, PDK1, PDPK1, RAF1 | 0.004 |
| Regulation of Telomerase | ATM, BLM, MAX, POT1, SAP18 | 0.005 |
| ErbB2/ErbB3 signaling events | KRAS, MAP2K1, MAPK10, RAF1 | 0.005 |
| Downstream signaling in naïve CD8+ T cells | FASLG, GZMB, KRAS, MAP2K1, RAF1 | 0.005 |
| CXCR4-mediated signaling events | CSK, HLA-DRA, ITGA6, PDK1, PDPK1, VPS4B | 0.006 |
| Signaling events mediated by VEGFR1 and VEGFR2 | MAP2K1, PDK1, PDPK1, RAF1, SH2D2A | 0.006 |
| LKB1 signaling events | ETV4, MAP2, SIK1, YWHAZ | 0.007 |
| Fanconi anemia pathway | ATM, BLM, FANCE, TOP3A | 0.007 |
| Class I PI3K signaling events | KRAS, PDK1, PDPK1, SGK1 | 0.007 |
| Signaling events mediated by Stem cell factor receptor (c-Kit) | MAP2K1, PDK1, PDPK1, RAF1 | 0.010 |
| IL8- and CXCR1-mediated signaling events | CXCR1, PDK1, PDPK1 | 0.011 |
| C-MYB transcription factor network | GSTM1, KRAS, MAD1L1, PIM1, SKI | 0.011 |
| TCR signaling in naïve CD8+ T cells | CSK, KRAS, PDK1, PDPK1 | 0.012 |
| Caspase Cascade in Apoptosis | FADD, FASLG, GZMB, SPTAN1 | 0.012 |
| Alpha6 beta4 integrin-ligand interactions | ITGA6, LAMA1 | 0.013 |
| HIV-1 Nef: Negative effector of Fas and TNF-alpha | FADD, FASLG, TRAF1 | 0.017 |
| IL8- and CXCR2-mediated signaling events | CXCR2, PDK1, PDPK1 | 0.017 |
| IL12-mediated signaling events | CCL3, FASLG, GZMB, HLA-DRA | 0.020 |
| Insulin Pathway | PDK1, PDPK1, SGK1 | 0.032 |
| Signaling events mediated by c-Met | MAP2K1, PDK1, PDPK1, RAF1 | 0.034 |
| a6b1 and a6b4 Integrin signaling | ITGA6, LAMA1, YWHAZ | 0.034 |
| Ceramide signaling pathway | FADD, MAP2K1, RAF1 | 0.039 |
| p38 signaling mediated by MAPKAP kinases | RAF1, YWHAZ | 0.042 |
| Canonical NF-kappaB pathway | ATM, XPO1 | 0.049 |
*Potential target pathways were determined using NCI pathway interaction database.
Targeting HDAC signaling enhanced dasatinib killing of pancreatic cancer cells
Silencin the HDAC pathway enhanced dasatinib activity. In our previous studies, we have shown anti-proliferative activity of HDAC inhibitors (belinostat, panobinostat, vorinostat) against pancreatic cancer cells [16, 17]. Effect of interaction between these inhibitors and dasatinib was examined in six pancreatic cancer cell lines (SU8686, Panc0203, Panc1005, BxPc3, MiaPaCa2, Panc1) (Figure 1). Cell proliferation was synergistically inhibited in the presence of dasatinib at low concentrations (25 or 100 nM) and each of the three HDAC inhibitors against SU8686, Panc1005, Panc0203, BxPc3, MiaPaCa2, and Panc1 (Table 2). These results suggested that concomitant inhibition of HDAC signaling may be an effective complement to dasatinib in pancreatic cancers.
Effect of combination of dasatinib with various HDAC inhibitors on cell proliferation of pancreatic cancer cells. Six human pancreatic cancer cell lines (SU8686, BxPc3, MiaPaCa2, Panc0203, Panc1, Panc1005) were treated with either single drug dasatinib, HDAC inhibitor (belinostat, panobinostat, vorinostat) or combination of both at various concentrations for 72h. Cell viability was measured (MTT assay); and data were normalized to controls without drug treatment. Das, dasatinib; Bel, belinostat; Pan, panobinostat; SAHA, vorinostat.
