Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(23):4449-4462. doi:10.7150/jca.25926 This issue Cite
Research Paper
Tenascin-C Modulates Cell Cycle Progression to Enhance Tumour Cell Proliferation through AKT/FOXO1 Signalling in Pancreatic Cancer
Jun Cai1, Wenli Lu2, Shaoxia Du1, Zhongkui Guo1, Hui Wang1, Wei Wei3, Xiaohong Shen1 ![]()
1. School of medicine, Nankai University, 94 Weijin Road, Tianjin 300071, China.
2. Department of Health Statistics, School of Public Health, Tianjin Medical University, Tianjin 300000, China.
3. Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center for Cancer, Huanhu West Road, Tianjin 300060, China.
Received 2018-3-7; Accepted 2018-7-27; Published 2018-10-31
Citation:
Cai J, Lu W, Du S, Guo Z, Wang H, Wei W, Shen X. Tenascin-C Modulates Cell Cycle Progression to Enhance Tumour Cell Proliferation through AKT/FOXO1 Signalling in Pancreatic Cancer. J Cancer 2018; 9(23):4449-4462. doi:10.7150/jca.25926. https://www.jcancer.org/v09p4449.htm
Other stylesAbstract
Pancreatic ductal adenocarcinoma (PDAC) is a disease with an extremely poor prognosis that is characterized by a rich extracellular matrix (ECM). Tenascin-C (TNC) is a component of the ECM and plays a role in tumour progression. In this study, we reported that TNC is overexpressed in PDAC tissues and is correlated with tumour stage and cyclin D1 expression. Cyclin D1 is key regulator of the cell cycle G1/S transition. Further experiments revealed that TNC promotes G1/S transition through AKT signalling. TNC/AKT increases the expression of cyclin D1 by enhancing the transcriptional activity of β-catenin, whereas the translocation of FOXO1 from the nucleus results in the downregulation of p27Kip1. Cyclin D1 and p27Kip1 regulate the activity of cyclin D1-CDK4 complexes and retinoblastoma (Rb), and then they stimulate the progression of G1/S transition and tumour cell proliferation. In conclusion, TNC exerts its activating effect on the proliferation of pancreatic cancer cells in vitro and in vivo through its functional target AKT/FOXO1/β-catenin. The molecular mechanisms that drive PDAC progression will be useful for the development of molecular markers and the evaluation of patient prognosis.
Keywords: TNC, FOXO1, cell cycle, pancreatic cancer, proliferation
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a lethal disease that is characterized by rapid disease progression, extreme metastatic potential and difficult early diagnosis and treatment. The epigenetic changes in PDAC create a unique process of growth-promoting signals that give pancreatic cancer cells a strong survival advantage. Desmoplastic stroma is one of the distinct features of PDAC, which is constituted by the accumulation of extracellular matrix (ECM) proteins synthesized by cancer cells and pancreatic stellate cells (PSCs) [1, 2]. Tenascin-C (TNC), a large glycoprotein located in the extracellular matrix, is secreted primarily by PSCs and tumour cells in PDAC [3]. Its expression is low in normal adult tissues, whereas it is highly expressed in the stroma of most solid tumours, such as breast, colorectal and pancreatic carcinomas, which might indicate a distinct value for cancer diagnosis [4-6]. A previous study also demonstrated that TNC is expressed at a higher frequency during the progression from intraepithelial neoplastic lesions to PDAC [3]. Therefore, the expression of TNC might be related to tumour cell growth and cancer progression. The underlying mechanism by which TNC affects the tumour grade in PDAC requires further investigation.
Cancer is a disease of inappropriate cell proliferation, and the disruption of normal cell cycle progression is a critical step in cancer development. The overexpression of several cell cycle-associated molecules (p27Kip1, cyclin D/E) was found to correlate with tumour grade, pathological stage, and patient survival [7-9]. Therefore, cell cycle dysfunction manipulates tumour cells' growth proceeding, which leads to cancer development. TNC and cyclin D1 are highly expressed in pancreatic cancer. It was demonstrated that TNC is able to affect the proliferation, migration and adhesion of pancreatic cancer cells through activation of the integrin/AKT pathway [10]. Activity AKT upregulates the expression of cyclin D1 through the promotion of the transcription of β-catenin and then enhances the activity of the cyclin D1-CDK4 complex [11, 12]. The cyclin D1-CDK4 complexes activity could also be inhibited by p27Kip1 [13, 14]. Cyclin D1-CDK4 complexes is a critical factor in the progression through the G1/S phase of the cell cycle. Furthermore, the forkhead box protein O1 (FOXO1) functions as a downstream molecular effector of the AKT pathway and is an important cell cycle-associated transcription factor that functions through the direct regulation of p27Kip1 [15-17]. Therefore, we hypothesized that TNC-driven cell cycle progression might effectively proceed via activation of AKT/FOXO1 signalling.
In the present study, we showed that the expression of TNC is correlated with tumour stage in pancreatic cancer tissues and that it promotes the proliferation of pancreatic cancer cells both in vivo and in vitro. Subsequent mechanistic studies were performed to confirm the function of TNC in the promotion of cell cycle G1/S transition via activation of the AKT signalling pathway as well as the expression of downstream target genes. The results suggested that the TNC/AKT/FOXO1 pathway plays an important role in cell proliferation in human pancreatic ductal adenocarcinoma.
Materials and Methods
Clinical samples and immunohistochemistry
Fresh pancreatic adenocarcinoma and paracarcinoma tissues were obtained from patients who underwent radical pancreatectomy for pancreatic adenocarcinoma without prior radiation or chemotherapy. The diagnosis was confirmed according to the standards of the American Joint Committee on Cancer (AJCC, 2010). Histologic slides were reviewed by two experienced pathologists who were blinded to the clinical data. The study was approved by the Human Research Committee of Nankai University and the China Anti-Cancer Association (CACA) and was performed in accordance with the Declaration of Helsinki. In all, 91 pancreatic cancer tissue samples were collected and arrayed by Alenabio Corporation (Xi'an, China) after authorization from the local medical ethics committee.
Samples were immunostained with antibodies against TNC, cyclin D1, Ki67, p27Kip1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), pAKT (Abcam, Cambridge, MA, USA), pFOXO1 and β-catenin (Cell Signaling Technology, Danvers, MA, USA) and were incubated at 4°C overnight. Samples were then exposed to the appropriate secondary antibodies at room temperature for 1 h. Protein expression was evaluated by calculating a total immunostaining score (TIS) as the product of a proportion score (PS) and an intensity score (IS). The proportion score (PS), which was expressed as the percentage of tumour cells that were stained out of the total number of tumour cells, was assigned to one of five categories: 0 for a percentage ≤5 %, 1 for 5-25 %, 2 for 25-50 %, 3 for 50-75 %, and 4 for ≥75 %. The intensity of staining (IS) was scored as 0, 1, 2 or 3 for negative (no staining), mild (weak but detectable above control), moderate (distinct) or intense (strong) staining, respectively. The scoring was performed by two independent evaluators who had no knowledge of the patients' pathological and clinical characteristics.
Cell culture
The human pancreatic cancer cell lines Capan-2, MiaPaCa-2 and PANC-1 were purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in RPMI 1640 (Capan-2) or DMEM (MiaPaCa-2 and PANC-1) supplemented with 10 % foetal bovine serum (FBS; Biological Industries, Kibbutz Beit-Haemek, Israel) at 37 °C with 5 % CO2. For the induction experiments, PANC-1 cells were treated with 5 μg/ml recombinant human TNC (rhTNC; Millipore, Billerica, MA, USA) for the indicated time. The AKT inhibitor LY294002 (20 μM) and the ERK inhibitor PD98059 (100 μM) (Selleck Chemicals, Houston, TX, USA) were applied to the culture medium for 1 h.
