Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(15):2678-2686. doi:10.7150/jca.25438 This issue Cite
Research Paper
Mutations in NOTCH1 and nucleotide excision repair genes are correlated with prognosis of hepatitis B virus-associated hepatocellular carcinoma
Wen-Hui Su1,2, ![]() , Yuh-Shan Jou3,4,5, Jia-Hao Zhang1, Chun-Ming Ho4,5,6, Dar-In Tai7,
, Yuh-Shan Jou3,4,5, Jia-Hao Zhang1, Chun-Ming Ho4,5,6, Dar-In Tai7, ![]()
1. Department of Biomedical Sciences, Graduate Institute of Biomedical Sciences, College of Medicine, Chang Gung Molecular Medicine Research Center, Chang Gung University, 259 Wen-Hua 1st Road, Kweishan, Taoyuan 333, Taiwan
2. Department of Otolaryngology, Chang Gung Memorial Hospital, Linkou, 5 Fu-Hsing Street, Kweishan, Taoyuan 333, Taiwan
3. Graduate Institute of Life Sciences, National Defense Medical Center, Taipei, Taiwan
4. Institute of Biomedical Sciences, Academia Sinica, 128 Academia Road, Section 2, Nankang, Taipei 115, Taiwan
5. Bioinformatics Program, Taiwan International Graduate Program, Academia Sinica, 128 Academia Road, Section 2, Nankang, Taipei 115, Taiwan
6. Institute of Bioinformatics and Systems Biology, National Chiao Tung University, 1001 University Road, Hsinchu 300, Taiwan
7. Division of Hepatology, Department of Gastroenterology and Hepatology, Chang Gung Memorial Hospital, Linkou, 5 Fu-Hsing Street, Kweishan, Taoyuan 333, Taiwan
Received 2018-2-8; Accepted 2018-5-8; Published 2018-6-23
Citation:
Su WH, Jou YS, Zhang JH, Ho CM, Tai DI. Mutations in NOTCH1 and nucleotide excision repair genes are correlated with prognosis of hepatitis B virus-associated hepatocellular carcinoma. J Cancer 2018; 9(15):2678-2686. doi:10.7150/jca.25438. https://www.jcancer.org/v09p2678.htm
Other stylesAbstract
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the second leading cause of cancer-related mortality because of its poor prognosis. Therefore, identifying targetable genetic mutations and mutational signatures associated with prognosis and treatment strategies are needed. Ultra-deep sequencing of 409 cancer genes using formalin-fixed paraffin-embedded tissue from 33 male patients with hepatitis B virus-associated HCC was performed to identify mutational signatures associated with the prognosis of HCC. A total of 47 genes were found to be mutated in more than 10% of patients. Chromatin remodeling genes were overrepresented in the mutation profile. We found patient survival was associated with mutations in NOTCH1 and the nucleotide excision repair genes which have not been described previously in HCC. From the mutation profile, six patients were eligible for Sorafenib treatment. Among the remaining patients, 7 patients had mutations in genes that are targets for other cancer drugs and 16 patients had mutations in potentially targetable genes. Only one patient carried no potential drug target. We identified mutational signatures associated with the patient survival of HCC. The findings may facilitate identifying subgroups of patients with a poor prognosis as well as potential drug targets for use in personalized strategies for HCC treatment.
Keywords: hepatocellular carcinoma, hepatitis B virus, mutation, prognosis, high-throughput nucleotide sequencing
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the second leading cause of cancer-related mortality [1]. Chemotherapy and radiotherapy are ineffective treatments for HCC. Surgical procedures offer the most effective means to cure HCC; however, most patients who receive a diagnosis of HCC are ineligible for surgery [2]. For targeted therapy, Sorafenib, a multi target tyrosine-kinase receptor inhibitor, is the only FDA-approved first-line drug for HCC treatment [3]. Therefore, new markers for prognosis and treatment strategies are urgently needed.
Hepatitis B virus (HBV) infection is the predominant risk factor in Asian countries, excluding Japan; 63%-77% of all HCC patients have HBV infection [4]. Chronic HBV infection may progress to cirrhosis and genetic alteration, leading to HCC. HBV-associated HCC has a higher rate of chromosomal alteration, which is caused by hepatitis B x (HBx) protein expression, genome integration and subsequent hepatitis-induced DNA damage [5]. Compared with tumors associated with other risk factors, HBV-associated HCC has a higher rate of chromosomal abnormalities, such as a higher frequency of TP53 mutation; lower frequencies of CTNNB1 and TERT promoter mutations; amplification of CCND1, FGF19, and TERT; and HBV integrations in TERT, KMT2B and KMT2D [6-12]. Several studies have described the mutational landscape of HBV-associated HCC [8, 11-14]. Mutations of TP53 are the most prominent characteristics of HBV-associated HCC; these mutations occur in 27%-40% of cases [8, 12, 14-16]. The major mutation sites include the WNT/β-catenin pathway genes (CTNNB1[11%-16%] [8, 12, 15-17], AXIN1[8%-17%] [8, 12, 16], and APC[1%-3%] [8, 12]), SWI/SNF chromosome remodeling complex genes (ARID1A[7%-13%] [8, 12, 14, 16] and ARID2[2%-6%] [8, 12, 15, 16]), and histone methyl transferases (KMT2A[4%], KMT2C[8%], and KMT2D[5%] [8]). However, only a few somatic mutations associated with prognosis have been reported. TP53 mutation has been found to be associated with poor prognosis in patients with HBV-associated HCC [18]. RB1 mutation was also found to be associated with the survival of patients with HCC [8]. Further research is still required to discover more markers associated with the prognosis of HCC.