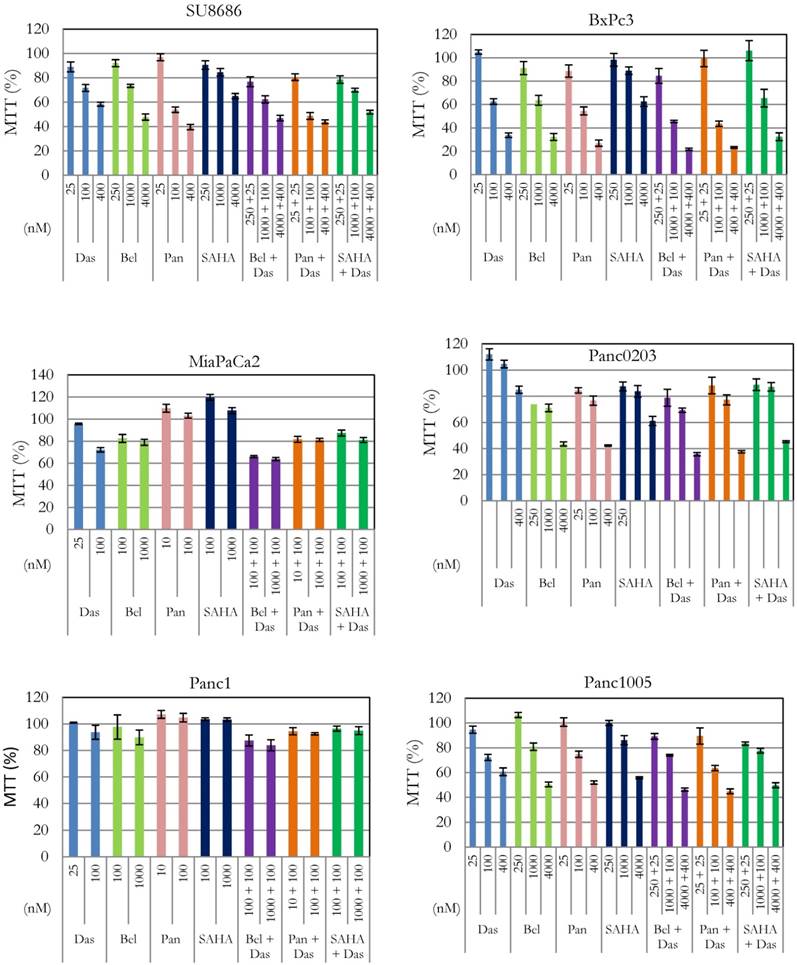
Analysis of drug interaction between dasatinib and HDAC inhibitors
| Das (nM) | Bel (nM) | Pan (nM) | SAHA (nM) | CI | |||||
|---|---|---|---|---|---|---|---|---|---|
| SU8686 | Panc0203 | Panc1005 | BxPc3 | MiaPaCa2 | Panc1 | ||||
| 25 | 250 | - | - | 0.56 | 1.33 | 0.67 | 0.8 | - | - |
| 100 | 1000 | - | - | 0.87 | 1.8 | 1.11 | 0.86 | - | - |
| 400 | 4000 | - | - | 1.6 | 0.7 | 1.98 | 1.87 | - | - |
| 25 | - | 25 | - | 0.75 | 1.3 | 0.73 | 1.92 | - | - |
| 100 | - | 100 | - | 0.61 | 1.83 | 0.79 | 0.95 | - | - |
| 400 | - | 400 | - | 2.03 | 0.98 | 2.01 | 2.13 | - | - |
| 25 | - | - | 250 | 0.52 | 1.12 | 0.41 | 2.87 | - | - |
| 100 | - | - | 1000 | 0.97 | 3.16 | 1.27 | 0.79 | - | - |
| 400 | - | - | 4000 | 1.14 | 0.62 | 2.12 | 1.74 | - | - |
| 100 | 100 | - | - | - | - | - | 0.93 | 0.84 | |
| 100 | 1000 | - | - | - | - | - | 1.27 | 0.8 | |
| 100 | - | 10 | - | - | - | - | 3.23 | 2.16 | |
| 100 | - | 100 | - | - | - | - | 2.14 | 1.84 | |
CI (Combination index) was calculated with Calcusyn as described in Materials and Methods. Additive effect (CI = 1), synergism (CI < 1), antagonist (CI >1). Das: dasatinib; Bel: belinostat; Pan: panobinostat; SAHA: vorinostat. Synergy is highlighted in red.