Plasmids, small interfering RNA, and transfection
The pcDNA3.1-TNC plasmid expressing TNC was obtained as previously described [18]. The CCND1 promoter regions from -47 to +216 (containing a candidate TCF/LEF binding site) or from -2 to +216 (lacking the candidate TCF/LEF binding site) relative to the transcription start site (TSS) were amplified from human genomic DNA by PCR. The PCR products were then inserted into the luciferase reporter pGL3-Basic vector (Promega, Madison, WI, USA). The mutated luciferase construct of the CCND1 promoter was constructed by the Fast Mutagenesis System (TransGen Biotech, Beijing, China). The pcDNA3-Flag-FOXO1 and pCMV5-human-p27Kip1 plasmids were a gift from Kunliang Guan (Addgene plasmid # 13507) and Joan Massague (Addgene plasmid #14049), respectively. Three small interfering RNAs (siRNAs) targeting independent sequences of the human TNC, AKT, β-catenin and FOXO1 genes were designed and synthesized by GenePharma (Shanghai, China). The siRNA that displayed optimal knockdown efficiency was selected for further experiments. Non-targeting siRNA was used as a control (siControl). The transfection of cells with plasmids or siRNAs was performed using Lipofectamine 2000 Reagent (Invitrogen, Gaithersburg, MD, USA) according to the manufacturer's protocol.
Cell proliferation assay
Pancreatic cancer cells were plated in 96-well plates in triplicate at a density of 2×103 cells per well and maintained in complete medium. The Cell Counting Kit-8 (CCK-8; Beyotime, Jiangsu, China) assay was performed at 24, 48 and 72 h according to the manufacturer's protocol.
Flow cytometry analysis
Following the culture of PANC-1 cells in the absence of FBS for 12 h, the cells were incubated with BrdU at a final concentration of 10 μM in cell culture medium for 2 h at 37 °C, and a cell cycle assay was performed using the Cytofix/Cytoperm™ kit (BD Biosciences, New York, USA) according to the manufacturer's instructions. A flow cytometry analysis was performed in a FACSCalibur cytometer (BD Biosciences), in which a minimum of 10,000 cells was assayed.
Western blot
Cultured cells were collected and solubilized using protein lysis buffer. The proteins were then separated according to size using SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore). The membranes were incubated with primary antibodies followed by the secondary antibody (Santa Cruz Biotechnology). The immunereactive proteins were detected using an enhanced chemiluminescence kit (Millipore). The primary antibodies used were against FOXO1, AKT, CDK4, E2F, Rb, pRb (Cell Signaling Technology), Histone H3 and β-actin (Santa Cruz Biotechnology). The digital images of the western blot bands were quantified by ImageJ software after background subtraction.
RT-qPCR
Total RNA isolation, RT-qPCR and the quantification of target gene expression were performed as previously described [18]. The housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal mRNA quantity control for target gene expression. The primer sequences of cyclin D1 were as follows: F: GCC GAG AAG CTG TGC ATC TAC; R: TCC ACT TGA GCT TGT TCA CCAG; those for GAPDH were as follows: F: GAA GGT GAA GGT CGG AGTC; R: GAA GAT GGT GAT GGG ATT TC. The results were confirmed by three independent experiments.
Chromatin immunoprecipitation assay
A chromatin immunoprecipitation (ChIP) assay was performed using a ChIP kit (Millipore) according to the manufacturer's protocol. The primers for amplification of the CCND1 promoter region containing a TCF/LEF putative binding site were as follows: F: CTC CCA TTC TCT GCC GGG CTT T; R: GGA CTC TGC TGC TCG CTG CTA. The primers for amplification of the CDKN1B promoter region containing a FOXO1 putative binding site from -1354 to -1347 relative to the TSS were as follows: F: GAG GGT TAA ACC ACA GGG TC; R: GGA AAC CAA CCT TCC GTT CT.
Dual-luciferase reporter assay
Cells were seeded into 24-well plates, cultured without antibiotics and grown to 80 % confluence. Then, the cells were incubated with LY294002 or DMSO for 1 h. Subsequently, siTNC, TNC, TNC+siβ-catenin/FOXO1 as well as their controls were cotransfected with the pGL3-CCND1 promoter luciferase reporter (or the TOP/FOP reporter for β-catenin activation) into the cells. To normalize transfection efficiency, cells were also co-transfected with 0.1 μg of the pRL-TK vector (Renilla luciferase). After 48 h, the luciferase activities of the cells were measured using a dual-luciferase reporter assay kit (Promega). Reporter luciferase activity was normalized to Renilla luciferase activity.
Immunofluorescence
Coverslips were coated with rhTNC/BSA as previously described [18]. PANC-1 cells were seeded onto the coverslips, cultured for 48 h, and then fixed in 4 % paraformaldehyde. After washing with PBS, the cells were permeabilized with 0.25 % Triton X-100 for 10 min, washed again with PBS, blocked with 3 % goat serum blocking solution, and then incubated with the primary antibodies overnight at 4 °C. The cells were then washed with PBS and incubated with a fluorescein isothiocyanate-conjugated secondary antibody (Santa Cruz Biotechnology). Nuclei were stained with DAPI. Confocal microscopy studies were performed with a laser scanning microscope (TCS-SP2-AOBS-MP, Leica Microsystems CMS, Wetzlar, Germany). Each image was obtained using identical microscope settings, including laser power, gain, and contrast.
Immunoprecipitation
Cell lysates were incubated with protein A/G-agarose beads (Millipore) for 1 h at 4 °C to minimize nonspecific binding. Lysates were transferred to clean tubes and incubated further with a CDK4 antibody. After incubation at 4 °C overnight, protein A/G-agarose beads were added and were rotated for 2 h. The complexes were washed three times with 1× immunoprecipitation buffer. Proteins were eluted by boiling in 2× SDS loading buffer and subjected to western blot using cyclin D1 and CDK4 antibodies.
Tumourigenicity
The PANC-1 cells that stably expressed TNC or empty vector (5×106 cells) were subcutaneously injected into the flank region of 5-week-old BALB/c athymic nude mice (Beijing HFK Bio-Technology, Beijing, China), with five mice per group. Tumour dimensions were measured weekly. The mice were sacrificed 7 weeks after injection, and the tumours were surgically isolated. Tumour volume (V) was calculated using the formula (S×S×L) × 0.5, where S and L are the short and long dimensions, respectively. All animal studies were approved by the Ethics Committee of the Tianjin Medical University Cancer Institute and Hospital and were conducted by skilled experimenters under an approved protocol in accordance with the principles and procedures outlined in the NIH Guide for the Care and Use of Laboratory Animals.
Statistical analysis
Data are presented as the mean ± the s.d. Student's t-test and ANOVA were used to compare differences between the experimental group and the control group. Statistical significance was defined as P < 0.05.