Although the exome sequencing and whole-genome sequencing approach can provide a complete mutational landscape, the detection sensitivity might be limited by the low sequencing depth of those approaches. The ultra-deep targeted sequencing of candidate genes has been shown to provide greater detection sensitivity for mutated genes. The use of formalin-fixed paraffin-embedded (FFPE) samples with deep sequencing of target genes provides a dependable method for identifying potential druggable targets for patients who are ineligible for surgery. Because Sorafenib is the only approved drug for HCC systemic therapy, identifying additional targetable genes for future targeted therapy would be useful. In this study, we explored the relationship between the mutations of 409 known cancer genes and the clinicopathological characteristics of HCC through ultra-deep sequencing FFPE tissues from 33 male patients with HBV-associated HCC. We identified previously unknown prognosis-related gene and pathway. The mutation profile of each patient was used to predict potentially druggable targets that might be applicable in personalized HCC treatment.
Materials and Methods
Patients and tissue samples
We retrospectively analyzed tumor samples from 33 male patients with HBV-associated HCC, who received surgical resection and treatment at Chang Gung Memorial Hospital between 2006 and 2008. The patients had not been previously treated and had no proven metastatic disease at the time of diagnosis. The study protocol was approved by the Institutional Review Broad of Chang Gung Memorial Hospital (CGMH 99-3817B and 100-3409C). FFPE tumor specimens were obtained from the Tissue Bank, Chang Gung Memorial Hospital, Linkou. Signed consent for research use of resected tissue was obtained from each patient before tissue collection. All samples used for DNA extraction must contained a minimum of 15% DNA derived from tumor cells. For each tumor, one roll of a 10-µm FFPE section was used for genomic DNA extraction and library preparation.
Sequencing and mutation detection
Eighty nanograms of genomic DNA was amplified using four pools of 15,992 primer pairs (Ion AmpliSeq Comprehensive Cancer Panel, Life Technologies) to target all coding exons of 409 most frequently mutated cancer related genes selected from Wellcome Trust Sanger Institute Cancer Gene Census. Sequencing was performed using an Ion Proton sequencer equipped with an Ion PI chip (Life Technologies) following the manufacturer's protocol. Raw reads generated by the sequencer were mapped to the hg19 reference genome. In this study, only tumor tissues were analyzed without comparing to paired non-tumor tissue. The data was filtered using dbSNP and 1000 genome project data to remove possible germline mutations. Variant Effect Predictor (v.74) was used to annotate every variant. Variant coverage lower than 25 or variant frequencies lower than 5% were excluded from the subsequent analysis. Druggable genes were selected by searching drug-gene interactions in the Drug Gene Interaction Database (DGIdb, http://dgidb.genome.wustl.edu/) [19]. Mutator phenotype were defined as 1) >1,000 mutations per tumor, 2) a higher number of frameshift mutations, and 3) mutations in DNA mismatch repair genes according to the feature of cancer mutator [20] and microsatellite instability (MSI) phenotype [21].
The Cancer Genome Atlas data
The Cancer Genome Atlas Liver Hepatocellular Carcinoma (TCGA-LIHC) data were downloaded from cBioPortal [22, 23]. HCC mutation data of 75 HBV-positive Asian patients and their survival data were obtained from TCGA-LIHC for statistical analysis.
Statistical analysis
Only non-synonymous mutations identified were included for statistical analysis. Samples classified as unknown for any variable were excluded. Overall survival was determined using the Kaplan-Meier method, and the survival curves were compared using the log-rank test. Independent variables were first analyzed by univariate methods. A multivariate analysis of different clinical pathological variables was performed using the Cox proportional hazard model. The primary endpoint, overall survival, was defined as the period in months from the date of surgery to the date of death from any causes or the date of the last follow-up. The hazard ratio (HR) along with 95% confidence interval (CI), were assessed for each factor. All tests were two-sided and p < 0.05 was considered statistically significant. Statistical analyses were performed using IBM SPSS Statistics version 1.0.0.355 (IBM Corp.) and GraphPad Prism 6.07 statistical software (GraphPad Software, Inc.).
Results
Clinical characteristics of HCC specimens
In this study, we included 33 patients with histology proven HCC who received tumor resection between 2006 and 2008. Complete clinical data for all 33 tumor samples are listed in Table 1. All patients were Han Chinese males with chronic HBV infection. The median follow-up of participants was 97.0 months (range 13.5-120.6).
Spectrum of mutations in mutator phenotype
In the 33 HBV-positive HCC cases, we searched for mutations by deep sequencing of all coding exons of 409 cancer-related genes (Supplementary Table S1). In total, we discovered 14,957 polymorphisms and 19,869 mutations with an average depth of 820.35 time per locus in total samples (Supplementary Table S2). Of the 33 samples, three were classified as mutator phenotype because they exhibited higher mutation counts (>1,000 mutations per tumor) (Supplementary Figure S1), a higher number of frameshift mutations (Supplementary Figure S2), and mutations in DNA mismatch repair genes (Supplementary Figure S3A) than the other samples did. Although it is not statistically significant, patients with the mutator phenotype had better prognosis than the non-mutators (Table 1 and Figure 1). Mutator cases were excluded from further mutation analysis because of their distinctive mutation patterns. Among the 30 patients with the non-mutator phenotype, we found a total of 579 nonsynonymous mutations with an average depth of 895.69 and an average variant frequency of 21.78%. On average, 16.17 nonsynonymous mutated genes were found in each non-mutator patient (Supplementary Table S2) and most of them (78%, 452/579) was deleterious or COSMID mutations (Supplementary Table S3).
Mutation profile of HBV-associated HCC from TCGA
The gene mutation profiles of 75 HBV-positive Asian patients were obtained from TCGA-LIHC dataset. There are no mutator phenotype patients in TCGA dataset. Among 175 genes with nonsynonymous mutations, only TP53 (26/75, 35%) and CYP2C19 (20/75, 27%) had nonsynonymous mutations greater than 10% in total series. There were no significant associations were found between mutations and overall survival in the TCGA series.