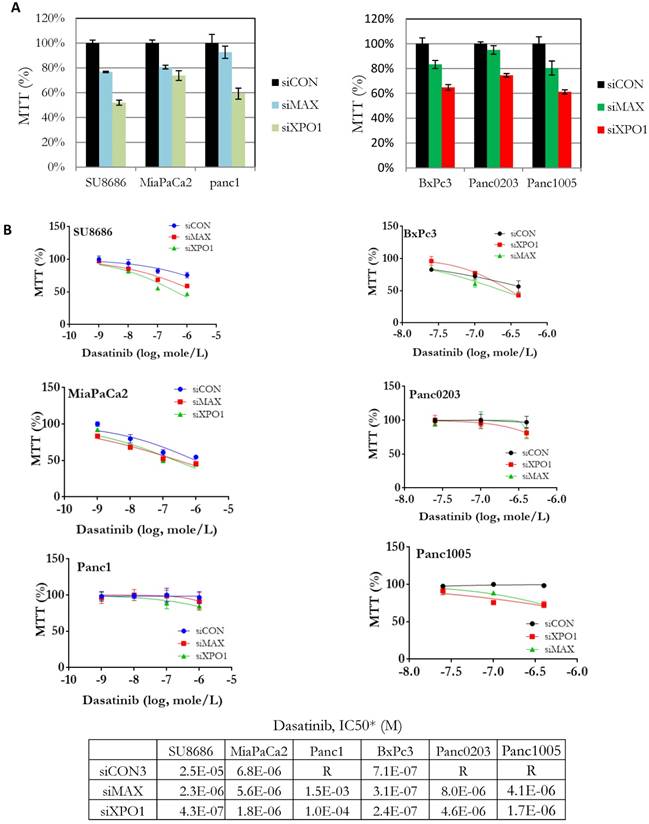
Components (MAX, SAP18, XPO1) of the HDAC signaling pathway were identified from our synthetic lethal screening assay (Table 1). These genes were over-expressed in tumors compared to normal pancreas using public data databases (Supplementary Figure S4A). High expression of XPO1 was associated with high risk pancreatic cancers (Supplementary Figure S4B). Endogenous expression levels of these genes in the pancreatic cancer cell lines did not correlate with their sensitivities to dasatinib (Supplementary Figure S5). Howeve, pancreatic cancer cell lines (SU8686, MiaPaCa2, Panc1, BxPc3, Panc0203, Panc1005) with silencing of both MAX (siMAX) and XPO1 (siXPO1) inhibited cell growth in pancreatic cancer cells compared to control cells (siCON) (Figure 2A). Additionally, the IC50 of dasatinib against pancreatic cancer cell lines was lower if these cells also had silencing of either MAX or XPO1 compared to controls (siCON) (Figure 2B). For example, the IC50 of dasatinib against the SU8686 cells was 2.5 x 10-5 M; but with silencing of either MAX or XPO1, the IC50 of dasatinib decreased to 2.3 x10-6 M and 4.3 x 10-7 M, respectively (Figure 2B). These observations indicate that silencing the HDAC pathway (e.g. MAX and XPO1) enhances the ability of dasatinib to inhibit proliferation of pancreatic cancer cell.
Effect of siRNA-mediated gene silencing of either MAX or XPO1 on human pancreatic cancer cell proliferation and dasatinib sensitivity. Six human pancreatic cancer cell lines (SU8686, MiaPaCa2, Panc1, BxPc3, Panc0203, Panc1005) were transfected with siRNAs against either MAX or XPO1 (siMAX or siXPO1) and treated with dastainib (10-9 - 10-6 M). A. Without dasatinib treatment, proliferation of siMAX-transfected and siXPO1-transfected cells was compared to siCON-transfected cells by MTT assays. B. siRNA transfected cells were treated with dasatinib (10-9 - 10-6 M), and cell viability was measure by MTT assays. For each cell line, cell growth was normalized to itself without dasatinib treatment. *Values of IC50 were extrapolated from dose curves using Graphpad Prism software.
Effect of XPO1 inhibitor KPT-330 (selinexor) in pancreatic cancer cells
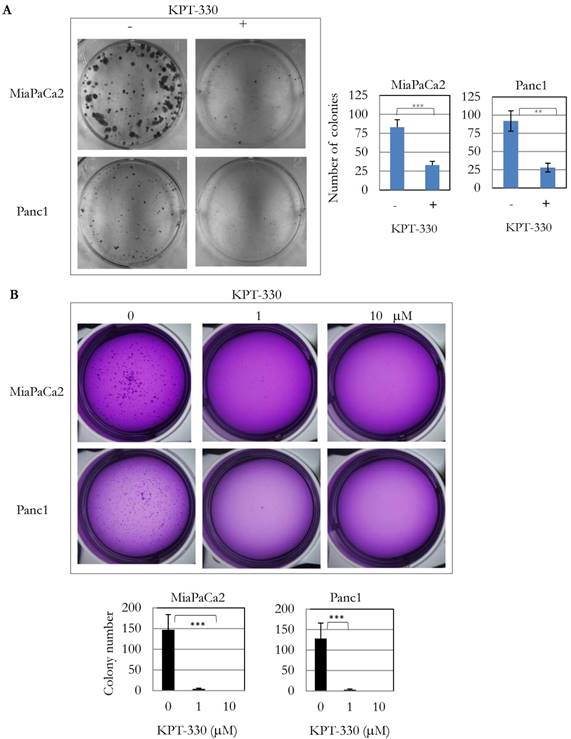
Because knockdown of XPO1 by siRNA inhibited pancreatic cancer cell proliferation, the anti-tumor activity of three XPO1 inhibitors (KPT-185, -276, -330) was investigated. Several pancreatic cancer cell lines were sensitive to these inhibitors at an IC50 from 1 x 10-7 to 1 x 10-5 M (Table 3). KPT-330 (selinexor) is undergoing clinical trials for various cancers including T cell leukemia and solid tumors [18]. Seven pancreatic cancer cell lines (SU8686, MiaPaCa2, Panc1, BxPc3, Panc0203, Panc1005, Panc0813) were sensitive to KPT-330 (selinexor) (Table 3). KPT-330 (selinexor) decreased clonal growth of pancreatic cancer cell lines (MiaPaCa2, Panc1) on plastic (Figure 3A). Likewise, anchorage-independent growth of these cells progressively decreased with increasing concentration of KPT-330 (selinexor) (1 to 10 μM) (Figure 3B). These results showed that KPT-330 (selinexor) effectively inhibited growth of pancreatic cancer cells.