Results
TNC and cyclin D1 are overexpressed in pancreatic cancer tissues
To investigate whether TNC and cyclin D1 play dominant roles in the progression of pancreatic cancer, we performed an immunohistochemical (IHC) analysis. Higher expression of TNC and cyclin D1 were found in tumour tissues compared with paracarcinoma tissues (Fig. 1A). Consistently, elevated TNC, pFOXO1 and cyclin D1, but decreased p27Kip1 expression, were observed in malignant areas compared with adjacent non-malignant areas by western blot (Fig. 1B). Next, an IHC assay of 91 pancreatic cancer tissues revealed that the expression of TNC and cyclin D1 were correlated with tumour stage (I-IV) in pancreatic cancer (Fig. 1C). A structured form was developed to extract the following information from the paper medical records: gender, age at diagnosis, histologic grade, anatomic stage, TNM stage (Table 1). In addition, the expression levels of TNC and cyclin D1 were significantly associated with each other (Fig. 1D). Cyclin D1 is critical for the progression of the cell cycle, specifically G1/S transition [19], and thus the above data indicated that TNC might be involved in pancreatic cancer cell proliferation and tumour development by regulation of cell cycle process. Then the related mechanism is detected.
Table 1
Characteristics of patients after pancreatic cancer resection for pancreatic adenocarcinoma (n = 91).
| Characteristics | N (%) |
|---|---|
| Gender | |
| Male | 56 (61.5%) |
| Female | 35 (38.5%) |
| Age at diagnosis (yr) | |
| ≤60 | 76 (83.5%) |
| >60 | 15 (16.5%) |
| Histologic grade * | |
| I | 23 (25.3%) |
| II | 31 (34.1%) |
| III | 14 (15.4%) |
| Anatomic stage | |
| Stage I | 29 (31.9%) |
| Stage IIA | 44 (48.4%) |
| Stage IIB | 12 (13.2%) |
| Stage III-IV | 6 (6.6%) |
| TNM stage | |
| T stages | |
| T1 (≤2 cm) | 3 (3.3%) |
| T2 (2.1-3.0 cm) | 32 (35.2%) |
| T3 (3.1-4.0 cm) | 54 (59.3%) |
| T4 (≥4.1 cm) | 2 (2.2%) |
| N stages | |
| N0 | 76 (83.5%) |
| N1 | 14 (15.4%) |
| N2 | 1 (1.1%) |
| M stages | |
| M0 | 89 (97.8%) |
| M1 | 2 (2.2%) |
* Histologic grade with Unknown (n = 23).
TNC promotes pancreatic cancer cell proliferation through acceleration of G1/S transition
TNC can be synthesized by cancer cells and then efficiently secreted into the stroma, but the protein level of TNC cannot be detected in normal pancreatic cell lines. Therefore, we tested the mRNA levels of TNC in all the tested pancreatic cancer cell lines by RT-qPCR (Fig. 1E). The expression levels of TNC were in accordance with those in a previous study [3], we then selected the Capan-2, MiaPaCa-2 and PANC-1 cell lines, which express moderate levels of TNC, for mechanistic experiments. To investigate the effect of TNC on the proliferation of pancreatic cancer cells, the proliferation rates of Capan-2, MiaPaCa-2 and PANC-1 cells transfected with TNC plasmid or siTNC were detected. As shown in Fig. 2A, the ectopic expression of TNC significantly induced cellular proliferation, whereas the knockdown of TNC resulted in marked growth inhibition.
Figure 1
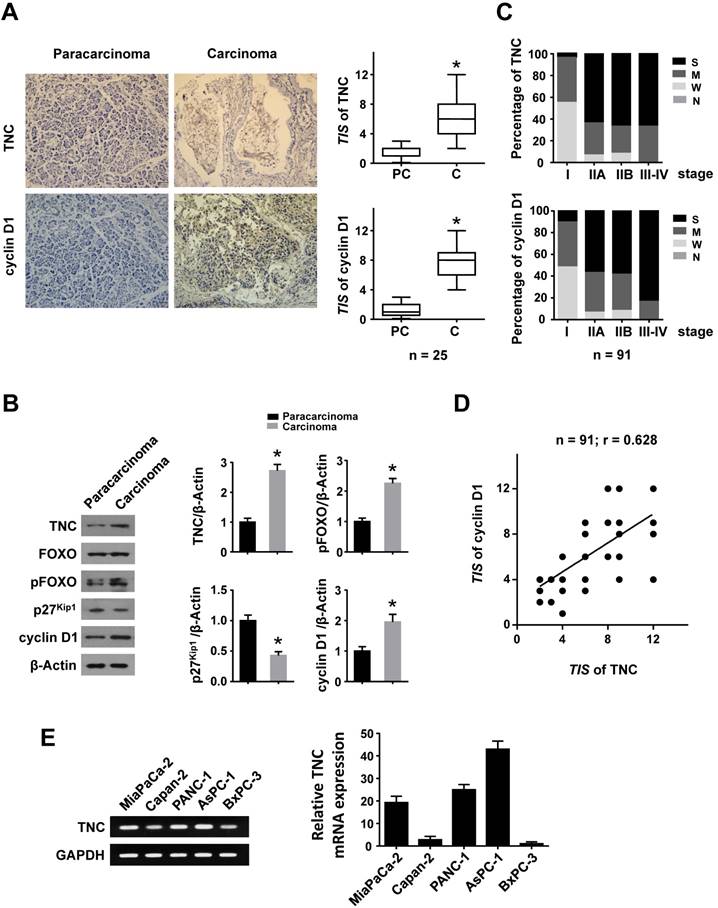
TNC and cyclin D1 are overexpressed in pancreatic cancer tissues. Immunohistochemical staining (A) and western blot (B) results of TNC and cyclin D1 in pancreatic adenocarcinoma and paracarcinoma tissues (n = 25) (original magnification ×40). (C) Percentage of different TNC/cyclin D1 expression levels (Negative, Weak, Moderate and Strong) in tissues representing different tumour stages [I - IV]. (D) The correlation between TNC and cyclin D1 protein levels in pancreatic cancer tissues (n = 91; r = 0.628; *P < 0.01). (E) The mRNA levels of TNC are shown in the tested cell lines.
Figure 2
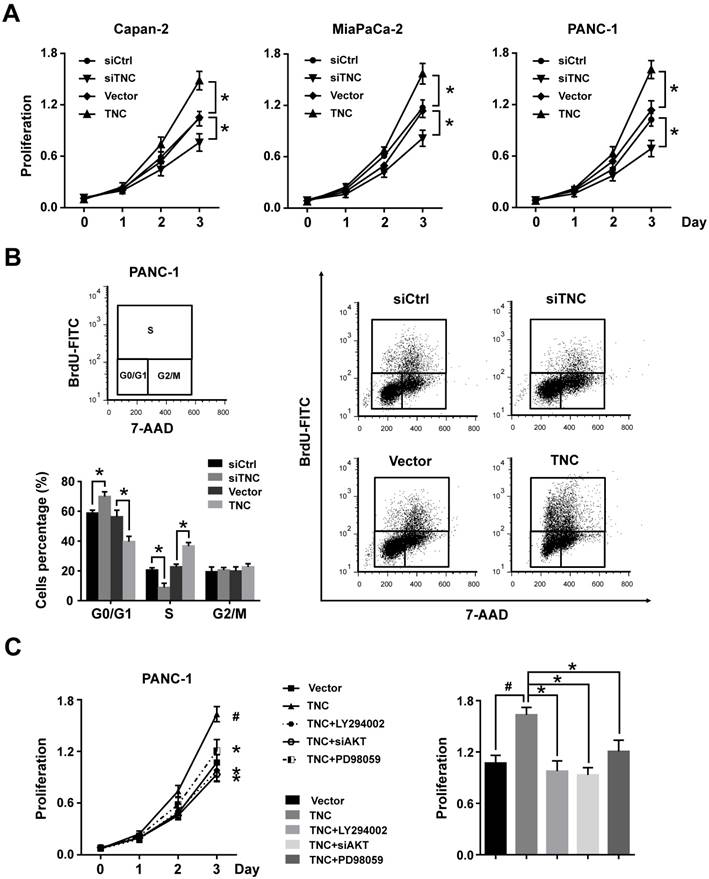
TNC promotes pancreatic cancer cell proliferation through the acceleration of G1/S transition. (A) Pancreatic cancer cell lines Capan-2, MiaPaCa-2 and PANC-1 were transfected with the TNC plasmid or siTNC, and then cell proliferation was determined by CCK-8 assay. (B) Representative FACS results are shown from PANC-1 cells that were stained for 7-AAD and BrdU-FITC. (n = 3, *P < 0.05). (C) The proliferation curves of PANC-1 cell growth were evaluated after treatment with TNC or TNC+LY294002/PD98059/siAKT. Data represent the mean ± s.d. (n = 3, #P < 0.05, transfection with TNC versus the Vector treatment; *P < 0.05, treatment with TNC+LY294002/PD98059/siAKT versus transfection with TNC alone).