Table 1
Clinical characteristics of patients included in the present study
| Characteristics | Mutator | Non-mutator | Total |
|---|---|---|---|
| Patients | 3 | 30 | 33 |
| Age at surgery (Median, Range ) | 53 (49-81) | 55 (28-79) | 54 (28-81) |
| <50 years | 1 (33%) | 9 (30%) | 10 (30%) |
| >=50 years | 2 (67%) | 21 (70%) | 23 (70%) |
| Tumor size (Median, Range) | 4.8 (1.1-5.0) | 3.5 (1.6-13.0) | 3.5 (1.1-13.0) |
| <5cm | 2 (67%) | 21 (70%) | 23 (70%) |
| >=5cm | 1 (33%) | 9 (30%) | 10 (30%) |
| Vascular invasion | |||
| Yes | 0 (0%) | 8 (27%) | 8 (24%) |
| No | 3 (100%) | 22(73%) | 25 (76%) |
| AFP level (Median, Range) | 3.6 (3-118) | 7.5 (2-10000) | 7(2-10000) |
| <10 | 2 (67%) | 16 (53%) | 18 (55%) |
| >=10 | 1 (33%) | 14 (47%) | 15 (45%) |
| Cirrhosis | |||
| Yes | 1 (33%) | 13 (43%) | 14 (42%) |
| No | 2 (67%) | 17 (57%) | 19 (58%) |
| Edmondson-Steiner grade | |||
| 1, 2 | 3 (100%) | 14 (47%) | 17 (52%) |
| 3, 4 | 0 (0%) | 16 (53%) | 16 (48%) |
| UICC Stage | |||
| I | 3 (100%) | 22 (73%) | 25 (76%) |
| II, III | 0 (0%) | 8 (27%) | 8 (24%) |
| Tumor number | |||
| Single | 3 (100%) | 28 (93%) | 31 (94%) |
| Multiple | 0 (0%) | 2 (7%) | 2 (6%) |
| Metastasis | |||
| Yes | 1 (33%) | 4 (13%) | 5 (15%) |
| No | 2 (67%) | 26(87%) | 28 (85%) |
| Recurrence | |||
| Yes | 0 (0%) | 4 (13%) | 4 (12%) |
| No | 3 (100%) | 26 (87%) | 29 (88%) |
Figure 1
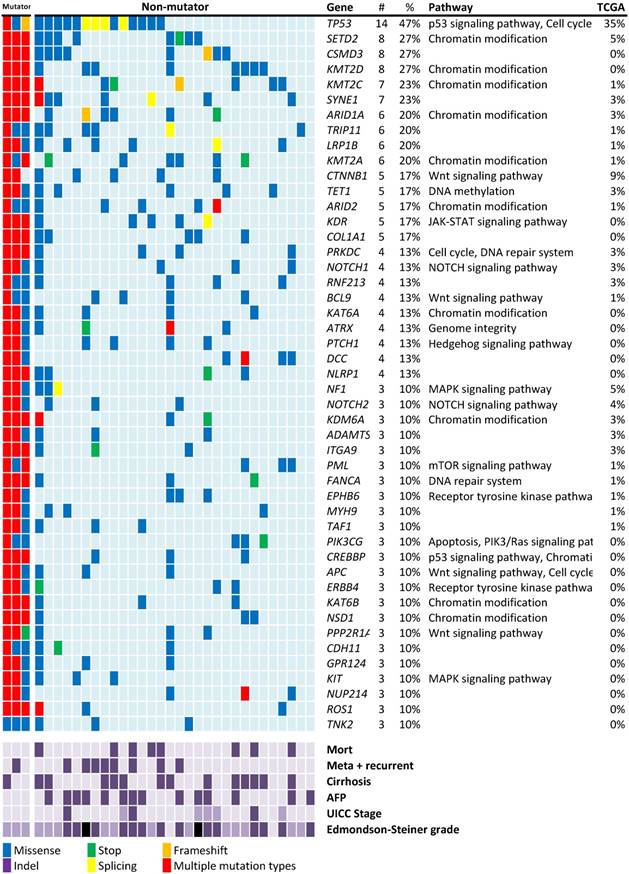
Recurrent mutations discovered in patients with HBV-associated HCC. Individual mutations in patient samples, color-coded according to the type of mutation. If more than one mutation types were detected in the one gene from the same patient sample, the mutation type will be described as “multiple mutation types”. Mortality, meta+recurrent, cirrhosis and AFP > 10ng/mL are represented in dark purple. UICC stages I, II, and III are represented using light, medium, and dark purple. For Edmondson-Steiner grade 1, 2, 3, and 4 are represented using light purple, medium purple, dark purple, and black, respectively. Numbers (#) and percentages (%) represent the fraction of 30 non-mutator tumors with at least 10% mutation in the specified gene. Mutation percentages of 75 Asian patients with HBV-associated HCC from the TCGA-LIHC dataset are also listed.
Mutated genes in the HCC cohort
Mutation screening in the rest of 30 non-mutators revealed 47 genes with nonsynonymous mutations in more than 10% of the patients with HCC (Figure 1). The mutation rates were higher than those reported in previous studies and the TCGA data of HBV-associated HCC in the Asian population.
The most frequently mutated gene observed in this study was the well-known tumor suppressor gene TP53 (47%; Figure 1). TP53 mutations have been linked to HBV-associated HCC [24]. Among the 16 nonsynonymous TP53 mutations identified in 14 patients of this study, only one aflatoxin B exposure-related R249W mutation was found. The lower mutation rate of CTNNB1 (17%) in this study is consistent with the fact that this gene is less frequently mutated in HBV-associated HCC [17].