Anti-tumor activity of XPO1 inhibitors against pancreatic cancer cells (IC50*, M)
| KPT-185 | KPT-276 | KPT-330 | |
|---|---|---|---|
| SU8686 | 3.5 x 10-6 | 6.7 x 10-6 | 9.2 x 10-7 |
| MiaPaCa2 | 2.4 x 10-6 | 1.1 x 10-5 | 7.2 x 10-7 |
| Panc1 | 1.1 x 10-6 | 2.9 x 10-6 | 1.1 x 10-6 |
| BxPc3 | 9.6 x 10-6 | 1.6 x 10-5 | 1.2 x 10-5 |
| Panc0203 | R | 2.2 x 10-5 | 5.6 x 10-6 |
| Panc1005 | 1.5 x 10-6 | 1.3 x 10-5 | 1.3 x 10-6 |
| Panc0813 | 1.7 x 10-6 | 2.3 x 10-6 | 4.1 x 10-7 |
| AsPc1 | 2.9 x 10-5 | R | R |
Cell lines were cultured with three XPO1 inhibitors for 72 and cell viability was measured by MTT assay. *IC50s were extrapolated from MTT data using Graphpad Prism software as described in Materials and Methods. R: resistant.
KPT-330 activity against human pancreatic cancer cells. A. Two pancreatic cancer cell lines (MiaPaCa2, Panc1) were treated with KPT-330 (10 μM) for 14 days, and the number of colonies present on plastic were counted and plotted on bar graphs (described in Materials and Methods). B. Soft agar colony formation with KPT-330 treatment. Two pancreatic cancer cell lines (MiaPaCa2, Panc1) were grown in soft agar with KPT-330 (1 or 10 μM) for 14 days. Colonies were stained and counted as shown in bar graphs (described in Materials and methods). **: p < 0.01; ***: p < 0.001.
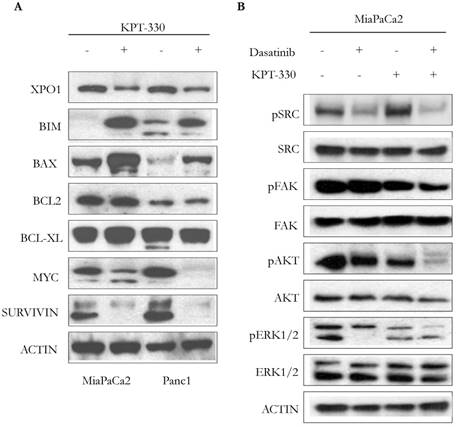
Incubation of pancreatic cancer cells (MiaPaCa2, Panc1) with KPT-330 (selinexor) (10 μM, 36 hr) decreased levels of XPO1, increased the apoptotic-related proteins BIM and BAX, but did not change the expression of the anti-apoptotic proteins BCL-2 or BCL-XL (Figure 4A). Protein levels of proliferation-related proteins, MYC and SURVIVIN, also decreased (Figure 4A). Because dasatinib is a multi-target ATP-competitive kinase inhibitor, we investigated the phosphorylation levels of 4 kinases (pSRC, pFAK, pERK, pAKT) since these 4 kinases are highly activated in human pancreatic cancer. Single agent dasatinib suppressed pSRC but KPT-330 did not. Combination of both dasatinib and KPT-330 slightly enhanced the suppression of pSRC (Figure 4B). Dasatinib did not inhibit pFAK but KPT-330 did. Combination treatment of dasatinib and KPT-330 showed enhanced inhibition of pFAK (Figure 4B). KPT-330 down-regulated pAKT and combined therapy prominently enhanced the down-regulation (Figure 4B). Either dasatinib or KPT-330 alone suppressed pERK1/2 and their combination enhanced suppression of pERK1 (Figure 4B). Taken together, combination of KPT-330 plus dasatinib enhanced de-phosporylation of the SRC, AKT, ERK, and FAK kinases.
Cooperative effect of KPT-330 and dasatinib on cell signaling. A. Western blot analysis of pancreatic cancer cells (MiaPaCa2, Panc1) treated with KPT-330 (10 μM, 36h). Changes in expression of XPO1 as well as proteins related to cell proliferation and apoptosis (BIM, BAX, BCL2, BCL-XL, MYC, SURVIVIN). B. MiaPaCa2 pancreatic cancer cell line was treated with dasatinib (200 nM) and/or KPT-330 (10 μM) for 16 h. Phosphorylation levels of SRC, FAK, AKT, and ERK1/2 were detected by Western blot analysis. Actin was used as loading control. pSRC, phosphorylated SRC; pFAK, phosphorylated FAK; pAKT, phosphorylated AKT; pERK1/2, phosphorylated ERK.