To further analyse the effect of TNC on cell cycle function, PANC-1 cells were double-stained with FITC-labelled BrdU, which incorporates within cells during S phase, and 7-amino-actinomycin (7-AAD), which stains total DNA content. The results shown in Fig. 2B indicated that a significantly higher number of TNC-expressing cells progressed to S phase, which suggested that TNC promotes faster G1/S phase transition in PANC-1 cells. Conversely, TNC knockdown caused G1 arrest (Fig. 2B). Overall, the present results demonstrated that TNC promotes pancreatic cancer cell growth via acceleration of G1 to S phase transition.
TNC promotes pancreatic cancer cell G1/S phase transition via the modulation of AKT/FOXO1 signals
Our previous study demonstrated that TNC induces resistance to apoptosis in pancreatic cancer cells through activation of the ERK/NF-κB pathway. Recently, emerging evidence has revealed that TNC promotes cell survival through AKT in human pancreatic cancer and chondrosarcoma cells [10, 20]. Thus, we examined the function of ERK and AKT on TNC-induced proliferation. The CCK-8 assay showed that inhibitors of ERK (PD98059) and AKT (LY294002) or siAKT could inhibit the TNC-induced proliferation, while the effect of AKT was more distinct (Fig. 2C). Therefore, we further verified whether AKT mediates the function of TNC with respect to cell G1/S transition, the flow cytometry analysis was performed in PANC-1 cells. Our data indicated that TNC enriched the pancreatic cancer cells that were in S phase, with a concomitant decrease in the number of cells in G1 phase. However, this effect was blocked by the AKT inhibitor LY294002 and siAKT (Fig. 3A). The results provided evidence that the promotion of G1/S transition by TNC is AKT-dependent. Additionally, CDK4 and cyclin D1 are key factors in cell cycle regulation, especially in the G1 to S phase transition [19], and thus we further tested the effect of TNC/AKT on the manipulation of CDK4/cyclin D1 expression in PANC-1 cells. As shown in Fig. 3B, the knockdown of TNC reduced the phosphorylation of AKT and the expression of cyclin D1 protein. However, the levels of pAKT and cyclin D1 were significantly increased in PANC-1 cells transfected with the TNC plasmid. In addition, the TNC treatment-induced increase in the levels of pAKT and cyclin D1 was reversed by LY294002 or siAKT (Fig. 3B). However, TNC had no effect on the expression of CDK4. Together, these results suggested that TNC regulates the expression of cyclin D1 through AKT signalling and thereby promotes cell cycle progression.
FOXO1 is a downstream substrate of AKT to inhibit G1/S transition through activates the expression of p27Kip1and p21Waf1, whereas phosphorylation of FOXO1 (pFOXO1) translocates to the cell cytoplasm to reduce its transcrpional activity [21], we then investigated whether TNC had an effect on the function of FOXO1 via activation of AKT signalling. As shown in Fig. 3C, stimulation of PANC-1 cells with exogenous TNC markedly increased the phosphorylation of AKT and FOXO1. Our study showed that pFOXO1 translocates from the nucleus to the cell cytoplasm after exogenous TNC treatment, which indicates that its transcriptional activity was inhibited. Notably, these effects were shown to be time-dependent. In addition, compared with control cells, the translocation of pFOXO1 to the cytoplasm could not be induced by TNC in LY294002-treated cells. The results showed that TNC induced the cytoplasmic translocation of pFOXO1 via regulation of AKT activation. Next, we determined whether FOXO1 is involved in G1/S transition regulated by TNC/AKT signalling. As shown in Fig. 3A, the ectopic expression of FOXO1 reversed the TNC-induced acceleration of G1/S transition, which was similar to the effect of LY294002/siAKT treatment, this indicated that TNC regulates G1/S transition mainly through the activation of the AKT/FOXO1 axis.
TNC/AKT transactivates the expression of cyclin D1 via FOXO1/β-catenin activity
Cyclin D1 levels can be regulated at both the transcriptional and post-transcriptional levels. We further confirmed that LY294002 and siAKT rescued the levels of cyclin D1 (CCND1) mRNA in cells with ectopic TNC expression, whereas siTNC downregulated the levels of cyclin D1 mRNA (Fig. 4A),which indicated that TNC/AKT could regulate the expression of cyclin D1 at the transcriptional level. Then, we investigated the mechanism by which TNC/AKT transcriptionally upregulates the expression of cyclin D1.
One of the most well studied activators of CCND1 transcription is β-catenin, which could be actived by AKT signalling to inducing G1/S transition. When β-catenin is translocated from the cytoplasm to the nucleus, it forms a complex with the ternary complex factor (TCF) and/or lymphoid enhancer-binding factor (LEF) and stimulates cyclin D1 gene transcription (Fig. 4C). To test the involvement of β-catenin in the TNC/AKT-induced upregulation of cyclin D1, we performed a western blot assay using PANC-1-TNC cells transfected with siAKT or cells treated with LY294002. The results showed that the β-catenin levels were significantly increased in the nuclear fraction of PANC-1-TNC cells but that the total β-catenin expression level was unaltered. However, siAKT and LY294002 significantly decreased the TNC-induced nuclear retention of β-catenin, which suggested that TNC upregulates β-catenin expression in the cell nucleus via AKT signalling. In addition, siTNC inhibited the expression of β-catenin in the nuclear fraction (Fig. 4B).
In agreement with the data described above, a chromatin immunoprecipitation (ChIP) assay confirmed that TNC regulates the binding of β-catenin to the TCF/LEF-binding site in the CCND1 promoter (Fig. 4C). Additionally, the β-catenin-binding activity with respect to the CCND1 promoter was much higher in TNC-overexpression PANC-1 cells than in the vector controls. In contrast, the knockdown of TNC inhibited the binding function of β-catenin. To investigate the effect of TNC on CCND1 promoter activity, dual luciferase activity assays were conducted. As shown in Fig. 4D, TNC transactivated the activity of the CCND1 promoter containing the TCF/LEF binding site, but TNC did not affect the activity of the CCND1 promoter that contained a deleted or a mutant TCF/LEF binding site. In addition, the effects of TNC activity relied on the AKT/β-catenin axis, as evidenced in the restored CCND1 promoter activity after treatment with LY294002 and siβ-catenin. Collectively, these data further supported our model that TNC/AKT transactivates the expression of cyclin D1 via β-catenin.