In addition to known HCC mutations, mutations in several histone modification genes, particularly histone lysine methyltransferases that target histone H3 to activate gene expression were discovered. Four histone lysine methyltransferases, namely SETD2, KMT2D, KMT2C, and KMT2A, were respectively ranked as the second (27%), fourth (27%), fifth (23%), and tenth (20%) most frequently mutated genes in this study (Figure 1). In total, 20 of the 30 patients (67%) carried at least one of these mutations. NOTCH1 mutations were found in 4 patients (13%). Other major mutation sites include the SWI/SNF chromosome remodeling complex genes (ARID1A: 6/30, 20%), ARID2 (5/30, 17%) and APC (3/30, 10%) are common mutation genes in this study.
Mutations associated with patient survival in the HCC
Kaplan-Meier analysis indicated that mutations in NOTCH1 were associated with significantly shorter overall survival (log-rank test; P = 0.0003) (Figure 2A). Cases with mutations in nucleotide excision repair genes had poor overall survival (log-rank test; P =0.0024) (Figure 2B). However, both associations did not remain significant after multiple testing corrections (P < 0.00023 for 205 mutated genes and 10 pathways tested). The larger tumor size as well as mutations in NOTCH1 and nucleotide excision repair genes were also significantly associated with short survival period using univariate analysis (Table 2). In multivariate analysis, the NOTCH1 and nucleotide excision repair genes mutations were also independently associated with an increased risk of mortality (Table 2).
Mutations in potential targetable genes
For the only FDA-approved HCC target-therapy drug, Sorafenib, 6 of the 30 patients carried its potential target genes (KIT, PDGFRA, and PDGFRB). Among the remaining 24 patients, 7 of them had potential target genes of FDA-approved drugs for cancers other than HCC, and 16 of them had potential targetable genes, which were obtained by searching Drug-Gene Interaction Database (DGIdb). On average, 7.24 targetable genes were found in 29 of the 30 patients (Table 3).
Figure 2
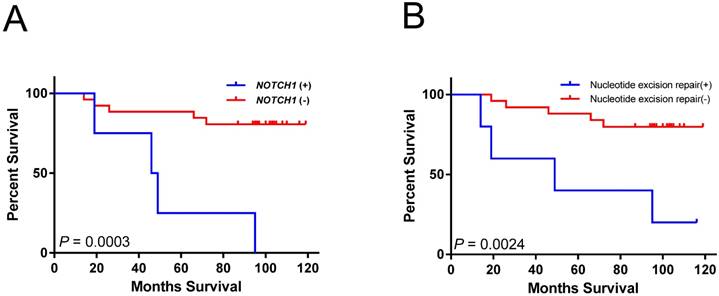
Overall survival associated with gene or pathway mutations. (A) NOTCH1 mutations are significantly associated with reduced overall survival. (B) mutations in nucleotide excision repair genes are significantly associated with reduced overall survival.
Table 2
Univariate and multivariate analysis for the selected mutations and clinical features of HCC
| Univariate analysis | Multivariate analysis | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| NOTCH1 | Nucleotide excision repair | ||||||||||
| HR | 95% CI | P-valuea | HR | 95% CI | P-valueb | HR | 95% CI | P-valueb | |||
| NOTCH1 | 8.20 | (2.11 - 31.74) | 0.002 | 12.3 | (1.27 - 113.22) | 0.030 | |||||
| Nucleotide excision repair | 6.00 | (1.60 - 22.54) | 0.008 | 11.10 | (1.40 - 88.17) | 0.023 | |||||
| Age | 1.04 | (0.99 - 1.10) | 0.151 | 1.07 | (0.99 - 1.15) | 0.066 | 1.11 | (1.02 - 1.21) | 0.014 | ||
| Tumor size | 3.79 | (1.01 - 14.22) | 0.049 | 4.95 | (0.67 - 36.78) | 0.118 | 11.42 | (1.23 - 106.34) | 0.032 | ||
| Cirrhosis | 3.17 | (0.79 - 12.74) | 0.105 | 1.08 | (0.11 - 11.03) | 0.943 | 4.38 | (0.53 - 36.47) | 0.172 | ||
| AFP | 0.97 | (0.26 - 3.62) | 0.965 | 1.23 | (0.16 - 9.64) | 0.843 | 3.13 | (0.35 - 28.34) | 0.311 | ||
| UICC Stage | 1.67 | (0.42 - 6.70) | 0.469 | 4.48 | (0.02 - 971.49) | 0.585 | 1.52 | (0.00 - 7669.56) | 0.924 | ||
| E-S stage | 0.74 | (0.20 - 2.76) | 0.657 | 0.65 | (0.05 - 8.07) | 0.738 | 2.65 | (0.15 - 47.60) | 0.509 | ||
| Vascular invasion | 1.67 | (0.42 - 6.70) | 0.469 | 1.19 | (0.00 - 340.44) | 0.951 | 0.20 | (0.00 - 1271.86) | 0.712 | ||
P-values obtained from univariatea and multivariateb Cox proportional hazards model are shown
Table 3
Patients with mutations of potential drug target genes