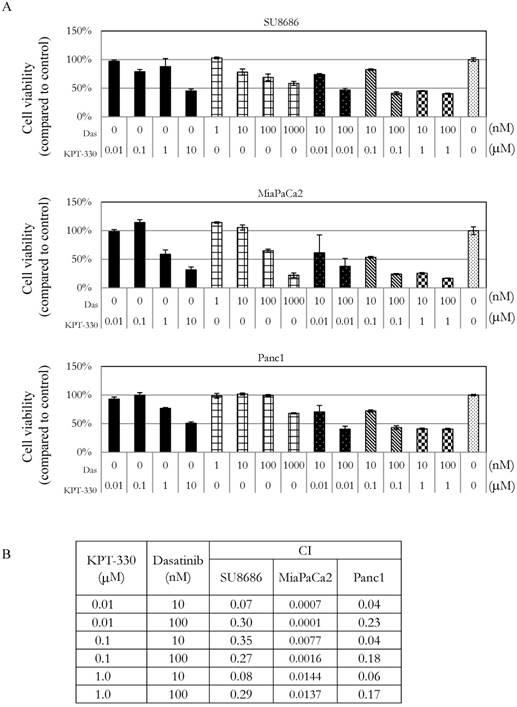
Sensitivity of pancreatic cancer cells to the combination of KPT-330 (selinexor) and dasatinib
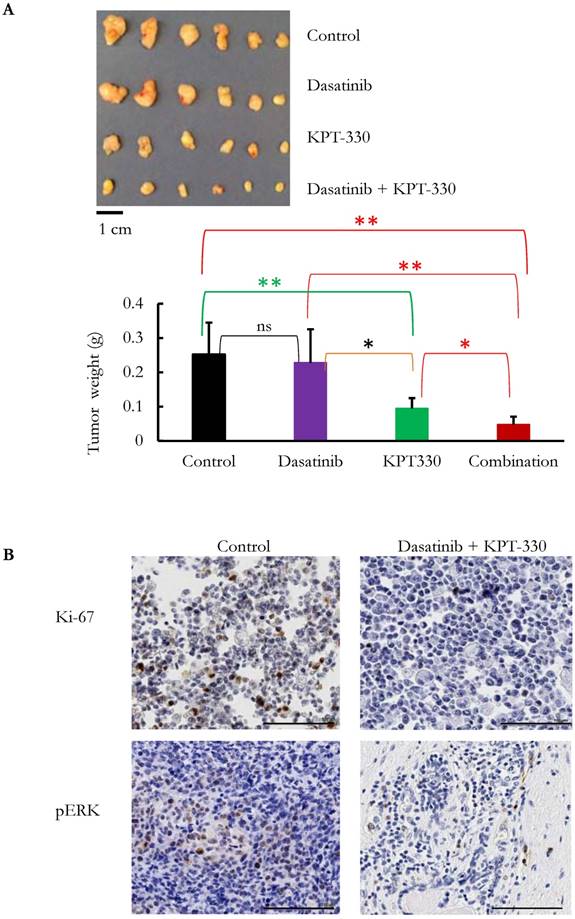
The ability of KPT-330 (selinexor) to potentiate the anti-tumor activity of dasatinib in pancreatic cancer cells was evaluated. Three pancreatic cancer cell lines (SU8686, MiaPaCa2, Panc1) were treated with either KPT-330 (selinexor) (0.01-10 μΜ), dasatinib (1-1000 nM) or a combination of both drugs (Figure 5A). The combination of both drugs synergistically decreased the pancreatic cancer cells as shown by their combination indices (CI) (Figure 5B). Xenografts using human pancreatic cancer cells MiaPaCa2 were established in NSG mice to evaluate the efficacy of the combination of KPT-330 (selinexor) and dasatinib in vivo. KPT-330 (selinexor) treatment by itself reduced average tumor weight from a mean of 0.252 ± 0.093 to a mean of 0.095 ± 0.030 grams compared to diluent control (p<0.01, Figure 6A). Average tumor weight with dasatinib treatment alone was 0.228 ±0.098 g; and in combination with KPT-330 (selinexor) the average tumor weight diminished to 0.048 ± 0.023 grams (p<0.05, Figure 6A). No significant change in either body weight or blood chemistry parameters occurred with drug treatments (Supplementary Figure S6). To investigate further the characteristics of tumors, the levels of cell proliferation (Ki-67) and phosphorylation levels of ERK1/2 were examined by IHC. Compared with control xenografts, a decreased cell proliferation (Ki-67) and phosphorylation of ERK1/2 was detected in xenografts from mice receiving the combined treatment of KPT-330 and dasatinib (Figure 6B). Likewise, the combination of KPT-330 and dasatinib decreased growth of the xenografts (Figure 6B).
Growth of pancreatic cancer cells in the presence of the combination of the XPO1 inhibitor (KPT-330) with dasatinib. A. MTT assays of three pancreatic cancer cell lines (SU8686, MiaPaCa2, Panc1) treated with either single drug (KPT-330 or dasatinib) or a combination of both. Cell viability was normalized to control without drug treatment. Das: dasatinib. B. Combination index (CI) was calculated for the drug combinations with Calcusyn as described in Materials and Methods. CI value > 1 (antagonist), = 1 (additive effect), < 1 (synergism).