Figure 3
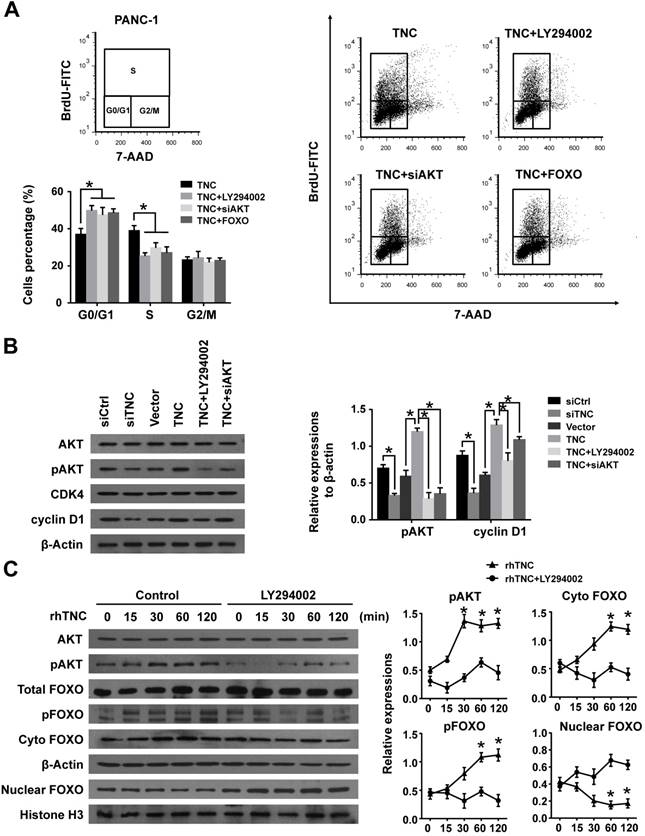
TNC promotes pancreatic cancer cell proliferation via the modulation of AKT/FOXO1 signals. (A) The flow cytometry cell cycle analyses of PANC-1 cell growth were evaluated after treatment with TNC or TNC+LY294002/siAKT/FOXO1. (B) Western blot analysis was performed to determine the expression of AKT, pAKT, CDK4 and cyclin D1 in PANC-1 cells after the indicated treatment. (C) PANC-1 cells were preincubated with LY294002 or DMSO for 1 h before exogenous TNC stimulation. Cells were harvested at the indicated time, and the levels of phosphorylated AKT, FOXO1 and the subcellular expression of pFOXO1 in the cell nuclear and cytoplasmic fractions were analysed by western blot. Data represent the mean ± the s.d. (n = 3, *P < 0.05).
Figure 4
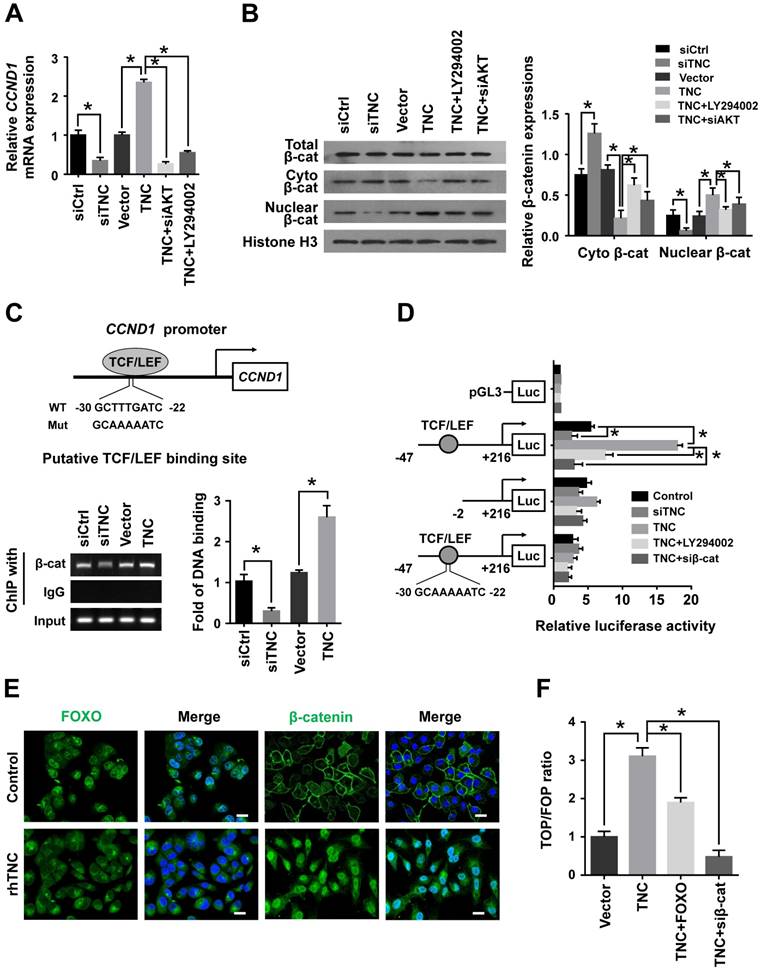
TNC/AKT transactivates cyclin D1 but is dependent on β-catenin. (A) RT-qPCR was used to detect the expression of cyclin D1 in PANC-1 cells with the indicated treatments. (B) The subcellular expression of β-catenin in the cell nuclear and cytoplasmic fractions was analysed by western blot. (C) ChIP of β-catenin bound to the promoter of CCND1. The sequence and position of the TCF/LEF binding site in the CCND1 promoter are shown. PANC-1 cells were transfected with siCtrl, siTNC, vector or TNC plasmid. PCR amplification from the total chromatin (bottom) was used as a positive control, anti-IgG (middle) served as a negative control, and anti-β-catenin (top) showed the interaction between β-catenin and the CCND1 promoter after the indicated treatment. (D) Dual luciferase activity assays were performed using the CCND1 promoter containing the WT, deleted, or mutant TCF/LEF binding site in PANC-1 cells transfected with the indicated treatments. (E) Confocal microscopy images of PANC-1 cells coated with rhTNC and their controls. The green signal represents the distribution of FOXO1 or β-catenin in the cytoplasm and nucleus, and the blue signal represents the nuclear DNA staining by DAPI. Scale bar, 20 μm. (F) TOPflash and FOPflash luciferase expression vectors were co-transfected with TNC or/and FOXO1/siβ-catenin, and the luciferase activity was measured. Data represent the mean ± the s.d. (n = 3, *P < 0.05).
In the above experiment ,we have found that TNC transactivated the expression of cyclin D1 via β-catenin. And phosphorylated AKT induce the translocation of FOXO1 to the cytoplasm. Therefore, we demonstrated that the activities of both FOXO1 and β-catenin were regulated by TNC/AKT activity. Both FOXO1 and β-catenin function as transcription factors in the nucleus, but they play opposite roles in tumour progression [16, 22]. To investigate the differential effects of TNC on the transcriptional activities of FOXO1 and β-catenin, immunofluorescence assays were conducted. As shown in Fig. 4E, TNC induced the nuclear exclusion of pFOXO1, but at the same time, TNC induced β-catenin translocation from the cytoplasm to the nucleus. Previous reports suggested that FOXO1 competes with TCF for interaction with β-catenin, which inhibits TCF/LEF transcriptional activity. To investigate whether FOXO1 functions as an inhibitor of TCF/LEF transcriptional activity, which then affects the regulation of β-catenin-cyclin D1 signal transduction induced by TNC, we performed a TOP/FOP reporter assay. As shown in Fig. 4F, the activation of β-catenin in PANC-1 cells, induced by TNC overexpression, resulted in the activation of TCF-dependent transcription. On the contrary, FOXO1 overexpression significantly downregulated TNC induced TCF transcriptional activity, which indicated that FOXO1 antagonizes TNC/β-catenin signalling. We indicated that FOXO1/β-catenin functions as an AKT downstream substrate plays a vital role in TNC-induced acceleration of G1/S transition. Next, we further detected the mechanism that TNC/FOXO1 function on the G1/S transition.