| Patient | Druggable genesa (drug example) | Tragetable genesb | Variant frequenciesc |
|---|---|---|---|
| B00088 | PDGFRA (Sorafenib) | PDGFRA, KDR, NOTCH2, NTRK1, PRKDC | 5.9% - 56.2% |
| B00099 | PDGFRA (Sorafenib) | PDGFRA, GRM8, KDR, MAP3K7, NLRP1, PTCH1, SDHB | 5.0% - 38.7% |
| B00120 | PDGFRB (Sorafenib),ROS1(Entrectinib) | PDGFRB, ROS1, APC, GNAS, GUCY1A2, MYC, PIK3C2B, PTCH1, RUNX1, TP53 | 42.3% - 93.0% |
| B00101 | KIT (Sorafenib) | KIT, CTNNB1, HSP90AA1, PTCH1, TP53 | 13.8% - 77.8% |
| B00118 | KIT (Sorafenib),RET(Regorafenib) | KIT, RET, AURKB, CTNNB1, IKBKB, RUNX1, TP53 | 21.3% - 62.6% |
| B00093 | KIT (Sorafenib), BTK (Ibrutinib), ERBB2 (Trastuzumab), FGFR1 (Ponatinib), FGFR2 (Ponatinib), FLT3 (Sunitinib), RET (Regorafenib), ROS1 (Entrectinib) | KIT, BTK, ERBB2, FGFR1, FGFR2, FLT3, RET, ROS1, APC, ATM, ATR, BCR, BIRC2, CBL, CCNE1, CDH2, CDH5, CDK12, COL1A1, CRBN, CTNNB1, EP300, EPHB4, ERBB4, FLT1, FLT4, IGF2R, IKBKB, IL7R, ITGB3, JAK1, JAK2, JAK3, KDM5C, KDR, LCK, LRP1B, MMP2, MPL, NLRP1, NOTCH1, NTRK1, NTRK3, PIK3R2, PPP2R1A, PRKDC, PTPRD, SYK, TNK2, TP53 | 5.0% - 58.8% |
| B00091 | FGFR3 (Ponatinib) | FGFR3, KDR, TP53 | 17.4% - 38.7% |
| B00095 | FGFR1 (Ponatinib) | FGFR1, COL1A1, TNK2 | 25.7% - 36.1% |
| B00098 | EGFR (Gefitinib) | EGFR, COL1A1, CREBBP, NLRP1, PIK3CG | 24.3% - 49.0% |
| B00105 | ERBB2 (Trastuzumab) | ERBB2, COL1A1, FLT1, JAK3, NFKB2, NLRP1, NOTCH2, NTRK3, PIK3R1, TP53 | 5.6% - 60.6% |
| B00106 | FGFR2 (Ponatinib) | FGFR2, APC, AR, CREBBP, DNMT3A, DPYD, ESR1, GRM8, IGF2R, MAP2K1, MTR, NCOA1, PARP1, PIK3CB, PPP2R1A, PTCH1, RRM1, SRC, UGT1A1 | 5.0% - 17.3% |
| B00121 | ROS1 (Entrectinib) | ROS1, CYP2D6, JAK1, LRP1B, MPL, NF2 | 5.1% - 52.5% |
| B00126 | ALK (Ibrutinib) | ALK, CTNNB1, NOTCH1, TP53 | 8.1% - 47.2% |
| B00090 | CTNNB1, ERBB4, LRP1B, PIK3CA, TSC1 | 13.4% - 49.5% | |
| B00092 | FLT4 | 53.6% | |
| B00096 | ERCC2, NOTCH1, PRKDC | 5.4% - 61.3% | |
| B00097 | HSP90AB1, PDGFB, TP53 | 9.8% - 47.7% | |
| B00100 | ATM, ERBB4, IGF1R, MTRR, NOTCH1, PIK3CG, SMARCA4, UGT1A1 | 22.6% - 50.4% | |
| B00102 | COL1A1, KEAP1, PIK3CA | 5.1% - 49.5% | |
| B00103 | NF1, TP53 | 15.6% - 22.9% | |
| B00107 | PIK3C2B | 47.7% | |
| B00108 | CSF1R, ERCC5, LRP1B, TP53 | 21.2% - 59.7% | |
| B00109 | CRBN, PRKDC, TP53 | 47.4% - 57.6% | |
| B00112 | KDR, LRP1B, TP53 | 7.9% - 61.0% | |
| B00119 | CREBBP, GNAS, KDM5C, LRP1B, NOTCH2, RUNX1, SDHB, TNK2, TP53 | 5.4% - 53.9% | |
| B00122 | CDKN2A, EZH2, PARP1, TP53 | 9.4% - 45.2% | |
| B00123 | BIRC2, KEAP1 | 7.5% - 8.7% | |
| B00124 | AR, MAP2K2, PDGFB, PIK3C2B, PIK3CG, TSC1 | 5.2% - 52.5% | |
| B00125 | HSP90AA1 | 7.5% |
a. Target genes of FDA approved drugs. b. Target genes from searching drug-gene interactions in The Drug Gene Interaction Database (DGIdb, http://dgidb.genome.wustl.edu/). c. Variant frequency was calculated by dividing the number of variant reads by the total number of sequence reads at a given position.
Discussion
To investigate potential prognosis markers for HBV-associated HCC, we performed ultra-deep sequencing in 409 potential cancer genes from FFPE samples. We found that mutations in NOTCH1 and nucleotide excision repair genes in HBV-associated HCC.
TP53 was reported to be the most frequently mutated tumor suppressor gene (27-40%) in previous studies [8, 10, 12, 15, 18]. The mutation rate of TP53 in this study (47%) was higher than that in the TCGA dataset (35%) and previous study (32%) [8] in HBV-associated HCC. Other major mutation sites include the WNT/β-catenin pathway genes (CTNNB1,17%), APC (10%), SWI/SNF chromosome remodeling complex genes (ARID1A,20%, ARID2, 17%) and histone lysine methyl transferases (KMT2A, 20% ) are generally higher in this study than others [8, 10, 12, 15, 17, 18]) This study was done with ultra-deep sequencing. Therefore, the percentages of mutated genes were considerably higher than those reported in previous studies.