KPT-330 was synergistic with dasatinib in the inhibition of pancreatic tumor growth in vivo. MiaPaCa2 cells were injected subcutaneously in immunodeficient mice. After the tumors became palpable, mice were treated with KPT-330 (10 mg/kg by oral gavage), dasatinib (10 mg/kg intraperitoneally), combination of KPT-330 and dasatinib, or diluent control, as described in Materials and Methods. A. At the end of the experiments, tumors were weighed. Student t-test was used to compare the difference among the tumors. ns: nonsignificant; *: p < 0.05; **: p < 0.01. B. Immunohistochemical staining with Ki-67 and phosphorylated ERK (pERK) on xenografts. Image magnification, 20x; scale bar, 100 μm. Representative slides are shown.
Discussion
Although dasatininb has some efficacy against pancreatic cancer, the majority of patients are either refractory to this therapy or quickly acquire resistance during treatment. Thus pancreatic cancer remains a clinical challenge if it cannot be cured by surgery. Using a large shRNA library screen and next-generation sequencing as a platform, 729 depleted shRNAs corresponding to 410 genes were identified in the dasatinib treated cells. Pathway analysis suggested targeting mTOR signaling (p = 0.000126), hedgehog signaling (p = 0.001), FOXO family (p = 0.001), or canonical NF-κB pathway (p = 0.049) may prominently inhibit pancreatic cancer cell proliferation when combined with dasatinib. We particularly focused on the synthetic lethal interaction between dasatinib and an inhibitor of either HDAC or XPO1.
HDAC inhibitors result in acetylation of both histones and proteins. For examples, acetylation of histone 3 lysine 27 is associated with increase gene expression. Acetylation of p53 enhances its activity to slow growth or induce apoptosis. HDAC inhibitors have shown anti-tumor activity against various cancer types including pancreatic cancer [17, 19]. We found that pharmacological levels of HDAC inhibitors in combination with dasatinib markedly suppressed human SU8686 pancreatic cancer cells even in the presence of a very low concentrations of dasatinib (25 or 100 nM). For other pancreatic cancer cell lines (BxPc3, Panc0203, MiaPaCa2, Panc1), similar but less robust anti-pancreatic cancer growth was noted with the combination of dasatinib and HDAC inhibitors.
One component required for signaling by HDACs as well as FOXO and canonical NF-κB involves transport across the nuclear membrane through XPO1 [20, 21]. Leptomycin B, an inhibitor for XPO1, was found to have anti-cancer activity but caused too much systemic toxicity [22]. This prompted the synthesis and development of a series of selective inhibitors of nuclear export (SINE) [KPT-185, -276, and -330] which have anti-proliferative activity against a wide variety of cancers including ovarian cancer [23], hepatocellular carcinoma [24], lung cancer [25], and several haematological malignancies [26]. KPT-330 (selinexor) has been and is used in several clinical trials in solid and hematopoietic malignancies including acute myeloid leukemia [27, 28], multiple myeloma [29], chronic myelogenous leukemia, and non-Hodgkin lymphoma [30] . Many key proteins (e.g. FOXOs, NPM, and p53) are aberrantly localized or altered in their abundance by this family of inhibitors [31-33]. Functionally, we showed that pancreatic cancer cells were sensitive to KPT-185, -276, and -330; and furthermore, the anticancer potency of KPT-330 (selinexor) was enhanced in hypoxic conditions (the normal condition of pancreatic cancer cells) (Supplementary Figure S7). KPT-330 (selinexor) suppressed colony formation of pancreatic cancer cells in vitro as well as those growing as xenografts in immuodeficient mice corroborating a previous study [21]. More importantly, KPT-330 (selinexor) over a wide range of concentrations was synergistic with dasatinib, the multi-targeted kinase inhibitor in their anti-proliferative activity against pancreatic cancer cells both in vitro and in a xenograft model. KPT-330 also synergized with gemcitabine in human pancreatic cancer cell lines (Supplementary Figure S8), corroborating a previous study [34]. In this study, KPT-330 up-regulated apoptotic molecules and down-regulated anti-apoptotic molecules in pancreatic cancer cells. The combination of dasatinib plus KPT-330 markedly decreased the phosphorylation of SRC, FAK, ARK, and ERK kinases.
Incidence of pancreatic cancer is increasing globally and an efficient non-surgical therapeutic treatment of is urgently needed. Phase I study of dasatinib when combined with gemcitabine showed partial response in 25% of patients [35] and phase II study indicates dasatinib does not have clinical activity as a single agent in metastatic pancreatic cancer [36]. Recent pre-clinical studies suggest targeting TGFβ together with dasatinib may be a novel therapeutic approach for pancreatic cancer [37, 38]. In this study, we identified two pathways that mediated synthetic lethality with dasatinib. The first is HDAC inhibitors which have been approved by the FDA for other cancers. The second is KPT-330 (selinexor) which blocks protein and RNA transport from the nucleus. The key anti-cancer targets of the drug are probably multiple; we showed that two pro-apoptotic proteins (BAX, BIM) markedly increased, while anti-apoptotic protein (SURVIVIN) as well as the pro-growth protein (MYC) decreased in the pancreatic cancer cells. In summary, either HDAC inhibitors or KPT-330 (selinexor) combined with dasatinib appeared should be explored in surgically failed pancreatic cancers.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was funded by the Singapore Ministry of Health's National Medical Research Council (NMRC) under its Singapore Translational Research (STaR) Investigator Award to H. Phillip Koeffler, the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative. This study was partially supported by a generous donation from the Melamed family and the Samuel Oschin Comprehensive Cancer Institute (SOCCI) at Cedars-Sinai Medical Center through the Eleanor and Glenn Padnick Discovery Fund in Cellular Therapy Project".