TNC regulates the cyclin D1-CDK4 complex formation and activity of Rb through regulation of FOXO1/β-catenin/p27Kip1
With the exception of the reduction in cyclin D1 expression, the CDK inhibitor p27Kip1 (CDKN1B), binds to the cyclin D1-CDK4 complex and causes G1 arrest. To date , we have found that TNC regulates the transcriptional activity of FOXO1. And p27Kip1 is one of the transcriptional targets of FOXO1 (Fig. 5A). We speculated that TNC could regulate the binding of FOXO1 to the CDKN1B promoter. As expected, siTNC decreased the binding activity of FOXO1 to the CDKN1B promoter, whereas the overexpression of TNC led to the opposite effect on FOXO1 binding (Fig. 5A). Notably, the expression of p27Kip1 is inverse to the phosphorylation of FOXO1, which is similar to the transcriptional activity of FOXO1. Moreover, the effect of TNC on p27Kip1 could be rescued by an AKT inhibitor and siRNA against AKT/FOXO1, which suggested that TNC regulates the expression of p27Kip1 through the AKT/FOXO1 signalling pathway (Fig. 5B).
The active cyclin D1-CDK4 complex phosphorylates the Rb protein at Ser780, which results in the release of the E2F transcription factor from the Rb-E2F complex. Subsequently, E2F activates its own promoter and its targets such as cyclin A2, cyclin E1 and Myc, which is a rate-limiting event during cell cycle progression through G1 phase [23]. We have showed that β-catenin/cyclin D1 and p27Kip1 are key factors in the modulation of G1/S transition and are regulated by TNC. To better understand the mechanism that TNC promotes the formation of the cyclin D1-CDK4 complex and Rb phosphorylation, we performed an immunoprecipitation assay using anti-CDK4 followed by a western blot with anti-cyclin D1. The results in Fig. 5C indicate that the ectopic expression of TNC enhanced the formation of the cyclin D1-CDK4 complex and Rb phosphorylation. And treatment of cells with LY294002 or transfection of cells with an siβ-catenin/p27Kip1 plasmid rescued the TNC-induced complex formation and the pRb levels. On the contrary, siTNC inhibited the formation of the cyclin D1-CDK4 complex and the phosphorylation of Rb. The results indicated that TNC enhances the formation of the cyclin D1-CDK4 complex via regulation of AKT/β-catenin/p27Kip1 to increase pRb levels (Fig. 5D).
TNC promotes tumourigenicity in vivo
To further determine the potent oncogenic activity of TNC in vivo, PANC-1-Vector and PANC-1-TNC cells were subcutaneously injected into nude mice. Unexpectedly, the tumours became palpable and grew rapidly after 3 weeks in mice inoculated with the PANC-1-TNC xenograft, but grew slowly in mice inoculated with the PANC-1-Vector xenograft (Fig. 6A). Tumours in the former group were significantly larger at 7 weeks than those in the latter group (Fig. 6B). In addition, IHC analyses confirmed that Ki67 expression was positively correlated with TNC expression in xenograft tissues (Fig. 6C). Ki67, which is expressed in the nucleus, is a useful marker for the evaluation of malignant proliferation. At the same time, the percentage of pAKT-positive tumour cells and the nuclear localization of β-catenin were higher in PANC-1-TNC xenograft animals compared with PANC-1-Vector xenograft animals. In contrast, pFOXO1 was localized to the cytoplasm, and p27Kip1 was weakly expressed in PANC-1-TNC xenograft animals, which indicated tumour cell proliferation driven by the regulation of TNC/pFOXO1/p27Kip1 in vivo (Fig. 6C). Taken together, these results suggested that TNC significantly enhances tumour size by promoting G1/S transition in vivo.
Figure 5
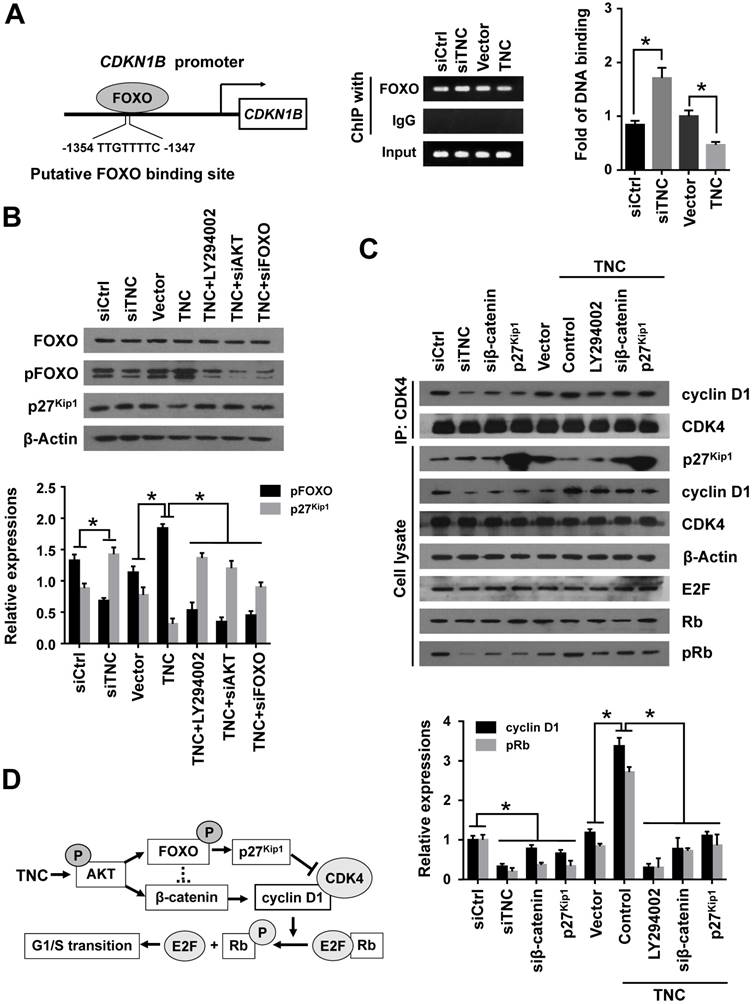
TNC regulates the formation of the cyclin D1-CDK4 complex via β-catenin and p27Kip1. (A) ChIP of FOXO1 bound to the promoter of CDKN1B. The sequence and position of the FOXO1 binding site in the CDKN1B promoter are shown. PANC-1 cells were transfected with siCtrl, siTNC, vector or TNC plasmid. PCR amplification from the total chromatin (bottom) was used as a positive control, anti-IgG (middle) served as a negative control, and anti-FOXO1 (top) showed the interaction between FOXO1 and the CDKN1B promoter after the indicated treatment. (B) Western blot assay was used to detect the expression of FOXO1, pFOXO1 and p27Kip1 in PANC-1 cells with the indicated treatments. (C) PANC-1 cell lysates were immunoprecipitated with anti-CDK4, and immunoblotting was performed with cyclin D1 and CDK4 antibodies. The expression levels of p27Kip1, cyclin D1, CDK4, β-actin, E2F, total Rb and phosphorylated Rb were determined in PANC-1 cell lysates by western blot. (D) A schematic diagram shows the effect of TNC on proliferation via activation of AKT signalling. Data represent the mean ± the s.d. (n = 3, *P < 0.05).