SETD2 was highly mutated (27% (8/30) in this study (Figure 1). This gene has not been extensive studied in HCC. Mutations in SETD2 are uncommon in other cancers. A low SETD2 mutation frequency was found in patients with HCC (3%) [8, 24] and HBV-associated HCC in the TCGA dataset (5%) [22]. SETD2 can activate expression of other genes through methylation in lysine-36 of histone H3 [25]. Depletion of SETD2 promotes DNA double-strand breaks and tumorigenesis [26]. SETD2 can interact with TP53 and selectively regulate its downstream genes [27]. Inactivating mutations of SETD2 in the SET catalytic domain and SRI interaction domain are critical events in the cancer progression of clear cell renal cell carcinomas [28]. SETD2 deletion may impair STAT1 interferon mediated immunity and enhance HBV replication [29]. The prolong HBV replication will increase risk of hepatocarcinogenesis.
Mutations in CSMD3 were found in 27% (8/30) in this series. A high mutation rate and low expression level of CSMD3 have been associated with lung cancer and has been proposed to have a tumor suppressor function [30]. CSMD3 has also been associated with the HBV pre-S/S gene in transgenic mice. CSMD3 mutation has only been described in HBV-associated HCC. Based on the findings of the CSMD3 mutation in our study and a previous Exome-Seq study (4%) [10] in HBV-associated HCC, CSMD3 mutations might be specific to HBV-associated HCC.
Mutations in NOTCH1 and nucleotide excision repair genes were associated with patient survival. These associations have not previously highlighted in HCC. Notch signaling is critical for cell proliferation, differentiation, and development [31]. Compared with the 13% (4/30) NOTCH1 mutation rate in our study (Figure 1), the mutation rate of NOTCH1 is relatively low (<5%) in the TCGA-LIHC dataset. The Notch pathway can be tumor suppressive or protooncogenetic in hepatic tumorigenesis. The tumor suppressor role of NOTCH1 in HCC is generally through crosstalk with other signaling pathways [32, 33]. Patients with HCC with higher expression levels of NOTCH1 gene are associated with better survival [33]. NOTCH1 could be oncogenic through the activation of the EMT pathway and could promote HCC progression [34]. The association of NOTCH1 mutation with poor prognosis in our study (Figure 2A and Table 2) could have resulted from disrupting the tumor suppressor role of NOTCH1. Because all the NOTCH1 mutations found in our study were located in the extracellular domain, those mutations could possibly be the gain-of-function mutations of NOTCH1, which activate NOTCH1 function by disrupting the formation of NOTCH1 heterodimers [35].
Present study show that mutations in nucleotide excision repair genes (DDB2, ERCC2, ERCC4, and ERCC5), are significantly correlated with poor survival in patients with HBV-associated HCC (Figure 2B and Table 2). DNA repair genes are responsible for protection against genomic instability and are considered to behave similar to tumor suppressor genes [36]. Their functions can be lost through somatic mutation and epigenetic silencing during the course of tumor progression [36, 37]. The nucleotide excision repair genes constitute the main DNA repair system responsible for defense against lesions due to radiation, alkylating agents, and viruses. The accumulation of mutations caused by defective DNA repair can lead to hepatic carcinogenesis and contribute to HCC progression [38]. Although the DNA repair genes are rarely mutated in HCC, polymorphism and expression alteration of DNA repair genes have been associated with hepatocarcinogenesis [38]. DNA repair gene expression levels are associated with HCC overall survival in the TCGA dataset [39]. Polymorphisms in XRCC1 and ERCC2 are also significantly associated with patient survival in HCC [40, 41]. Multivariate analysis also indicated that SNPs on XRCC1, ERCC5, and XRCC4 have a significant effect on the survival of patients with HBV-associated HCC [42]. By contrast, numerous studies has correlated the polymorphisms of nucleotide excision repair genes with chemotherapy resistance, because the nucleotide excision repair process is responsible for the elimination of DNA damage caused by carcinogens [38]. Further research on the prediction value of mutations in DNA repair genes in HCC prognosis or chemotherapy resistance is warranted.
In this study, three patients (10%) were identified as having the mutator phenotype and were excluded from the mutation profile analysis. The mutator phenotype could be associated with the MSI phenotype, frequently found in cirrhosis-associated HBV infection [38]. MSI has been implicated in the pathogenesis of several gastroinstestinal cancers [43] and could be a target for immunotherapy [44]. However, studies have indicated that the MSI phenotype is rarely observed in Chinese HCC tumors [45]. Further investigation is necessary to elucidate the relationship between mutator and MSI phenotypes in HBV-associated HCC.
From our study, six patients who carried RAF/MET/ERK signaling pathway mutations might benefit from Sorafenib treatment (Table 3). Another 7 patients carried mutations in target genes of FDA approved drugs against other cancers. Among the remaining patients, 16 patients carried mutations in potentially targetable genes in the DGIdb, leaving only one patient without any potential target genes. Although most of the drugs were not applicable to HCC treatment, the technology used here has potential for applications aimed at discovering new therapies for HCC.
The non-synonymous mutations identified in this study were identified in the absence of their corresponding normal tissue controls and therefore could be a mixture of somatic and germline mutations. Polymorphisms and synonymous mutations were excluded from statistical analysis. For all the non-synonymous mutations identified, only 2.3% (309/13,131) were with variant frequency >40% which could be an indication of germline mutations. Therefore, the higher mutation rates discovered might mostly due to the higher sequencing depth used in the present study. Both germline and somatic non-synonymous mutations were considered with deleterious effect to the biological functions of mutated gene. The association between non-synonymous mutations and clinicopathological features could still be valid, regardless of somatic or germline origin.