Competing Interests
Erkan Baloglu, Sharon Shacham and Michael Kauffman are employed by Karyopharm Therapeutics. The remaining authors declare no competing financial interest.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: A Cancer Journal for Clinicians. 2013;63:11-30
2. Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Practice & Research Clinical Gastroenterology. 2010;24:349-58
3. Huxley R, Ansary-Moghaddam A, Berrington de Gonzalez A, Barzi F, Woodward M. Type-II diabetes and pancreatic cancer: a meta-analysis of 36 studies. Br J Cancer. 2005;92:2076-83
4. Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M. et al. Presence of Somatic Mutations in Most Early-Stage Pancreatic Intraepithelial Neoplasia. Gastroenterology. 2012;142:730-3.e9
5. Barton CM, Staddon SL, Hughes CM, Hall PA, O'Sullivan C, Kloppel G. et al. Abnormalities of the p53 tumour suppressor gene in human pancreatic cancer. Br J Cancer. 1991;64:1076-82
6. Goldstein AM, Chan M, Harland M, Gillanders EM, Hayward NK, Avril M-F. et al. High-risk Melanoma Susceptibility Genes and Pancreatic Cancer, Neural System Tumors, and Uveal Melanoma across GenoMEL. Cancer Research. 2006;66:9818-28
7. Wilentz RE, Iacobuzio-Donahue CA, Argani P, McCarthy DM, Parsons JL, Yeo CJ. et al. Loss of Expression of Dpc4 in Pancreatic Intraepithelial Neoplasia: Evidence That DPC4 Inactivation Occurs Late in Neoplastic Progression. Cancer Research. 2000;60:2002-6
8. Jones S, Zhang X, Parsons DW, Lin JC-H, Leary RJ, Angenendt P. et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science. 2008;321:1801-6
9. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M. et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. New England Journal of Medicine. 2013;369:1691-703
10. da Cunha Santos G, Dhani N, Tu D, Chin K, Ludkovski O, Kamel-Reid S. et al. Molecular predictors of outcome in a phase 3 study of gemcitabine and erlotinib therapy in patients with advanced pancreatic cancer. Cancer. 2010;116:5599-607
11. Trevino JG, Summy JM, Lesslie DP, Parikh NU, Hong DS, Lee FY. et al. Inhibition of Src Expression and Activity Inhibits Tumor Progression and Metastasis of Human Pancreatic Adenocarcinoma Cells in an Orthotopic Nude Mouse Model. The American Journal of Pathology. 2006;168:962-72
12. Waddell N, Pajic M, Patch A-M, Chang DK, Kassahn KS, Bailey P. et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495-501
13. Chien W, Sun Q-Y, Lee KL, Ding L-W, Wuensche P, Torres-Fernandez LA. et al. Activation of protein phosphatase 2A tumor suppressor as potential treatment of pancreatic cancer. Molecular Oncology. 2015;9:889-905
14. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621-81
15. Xiao J-F, Sun Q-Y, Ding L-W, Chien W, Liu X-Y, Mayakanda A. et al. The MYC/BMI1 axis is essential for SETDB1-mediated breast tumorigenesis. The Journal of Pathology. 2018;246(1):89-102
16. Kumagai T, Wakimoto N, Yin D, Gery S, Kawamata N, Takai N. et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. International Journal of Cancer. 2007;121:656-65
17. Chien W, Lee DH, Zheng Y, Wuensche P, Alvarez R, Wen DL. et al. Growth inhibition of pancreatic cancer cells by Histone Deacetylase inhibitor belinostat through suppression of multiple pathways including HIF, NFkB, and mTOR signaling in vitro and in vivo. Molecular Carcinogenesis. 2014;53(9):722-35
18. London CA, Bernabe LF, Barnard S, Kisseberth WC, Borgatti A, Henson M. et al. Preclinical Evaluation of the Novel, Orally Bioavailable Selective Inhibitor of Nuclear Export (SINE) KPT-335 in Spontaneous Canine Cancer: Results of a Phase I Study. PLOS ONE. 2014;9:e87585
19. Banerji U, van Doorn L, Papadatos-Pastos D, Kristeleit R, Debnam P, Tall M. et al. A Phase I Pharmacokinetic and Pharmacodynamic Study of CHR-3996, an Oral Class I Selective Histone Deacetylase Inhibitor in Refractory Solid Tumors. Clinical Cancer Research. 2012;18:2687-94
20. Darb-Esfahani S, Sinn BV, Weichert W, Budczies J, Lehmann A, Noske A. et al. Expression of classical NF-κB pathway effectors in human ovarian carcinoma. Histopathology. 2010;56:727-39