Figure 6
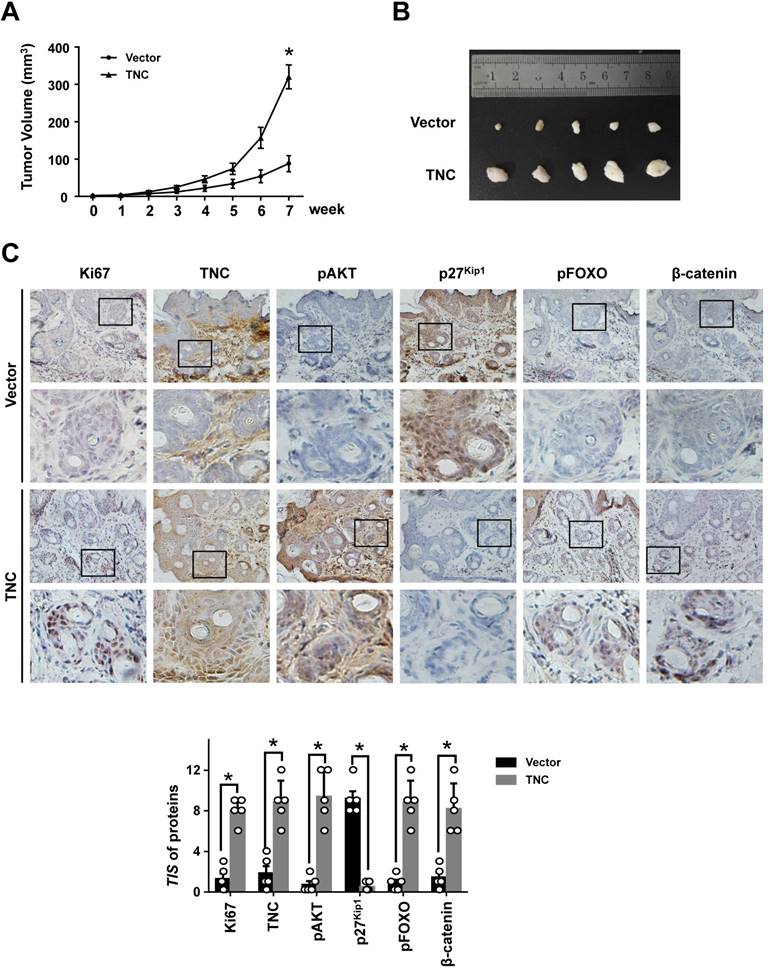
TNC promotes tumourigenicity in vivo. (A) Tumour sizes were measured at the indicated times after PANC-1-Vector and TNC cells were injected into the mice. (B) The photograph shows representative images of tumour size. (C) IHC staining was performed to evaluate Ki67, TNC, pAKT, p27Kip1, pFOXO1 and β-catenin expression in the indicated primary tumours. (original magnification ×40).
Discussion
In this study, we demonstrated that TNC was frequently upregulated in human pancreatic cancer tissues relative to adjacent noncancerous tissues. Moreover, TNC overexpression was correlated with tumour stage in patients with pancreatic cancer. These clinical data indicated that TNC contributes to the malignant progression of pancreatic cancer and may be a useful prognostic biomarker. In addition, increased TNC expression was positively correlated with the upregulation of cyclin D1 in pancreatic cancer tissues compared adjacent pancreatic tissues. Cyclin D1 is a critical regulator that is involved in cell cycle progression from G1 phase into S phase, and thus it contributes to cell proliferation [19, 24]. We demonstrated that TNC promotes the progression of tumour stage via the induction of cell cycle progression in pancreatic cancer.
We further found that the cell cycle-regulating protein cyclin D1/ FOXO1/p27Kip1 was shown to be abnormally expressed in malignant pancreatic cancer tissues, which was consistent with the high expression of TNC in tumours. This indicates the mechanism that TNC might be involved in cell cycle regulation by its action on cyclin D1/FOXO1/p27Kip1. First, we demonstrated that TNC/AKT promoted cell proliferation by accelerating G1/S transition. Previous findings have shown that TNC can activate both the ERK1/2 and AKT molecules [6, 10]. So we treated pancreatic cancer cells with ERK/AKT inhibitor and showed that both of them can inhibited the TNC-induced proliferation, while the effect of AKT was more notable. We further proved that AKT inhibitor LY294002 and siAKT could reverse the effect of TNC on cyclin D1 expression and the promotion of G1/S transition. Our data indicated that AKT mediates the key function of TNC on cell proliferation and G1/S transition in pancreatic cancer.
Moreover, A previous study has shown that FOXO1 acts downstream of active AKT to downregulate p27Kip1, p21Waf1 and Bim at the transcriptional level [15, 25, 26]. We observed that TNC/AKT caused a remarkable pFOXO1 activity and cytoplasm translocation, which indicates that TNC inhibited FOXO1 transcriptional activity. In addition, we showed that TNC reduced p27Kip1 transcription through the promotion of the binding activity of FOXO1 on its promoter, and p27Kip1 overexpression rescued the TNC-induced activities of the cyclin D1-CDK4 complex and Rb. We indicated that FOXO1 plays a critical role in TNC-induced cell G1/S transition. Another study showed that p21Waf1, which has a similar function to p27Kip1, was also downregulated by TNC in an integrin α9-dependent manner during the TNC-induced proliferation of haematopoietic stem and progenitor cells [27]. Further tests might be performed to demonstrate that TNC/AKT signalling drives the FOXO1 transcriptional programme to promote p21Waf1 expression in pancreatic cancer.
Additionally, Several researchers have shown that AKT/β-catenin signalling regulates cyclin D1 expression [11, 12]. In the present study, we proved that TNC/AKT had a significant influence on β-catenin and FOXO1 transcriptional activity, which regulated the expression of cyclin D1 and p27Kip1. Previous findings have provided evidence that β-catenin and FOXO1 share a functional interaction [28, 29]. According to the TOP/FOP analysis, we showed that the activation of β-catenin was induced by TNC overexpression and resulted in the activation of TCF-dependent transcription. However, this effect was impaired by FOXO1 overexpression, which indicated that FOXO1 antagonizes TNC/β-catenin signalling. Taken together, these data show that TNC regulates cyclin D1 and p27Kip1 through the AKT/FOXO1/β-catenin cascade, and then promotes the formation of the cyclin D1-CDK4 complex. The cyclin D1-CDK4 complex inactivates Rb to promote G1/S transition and cell proliferation. Consequently, we indicated the TNC's function on the cell cycle progression was shown to be a multiple form of a biological cascade effect and a coordinated, molecular event. And in the signal transduction cascade, FOXO1 is the vital molecular to promote TNC-induced tumor cell proliferation. Therefore, FOXO1 might be an effective therapeutic agent in patient of pancreatic cancer with high TNC's expression.