In conclusion, this study provides a comprehensive analysis of gene mutations and clinicopathological features in patients with HBV-associated HCC. Our data suggest that poor survival is associated with mutations in NOTCH1 and the nucleotide excision repair genes. Since NOTCH1 can be tumor suppressive or oncogenic in hepatocarcinogenesis, further exploration of the function of NOTCH1 mutations in HCC could be important. On the other hand, several studies have suggested that polymorphisms in nucleotide excision repair genes could be prediction markers for chemotherapy resistance in many cancers. Therefore, further exploration of the association between mutations in nucleotide excision repair genes and chemotherapy resistance in HCC is necessary. Although statistically significant results were obtained in our study, our findings are based on relatively small numbers and are limited by the small number of patients included in this study. Studies with a larger sample size are required.
Abbreviations
HCC: hepatocellular carcinoma
HBV: hepatitis B virus
FFPE: formalin-fixed paraffin-embedded
HR: hazard ratio
CI: confidence interval
MSI: microsatellite instability
TCGA-LIHC: The Cancer Genome Atlas Liver Hepatocellular Carcinoma
DGIdb: Drug Gene Interaction Database
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The results published here are in part based upon data generated by The Cancer Genome Atlas Research Network: http://cancergenome.nih.gov/. The authors wish to thank Tissue Bank, Chang Gung Memorial Hospital, Linkou for providing the study materials. This work was supported by the Ministry of Education (funding granted to Chang Gung University), the Ministry of Science and Technology (NSC 100-2320-B-182-005, MOST104-2314-B-182-033-MY3), and Chang Gung Memorial Hospital (BMRPC18, CMRPD3D0121, CMRPD3D0122, CMRPD3D0123, CMRPG3E1121), Taiwan. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ethics Committee Approval and Patient Consent
The study protocol was approved by the Institutional Review Broad of Chang Gung Memorial Hospital (CGMH 99-3817B and 100-3409C). FFPE tumor specimens were obtained from the Tissue Bank, Chang Gung Memorial Hospital, Linkou. Signed consent for research use of resected tissue was obtained from each patient before tissue collection.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-86
2. Brito AF, Abrantes AM, Tralhao JG, Botelho MF. Targeting Hepatocellular Carcinoma: What did we Discover so Far? Oncol Rev. 2016;10:302
3. Ohri N, Kaubisch A, Garg M, Guha C. Targeted Therapy for Hepatocellular Carcinoma. Semin Radiat Oncol. 2016;26:338-43
4. Park JW, Chen M, Colombo M, Roberts LR, Schwartz M, Chen PJ. et al. Global patterns of hepatocellular carcinoma management from diagnosis to death: the BRIDGE Study. Liver Int. 2015;35:2155-66
5. McKillop IH, Moran DM, Jin X, Koniaris LG. Molecular pathogenesis of hepatocellular carcinoma. J Surg Res. 2006;136:125-35
6. Neuveut C, Wei Y, Buendia MA. Mechanisms of HBV-related hepatocarcinogenesis. J Hepatol. 2010;52:594-604
7. Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol. 2016;64:S84-101
8. Ahn SM, Jang SJ, Shim JH, Kim D, Hong SM, Sung CO. et al. Genomic portrait of resectable hepatocellular carcinomas: implications of RB1 and FGF19 aberrations for patient stratification. Hepatology. 2014;60:1972-82
9. Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z. et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422-33
10. Huang J, Deng Q, Wang Q, Li KY, Dai JH, Li N. et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat Genet. 2012;44:1117-21
11. Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M. et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267-73
12. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB. et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694-8
13. Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z. et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422-33
14. Huang A, Zhang X, Zhou SL, Cao Y, Huang XW, Fan J. et al. Plasma Circulating Cell-free DNA Integrity as a Promising Biomarker for Diagnosis and Surveillance in Patients with Hepatocellular Carcinoma. J Cancer. 2016;7:1798-803
15. Li M, Zhao H, Zhang X, Wood LD, Anders RA, Choti MA. et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet. 2011;43:828-9
16. Amaddeo G, Cao Q, Ladeiro Y, Imbeaud S, Nault JC, Jaoui D. et al. Integration of tumour and viral genomic characterizations in HBV-related hepatocellular carcinomas. Gut. 2015;64:820-9
17. Pezzuto F, Izzo F, Buonaguro L, Annunziata C, Tatangelo F, Botti G. et al. Tumor specific mutations in TERT promoter and CTNNB1 gene in hepatitis B and hepatitis C related hepatocellular carcinoma. Oncotarget. 2016;7:54253-62
18. Amaddeo G, Cao Q, Ladeiro Y, Imbeaud S, Nault JC, Jaoui D. et al. Integration of tumour and viral genomic characterizations in HBV-related hepatocellular carcinomas. Gut. 2014;64:820-9
19. Griffith M, Griffith OL, Coffman AC, Weible JV, McMichael JF, Spies NC. et al. DGIdb: mining the druggable genome. Nat Methods. 2013;10:1209-10
20. Loeb LA. Human cancers express mutator phenotypes: origin, consequences and targeting. Nat Rev Cancer. 2011;11:450-7
21. Roberts SA, Gordenin DA. Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer. 2015;14:786-800
22. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
23. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
24. Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S. et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505-11
25. Li J, Duns G, Westers H, Sijmons R, van den Berg A, Kok K. SETD2: an epigenetic modifier with tumor suppressor functionality. Oncotarget. 2016;7:50719-34
26. Pfister SX, Ahrabi S, Zalmas LP, Sarkar S, Aymard F, Bachrati CZ. et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014;7:2006-18
27. Xie P, Tian C, An L, Nie J, Lu K, Xing G. et al. Histone methyltransferase protein SETD2 interacts with p53 and selectively regulates its downstream genes. Cell Signal. 2008;20:1671-8
28. Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A. et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360-3
29. Chen K, Liu J, Liu S, Xia M, Zhang X, Han D. et al. Methyltransferase SETD2-Mediated Methylation of STAT1 Is Critical for Interferon Antiviral Activity. Cell. 2017;170:492-506 e14
30. Liu P, Morrison C, Wang L, Xiong D, Vedell P, Cui P. et al. Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis. 2012;33:1270-6
31. Yuan X, Wu H, Xu H, Xiong H, Chu Q, Yu S. et al. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett. 2015;369:20-7
32. Qi R, An H, Yu Y, Zhang M, Liu S, Xu H. et al. Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res. 2003;63:8323-9
33. Viatour P, Ehmer U, Saddic LA, Dorrell C, Andersen JB, Lin C. et al. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J Exp Med. 2011;208:1963-76
34. Jue C, Lin C, Zhisheng Z, Yayun Q, Feng J, Min Z. et al. Notch1 promotes vasculogenic mimicry in hepatocellular carcinoma by inducing EMT signaling. Oncotarget. 2017;8:2501-13
35. Rand MD, Grimm LM, Artavanis-Tsakonas S, Patriub V, Blacklow SC, Sklar J. et al. Calcium depletion dissociates and activates heterodimeric notch receptors. Mol Cell Biol. 2000;20:1825-35
36. Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12:801-17
37. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
38. Yang SF, Chang CW, Wei RJ, Shiue YL, Wang SN, Yeh YT. Involvement of DNA damage response pathways in hepatocellular carcinoma. Biomed Res Int. 2014;2014:153867
39. Lin Z, Xu SH, Wang HQ, Cai YJ, Ying L, Song M. et al. Prognostic value of DNA repair based stratification of hepatocellular carcinoma. Sci Rep. 2016;6:25999
40. Wu JS, Chen YP, Wang LC, Yang YJ, Deng CW, Hou BX. et al. Implication of polymorphisms in DNA repair genes with an increased risk of hepatocellular carcinoma. Genet Mol Res. 2014;13:3812-8
41. Yu L, Liu X, Han C, Lu S, Zhu G, Su H. et al. XRCC1 rs25487 genetic variant and TP53 mutation at codon 249 predict clinical outcomes of hepatitis B virus-related hepatocellular carcinoma after hepatectomy: A cohort study for 10 years' follow up. Hepatol Res. 2016;46:765-74
42. Jung SW, Park NH, Shin JW, Park BR, Kim CJ, Lee JE. et al. Polymorphisms of DNA repair genes in Korean hepatocellular carcinoma patients with chronic hepatitis B: possible implications on survival. J Hepatol. 2012;57:621-7
43. Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc Natl Acad Sci U S A. 2003;100:776-81
44. Lordick F, Janjigian YY. Clinical impact of tumour biology in the management of gastroesophageal cancer. Nat Rev Clin Oncol. 2016;13:348-60
45. Sheu JC, Lin YW, Chou HC, Huang GT, Lee HS, Lin YH. et al. Loss of heterozygosity and microsatellite instability in hepatocellular carcinoma in Taiwan. Br J Cancer. 1999;80:468-76
Author contact
![]() Corresponding authors: Prof. Wen-Hui Su, Department of Biomedical Sciences, Chang Gung University, 259 Wen-Hua 1st Road, Kweishan, Taoyuan 333, Taiwan. Tel: +886-3-2118800 ext. 3564; Fax: +886-3-2118700; E-mail: whsucgu.edu.tw and Prof. Dar-In Tai, Division of Hepatology, Department of Gastroenterology and Hepatology, Chang Gung Memorial Hospital, Linkou, 5 Fu-Hsing Street, Kweishan, Taoyuan 333, Taiwan. Tel: +886-3-3281200 ext. 8101; Fax: +886-3-3272236; E-mail: tai48978cgmh.org.tw
Corresponding authors: Prof. Wen-Hui Su, Department of Biomedical Sciences, Chang Gung University, 259 Wen-Hua 1st Road, Kweishan, Taoyuan 333, Taiwan. Tel: +886-3-2118800 ext. 3564; Fax: +886-3-2118700; E-mail: whsucgu.edu.tw and Prof. Dar-In Tai, Division of Hepatology, Department of Gastroenterology and Hepatology, Chang Gung Memorial Hospital, Linkou, 5 Fu-Hsing Street, Kweishan, Taoyuan 333, Taiwan. Tel: +886-3-3281200 ext. 8101; Fax: +886-3-3272236; E-mail: tai48978cgmh.org.tw
Citation styles
APA
Su, W.H., Jou, Y.S., Zhang, J.H., Ho, C.M., Tai, D.I. (2018). Mutations in NOTCH1 and nucleotide excision repair genes are correlated with prognosis of hepatitis B virus-associated hepatocellular carcinoma. Journal of Cancer, 9(15), 2678-2686. https://doi.org/10.7150/jca.25438.
ACS
Su, W.H.; Jou, Y.S.; Zhang, J.H.; Ho, C.M.; Tai, D.I. Mutations in NOTCH1 and nucleotide excision repair genes are correlated with prognosis of hepatitis B virus-associated hepatocellular carcinoma. J. Cancer 2018, 9 (15), 2678-2686. DOI: 10.7150/jca.25438.
NLM
Su WH, Jou YS, Zhang JH, Ho CM, Tai DI. Mutations in NOTCH1 and nucleotide excision repair genes are correlated with prognosis of hepatitis B virus-associated hepatocellular carcinoma. J Cancer 2018; 9(15):2678-2686. doi:10.7150/jca.25438. https://www.jcancer.org/v09p2678.htm
CSE
Su WH, Jou YS, Zhang JH, Ho CM, Tai DI. 2018. Mutations in NOTCH1 and nucleotide excision repair genes are correlated with prognosis of hepatitis B virus-associated hepatocellular carcinoma. J Cancer. 9(15):2678-2686.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.