21. Azmi AS, Aboukameel A, Bao B, Sarkar FH, Philip PA, Kauffman M. et al. Selective Inhibitors of Nuclear Export Block Pancreatic Cancer Cell Proliferation and Reduce Tumor Growth in Mice. Gastroenterology. 2013;144:447-56
22. Mutka SC, Yang WQ, Dong SD, Ward SL, Craig DA, Timmermans PBMWM. et al. Identification of Nuclear Export Inhibitors with Potent Anticancer Activity In vivo. Cancer Research. 2009;69:510-7
23. Chen Y, Camacho C, Silvers TR, Razak ARA, Gabrail NY, Gerecitano JF. et al. INHIBITION OF THE NUCLEAR EXPORT RECEPTOR XPO1 AS A THERAPEUTIC TARGET FOR PLATINUM RESISTANT OVARIAN CANCER. Clinical Cancer Research. 2016
24. Zheng Y, Gery S, Sun H, Shacham S, Kauffman M, Koeffler HP. KPT-330 inhibitor of XPO1-mediated nuclear export has anti-proliferative activity in hepatocellular carcinoma. Cancer Chemotherapy and Pharmacology. 2014;74:487-95
25. Sun H, Lin D-C, Cao Q, Guo X, Marijon H, Zhao Z. et al. CRM1 Inhibition Promotes Cytotoxicity in Ewing Sarcoma Cells by Repressing EWS-FLI1-Dependent IGF-1 Signaling. Cancer Research. 2016;76:2687-97
26. Hing ZA, Fung HYJ, Ranganathan P, Mitchell S, El-Gamal D, Woyach JA. et al. Next-generation XPO1 inhibitor shows improved efficacy and in vivo tolerability in hematological malignancies. Leukemia. 2016
27. Alexander TB, Lacayo NJ, Choi JK, Ribeiro RC, Pui C-H, Rubnitz JE. Phase I Study of Selinexor, a Selective Inhibitor of Nuclear Export, in Combination With Fludarabine and Cytarabine, in Pediatric Relapsed or Refractory Acute Leukemia. Journal of Clinical Oncology. 2016
28. Ranganathan P, Yu X, Santhanam R, Hofstetter J, Walker A, Walsh K. et al. Decitabine priming enhances the antileukemic effects of exportin 1 (XPO1) selective inhibitor selinexor in acute myeloid leukemia. Blood. 2015;125:2689-92
29. Turner JG, Dawson JL, Grant S, Shain KH, Dalton WS, Dai Y. et al. Treatment of acquired drug resistance in multiple myeloma by combination therapy with XPO1 and topoisomerase II inhibitors. Journal of Hematology & Oncology. 2016;9:73
30. Muqbil I, Aboukameel A, Elloul S, Carlson R, Senapedis W, Baloglu E. et al. Anti-tumor activity of selective inhibitor of nuclear export (SINE) compounds, is enhanced in non-Hodgkin lymphoma through combination with mTOR inhibitor and dexamethasone. Cancer Letters. 2016;383(2):309-317
31. Cai X, Liu X. Inhibition of Thr-55 phosphorylation restores p53 nuclear localization and sensitizes cancer cells to DNA damage. Proceedings of the National Academy of Sciences. 2008;105:16958-63
32. Falini B, Nicoletti I, Martelli MF, Mecucci C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): biologic and clinical features. Blood. 2007;109(3):874-85
33. Kau TR, Schroeder F, Ramaswamy S, Wojciechowski CL, Zhao JJ, Roberts TM. et al. A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell. 2003;4:463-76
34. Kazim S, Malafa MP, Coppola D, Husain K, Zibadi S, Kashyap T. et al. Selective Nuclear Export Inhibitor KPT-330 Enhances the Antitumor Activity of Gemcitabine in Human Pancreatic Cancer. Molecular Cancer Therapeutics. 2015;14:1570-81
35. Hong DS, Choe JH, Naing A, Wheler JJ, Falchook GS, Piha-Paul S. et al. A phase 1 study of gemcitabine combined with dasatinib in patients with advanced solid tumors. Investigational New Drugs. 2013;31:918-26
36. Chee CE, Krishnamurthi S, Nock CJ, Meropol NJ, Gibbons J, Fu P. et al. Phase II Study of Dasatinib (BMS-354825) in Patients With Metastatic Adenocarcinoma of the Pancreas. The Oncologist. 2013;18:2
37. Deharvengt S, Marmarelis M, Korc M. Concomitant Targeting of EGF Receptor, TGF-beta and Src Points to a Novel Therapeutic Approach in Pancreatic Cancer. PLoS ONE. 2012;7:e39684
38. Bartscht T, Rosien B, Rades D, Kaufmann R, Biersack H, Lehnert H. et al. Dasatinib blocks transcriptional and promigratory responses to transforming growth factor-beta in pancreatic adenocarcinoma cells through inhibition of Smad signalling: implications for in vivo mode of action. Molecular Cancer. 2015;14:199
Author contact
![]() Corresponding author
Corresponding author