In summary, our study found that the expression level of TNC in PDAC tissues might be a potential prognostic biomarker. The present results provide compelling evidence that TNC functions as an active regulator of cell cycle G1/S transition to promote pancreatic cancer proliferation in vitro and in vivo; we also implicated the AKT/FOXO1 signalling axis in this process. These findings improve our understanding of the molecular mechanisms that underlie the activation of TNC and tumour progression and provide a potential target for cancer therapy.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 31370861) and the Tianjin Basic Research Plan Project (No. 13JCZDJC31300).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Linder S, Castanos-Velez E, von Rosen A, Biberfeld P. Immunohistochemical expression of extracellular matrix proteins and adhesion molecules in pancreatic carcinoma. Hepatogastroenterology. 2001;48:1321-7
2. Liu Q, Liao Q, Zhao Y. Chemotherapy and tumor microenvironment of pancreatic cancer. Cancer cell international. 2017;17:68
3. Esposito I, Penzel R, Chaib-Harrireche M, Barcena U, Bergmann F, Riedl S. et al. Tenascin C and annexin II expression in the process of pancreatic carcinogenesis. J Pathol. 2006;208:673-85
4. Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG. et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med. 2011;17:867-74
5. Li M, Peng F, Li G, Fu Y, Huang Y, Chen Z. et al. Proteomic analysis of stromal proteins in different stages of colorectal cancer establishes Tenascin-C as a stromal biomarker for colorectal cancer metastasis. Oncotarget. 2016;7:37226-37
6. Shi M, He X, Wei W, Wang J, Zhang T, Shen X. Tenascin-C induces resistance to apoptosis in pancreatic cancer cell through activation of ERK/NF-kappaB pathway. Apoptosis. 2015;20:843-57
7. Akli S, Zhang XQ, Bondaruk J, Tucker SL, Czerniak PB, Benedict WF. et al. Low molecular weight cyclin E is associated with p27-resistant, high-grade, high-stage and invasive bladder cancer. Cell Cycle. 2012;11:1468-76
8. Ahlin C, Lundgren C, Embretsen-Varro E, Jirstrom K, Blomqvist C, Fjallskog M. High expression of cyclin D1 is associated to high proliferation rate and increased risk of mortality in women with ER-positive but not in ER-negative breast cancers. Breast cancer research and treatment. 2017;164:667-78
9. Meng Y, He L, Guo X, Tang S, Zhao X, Du R. et al. Gankyrin promotes the proliferation of human pancreatic cancer. Cancer Lett. 2010;297:9-17
10. Paron I, Berchtold S, Voros J, Shamarla M, Erkan M, Hofler H. et al. Tenascin-C enhances pancreatic cancer cell growth and motility and affects cell adhesion through activation of the integrin pathway. PLoS One. 2011;6:e21684
11. Roy A, Ansari SA, Das K, Prasad R, Bhattacharya A, Mallik S. et al. Coagulation factor VIIa-mediated protease-activated receptor 2 activation leads to beta-catenin accumulation via the AKT/GSK3beta pathway and contributes to breast cancer progression. The Journal of biological chemistry. 2017;292:13688-701
12. Wang R, Zhang Q, Peng X, Zhou C, Zhong Y, Chen X. et al. Stellettin B Induces G1 Arrest, Apoptosis and Autophagy in Human Non-small Cell Lung Cancer A549 Cells via Blocking PI3K/Akt/mTOR Pathway. Scientific reports. 2016;6:27071
13. Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253-67
14. Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Developmental cell. 2008;14:159-69
15. Boreddy SR, Pramanik KC, Srivastava SK. Pancreatic tumor suppression by benzyl isothiocyanate is associated with inhibition of PI3K/AKT/FOXO pathway. Clin Cancer Res. 2011;17:1784-95
16. Coomans de Brachene A, Demoulin JB. FOXO transcription factors in cancer development and therapy. Cellular and molecular life sciences: CMLS. 2016;73:1159-72
17. Prasad SB, Yadav SS, Das M, Govardhan HB, Pandey LK, Singh S. et al. Down Regulation of FOXO1 Promotes Cell Proliferation in Cervical Cancer. Journal of Cancer. 2014;5:655-62
18. Cai J, Du S, Wang H, Xin B, Wang J, Shen W. et al. Tenascin-C induces migration and invasion through JNK/c-Jun signalling in pancreatic cancer. Oncotarget. 2017;8:74406-22
19. Qie S, Diehl JA. Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med (Berl). 2016;94:1313-26
20. Jang JH, Chung CP. Tenascin-C promotes cell survival by activation of Akt in human chondrosarcoma cell. Cancer Lett. 2005;229:101-5
21. Burgering BM, Kops GJ. Cell cycle and death control: long live Forkheads. Trends in biochemical sciences. 2002;27:352-60
22. Shang S, Hua F, Hu ZW. The regulation of beta-catenin activity and function in cancer: therapeutic opportunities. Oncotarget. 2017;8:33972-89
23. Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nature reviews Molecular cell biology. 2013;14:297-306
24. Sherr CJ. A New Cell-Cycle Target in Cancer - Inhibiting Cyclin D-Dependent Kinases 4 and 6. The New England journal of medicine. 2016;375:1920-3
25. Moeinifard M, Hassan ZM, Fallahian F, Hamzeloo-Moghadam M, Taghikhani M. Britannin induces apoptosis through AKT-FOXO1 pathway in human pancreatic cancer cells. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2017;94:1101-10
26. Zhu J, Li Y, Tian Z, Hua X, Gu J, Li J. et al. ATG7 Overexpression Is Crucial for Tumorigenic Growth of Bladder Cancer In Vitro and In Vivo by Targeting the ETS2/miRNA196b/FOXO1/p27 Axis. Molecular therapy Nucleic acids. 2017;7:299-313
27. Nakamura-Ishizu A, Okuno Y, Omatsu Y, Okabe K, Morimoto J, Uede T. et al. Extracellular matrix protein tenascin-C is required in the bone marrow microenvironment primed for hematopoietic regeneration. Blood. 2012;119:5429-37
28. Iyer S, Ambrogini E, Bartell SM, Han L, Roberson PK, de Cabo R. et al. FOXOs attenuate bone formation by suppressing Wnt signaling. J Clin Invest. 2013;123:3409-19
29. Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181-4
Author contact
![]() Corresponding author: Xiaohong Shen, Department of Pathophysiology, School of Medicine, Nankai University, Tianjin 300071, China. Tel.: +86-22-23501649; Fax: +86-22-23502554; E-mail: zebal2014com
Corresponding author: Xiaohong Shen, Department of Pathophysiology, School of Medicine, Nankai University, Tianjin 300071, China. Tel.: +86-22-23501649; Fax: +86-22-23502554; E-mail: zebal2014com
Citation styles
APA
Cai, J., Lu, W., Du, S., Guo, Z., Wang, H., Wei, W., Shen, X. (2018). Tenascin-C Modulates Cell Cycle Progression to Enhance Tumour Cell Proliferation through AKT/FOXO1 Signalling in Pancreatic Cancer. Journal of Cancer, 9(23), 4449-4462. https://doi.org/10.7150/jca.25926.
ACS
Cai, J.; Lu, W.; Du, S.; Guo, Z.; Wang, H.; Wei, W.; Shen, X. Tenascin-C Modulates Cell Cycle Progression to Enhance Tumour Cell Proliferation through AKT/FOXO1 Signalling in Pancreatic Cancer. J. Cancer 2018, 9 (23), 4449-4462. DOI: 10.7150/jca.25926.
NLM
Cai J, Lu W, Du S, Guo Z, Wang H, Wei W, Shen X. Tenascin-C Modulates Cell Cycle Progression to Enhance Tumour Cell Proliferation through AKT/FOXO1 Signalling in Pancreatic Cancer. J Cancer 2018; 9(23):4449-4462. doi:10.7150/jca.25926. https://www.jcancer.org/v09p4449.htm
CSE
Cai J, Lu W, Du S, Guo Z, Wang H, Wei W, Shen X. 2018. Tenascin-C Modulates Cell Cycle Progression to Enhance Tumour Cell Proliferation through AKT/FOXO1 Signalling in Pancreatic Cancer. J Cancer. 9(23):4449-4462.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.